Experimental model and hemodynamic measurements
Experiments were performed on adult male C57BL/6 mice at an age of 2 months and an average weight of 21.6±1.3 g (Charles River, Sulzfeld, Germany). Animals were kept in individually ventilated cages in a room with a 12 h light/dark cycle and maintained at 20±1°C, with free access to standard mouse chow and water. Vehicle or PTHrP(107-111) (Fa. Bachem, Bubendorf, Switzerland) was administered via implanted osmotic minipumps (Alzet 1002, Alzet Osmotic Pumps, Cupertino, CA, USA) which released their contents in a controlled way over the next 14 days. Initial dose-finding experiments were performed with 25 µg and 50 µg of the peptide. No effects were seen with 25 µg. Therefore, all subsequently used pumps were filled with either 50 µg PTHrP(107-111) in 100 µl isotonic NaCl solution which was released at a rate of 125 ng/h (treatment group, 6.25 µg/kg) or vehicle (placebo). The concentration corresponds to that used in another in vivo study (8). Implantation of osmotic minipumps filled with an equal volume of NaCl was used in control (placebo). Pressure overload was induced by transverse aortic constriction (TAC), as previously described in detail (9). Briefly, mice were anesthetized with isoflurane (2.5% vol/vol; Forene®, Abott GmbH, Wiesbaden, Germany), intubated and ventilated with the suitable parameters. After opening the thorax a suture was passed underneath the aorta and tied down on a 27G needle to achieve a standardised decreased diameter of the aorta.
Animals were evaluated after 14 days under anaesthesia. Preparation was performed under isoflurane (2.5% vol/vol), and recording under isoflurane 1% (vol/vol). Haemodynamic parameters were recorded with a Millar pressure-transducer catheter (Millar Instruments, Houston, TX, USA) connected to a computerized data acquisition system (Lower Lab., ADI Instruments, Melbourne , Australia) as described previously in greater detail (9). At first, ECG electrodes were placed at the extremities and ECG recordings were obtained for 10 min. Thereafter, a Millar-catheter was introduced into the right carotid artery and then moved forward to a position 4 mm in front of the aortic valve to record peripheral blood pressure. Finally, it was advanced into the left ventricle to assess ventricular function. After haemodynamic measurements the animals were sacrificed by exsanguinations, the hearts were excised and all external fluids were completely removed before weighing. The atria were then removed, separated into right and left atria and together with the left ventricle plus septum all tissue samples were frozen prior to further analysis.
The animals were handled according to the principles of laboratory animal care (NIH publication No. 86-23, revised 1985) and the animal protection law stated in German Civil Code. The experiments were approved by the district government.
Quantification of mRNA expression
The left ventricles were homogenized and RNA was extracted according to the manufacturer's protocol to obtain total cellular RNA as described previously (10). Aliquots (1 µg) were used for real-time polymerase chain reactions (PCRs) using the I-cycler (Biorad, München, Germany) and SYBR-green as the fluorescence signal. The expression of TGF-ß1, ODC, and ANF were normalized to ß-actin, as a housekeeping gene used for loading control. The primers used in this study have been reported previously (10, 11) except for connexin 40, -43, and -45. These primers had the following sequence: connexin 40 forward: CCT GGC TGA ACT CTA CC; reverse: ATT ACT GAA GTC GCT GAA G; connexin 43 forward: GGT GGT GTC CTT GGT GTC TC; reverse: CGG TGG TGG CGT GGT AAG; connexin 45 forward: ACA CCC TCT GCT CCC CCT GG; reverse: CCT CGT GGC TGC CGT ACT GC. Relative changes in gene expression from real-time RT-PCR were calculated by the 2-
Immunoblotting
To determine protein expression of connexin 43 and its phosphorylation, ventricles were homogenized and transferred to a lysis buffer (composition in mmol/l: cacodylate 10, NaCl 150, CaCl2 20, sodium acetate 1.5, ZnCl2 0.001, Triton X100 (0.01% vol/vol), pH 5.0). After centrifugation the supernatant was diluted with Laemmli buffer. Sodium dodecyl sulphate (SDS-gel) electrophoresis was performed as described before (13). Proteins were separated by a 10% (w/v) SDS-polyacrylamine gel electrophoresis (acrylamide:bisacrylamide 30:1). After SDS-gel electrophoresis, proteins were transferred onto reinforced nitrocellulose sheets by semi-dry blotting. The sheets were saturated with 2% (wt/vol) bovine serum albumine (BSA) and incubated for 2 h with 0.2 µg/ml primary antibody (anti phosphorylated connexin 43 (ser 368); NEB #3511 (Herts; United Kingdom); anti connexin 43 Zymed Lab #71-0700; Invitrogen, Karlsruhe, Germany). After sheets were washed with phosphatase-labeled goat anti-rabbit IgG antibodies were added for another 2 h. Bands were visualized by alkaline phosphatase activity using 5-bromo-4-chloro-3-indolyl phosphate and nitro blue tetrazolium.
Statistical analysis
Data are presented as means ±S.E. from n experiments. Analysis of Variance was performed, followed by Tukey post hoc analysis when appropriate. Student's t-test was performed in cases where only two groups were compared. A p value <0.05 was considered statistically significant. The statistical analysis was performed via SPSS version 17.0.
Effect of osteostatin on cardiac hypertrophy and function in vivo
In order to demonstrate possible effects of osteostatin on cardiac function or myocardial hypertrophy in vivo, mice received either osteostatin (treatment group) or saline (placebo group) via osmotic minipumps for 14 days. Mice used in this study did not differ in regard to body weight at the beginning of the study (Table 1). After 14 days, there was a slight increase in body weight in both groups. There were no group differences in body weight-to-tibia length, heart-to-body weight, lung weight-to-tibia length, or lung-to body weight, too (Table 1). However, left ventricular mRNA expression of ANF was moderately but significantly higher in osteostatin treated mice (+29%, p=0.033; Table 1). On the functional level osteostatin did not significantly modify any of the parameters under investigation but osteostatin significantly shortened P-wave duration by 9.3% (Table 1, Fig. 1). No other parameter investigated by ECG recording, such as heart rate, amplitudes of P wave, QRS complex, T wave, and QRS duration was altered.
| Table 1. In vivo
effect of osteostatin in mice. Data are means ±S.E. BW0= body weight at day 0; BW14= body weight at day 14; TL= tibia length; HW= heart weight; LW= lung weight; LVSP= left ventricular systolic pressure; LVDP= left ventricular diastolic pressure; HR= heart rate; *, p<0.05 vs. placebo (Tukey-Test). |
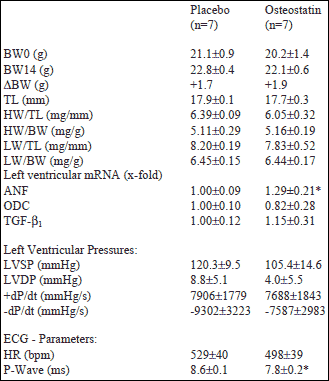 |
Effect of osteostatin on cardiac hypertrophy and function in pressure overloaded hearts
Next, we investigated the effect of osteostatin on functional and hypertrophic adaptations in hearts with chronic pressure overload. Placebo and treatment groups did not differ in body weight and tibia length at the beginning of the experiments. However, after 14 days TAC/placebo mice had lost 4.5 g/body weight, whereas the treatment group lost only 1.1 g (Table 2). Furthermore, osteostatin-treated mice had an increased right ventricular weight-to-tibia length (Table 2). Left ventricular mRNA expression of ANF, ODC, and TGF-ß was not different between TAC/placebo and TAC/osteostatin but ANF (placebo: +9.7±4.5-fold; osteostatin: +10.4±3.9-fold; each p<0.05 vs. shams) and ODC (placebo: 250.8±25.8-fold; osteostatin: 227.6±23.8-fold; each p<0.05 vs. shams) were strongly up-regulated compared to hearts without pressure overload (shams). On the functional level, no significant differences did occur between placebo and osteostatin-treated TAC mice. However, in the TAC/osteostatin group there was a trend to higher mean systolic pressures and a better relaxation index (-dP/dt). Of note, left ventricular end-diastolic pressures, +dP/dt, and -dP/dt were all significantly different from shams but no treatment-dependent effect was noticed. On ECG recordings, osteostatin-treated hearts had again lower P-wave durations (-23.1%, Table 2, Fig. 1).
| Table 2. Effects
of osteostatin on pressure overloaded hearts. Data are means±S.E. BW0= body weight at day 0; BW14= body weight at day 14; TL= tibia length; HW= heart weight; LW= lung weight; LVSP= left ventricular systolic pressure; LVDP= left ventricular diastolic pressure; HR= heart rate; *, p<0.05 vs. placebo (Tukey-Test). |
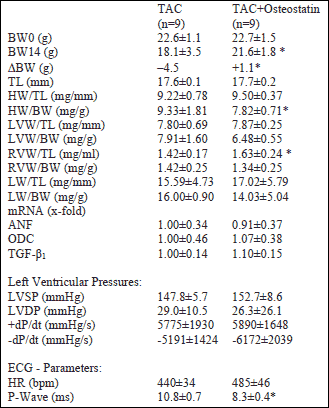 |
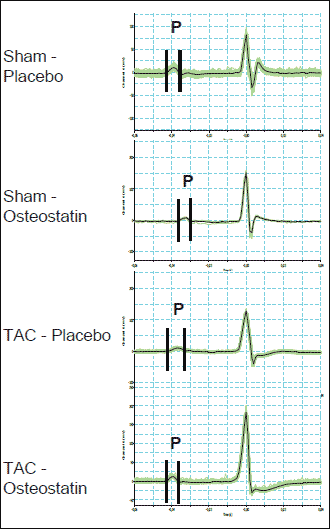 |
Fig. 1. Original registration of CG recordings showing the duration of P waves (P) in sham-placebo, sham-osteostatin, TAC-placebo, and TAC-osteostatin mice. Average P wave durations are given in Table 1 and 2. |
Effect of osteostatin on connexin expression and phosphorylation
Of note, osteostatin decreased P wave duration in non-banded mice and banded mice by 9.3% and 23.1%, respectively. Since shortening of P wave duration was the only effect caused by osteostatin in both experimental series, sham and TAC, we concluded that the effect of osteostatin on conduction acceleration in the atria is the most important in vivo effect of the peptide. As P-wave duration depends on connexin 40 expression (14, 15) but conduction velocity also depends on connexin 43 phophorylation (16), we finally investigated these parameters in mice. As indicated in Fig. 2, osteostatin-treated mice displayed lower mRNA expression of connexin 43 and -40, but unaltered expression of connexin 45. However, in osteostatin-treated mice phosphorylation of connexin 43 was robust whereas it was nearly undetectable in TAC mice without osteostatin (Fig. 3). No significant effect was obtained with respect to connexin 43 protein expression.
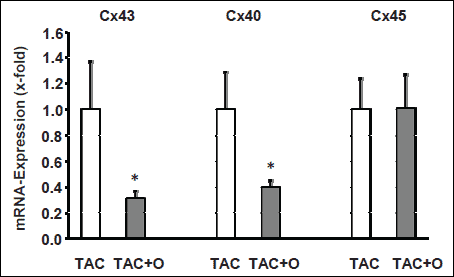 |
Fig. 2. mRNA expression of different connexins (Cx) in the right atria of mice. Data are normalized to TAC banded mice without administration of osteostatin. Data are means ±S.E. from n=4-6 samples. * p<0.05 vs. TAC. |
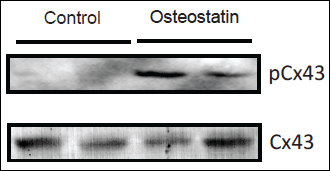 |
Fig. 3. Immunoblot indicating phosphorylation of connexin 43 in right atria of TAC-banded mice without treatment or with osteostatin treatment. Upper panel) anti phosphoconnexin 43 (pCx43); lower panel) anti connexin 43 (Cx43). |
This is the first study that investigated cardiac effects of osteostatin in vivo. The main results of this study are first, that osteostatin moderately induces cardiac hypertrophy under specific conditions and second, that it accelerates the atrial electrical propagation as indicated by a reduction of P-wave duration. The most likely mechanism by which osteostatin affects atrial conduction is the preserved PKC-dependent phosphorylation of connexin 43 in TAC-banded mice. Overall, these moderate effects on cardiac parameters in vivo are sufficient to attenuate the loss of body weight in mice exposed to chronic pressure overload.
In vitro studies have been used to identify signal transduction pathways that are induced by osteostatin and to identify physiological responsiveness of cells to osteostatin. The pentapeptide 107-111 represents an essential PKC-activating part in C-terminal fragments of PTHrP (5, 7). We have recently shown that the pentapeptide is sufficient to increase protein synthesis in cardiomyocytes and to induce a fetal type re-expression of proteins in a PKC-dependent way (7). However, in vivo studies using osteostatin have rarely been performed. If so, they did not analyze cardiac specific effects. A previous study on ovariectomized rats treated with osteostatin showed that the pentapeptide is sufficient to antagonize bone-loss that normally occurs in ovariectomized rats (8). Thus it is likely that the pentapeptide is able to exert in vivo effects. This study is the first one that investigates effects of osteostatin on cardiac function in vivo. Based on our previous in vitro study, we expected that osteostatin independently of other stimuli directly induces cardiac hypertrophy. In the in vivo study performed here, we were unable to find a similar pro-hypertrophic effect in vivo. However, in non pressure-overloaded hearts osteostatin significantly increased ventricular expression of ANF, a peptide strongly associated with cardiac hypertrophy rather than with hypertension (17) and representing the induction of a fetal type protein expression. Furthermore, in TAC banded mice there was a pro-hypertrophic response in right ventricles, which do not become overloaded by the procedure of aortic banding. Collectively, these data argue that osteostatin may modify hypertrophic responses in vivo but it is certainly not a major factor contributing to hypertrophic growth. As indicated in this study, osteostatin does not modify the hypertrophic responsiveness to pressure overload. One may argue that the observed right ventricular hypertrophy is not directly induced by osteostatin but rather indirectly by an increased afterload in these mice due to the pressure overload of the left ventricles. However, it is unlikely that this explanation is true because the corresponding increase of left ventricular end-diastolic pressure is not different between TAC-sham and TAC-osteostatin mice and the lung wet weight, another marker of left heart failure leading to increased afterload for the right ventricle is slightly lower in TAC-osteostatin mice compared with TAC-placebo mice. Therefore, direct effects caused by osteostatin are more likely to induce right heart hypertrophy under such conditions instead of hemodynamic differences between the two groups.
Interestingly, osteostatin caused small effects on cardiac functional parameters such as a trend to higher -dP/dt values in TAC banded mice. These findings indicate again a possible modifier role for osteostatin. Of note, the small effects on hypertrophy and cardiac function were sufficient to lower the loss of body weight in TAC banded mice, a parameter clearly associated with a general loss of health status in small rodents.
The most robust and reproducible effect of osteostatin in vivo was the shortening of P-wave duration. In TAC banded mice a reduction of connexin 43 phosphorylation has recently been shown (16, 18) and interpreted as an indication of gap junction remodelling (GJR). Phosphorylation of connexin 43 is required for proper synthesis and assembly of connexins into gap junctions (19, 20). Here we confirmed the non-phosphorylated status of connexin 43 in the atria of TAC-banded mice. Phosphorylation of connexin 43 depends on PKC-activation, mainly by PKC-
PTHrP is target of proteolysis in vivo leading to the formation of C-terminal fragments that occur in plasma. These peptides are released by glomerular filtration. Therefore the concentration of such peptides is elevated in patients with a significant impairment of the glomerular filtration rate (
In conclusion this is the first study that demonstrates cardiovascular in vivo effects of PTHrP(107-111) (=osteostatin). Although the changes evoked by the pentapeptide were only moderate the overall effects are arguing for a protective role of C-terminal peptides covering the amino acids 107-111. Future work is required to address the point under which conditions osteostatin antagonizes the progression of heart failure.
Conflict of interest: None declared.
- Arakelyan KP, Sahakyan YA, Hayrapetyan LR, et al. Calcium-regulating peptide hormones and blood electrolyte balance in chronic heart failure. Regul Pept 2007; 142: 95-100.
- Budyar AA, Nissenson RA, Klein RF, et al. Increased serum levels of parathyroid hormone-like protein in malignancy-associated hypercalcaemia. Ann Int Med 1989; 111: 807-812.
- Burtis WJ, Brady TG, Orloff JJ, et al. Immunochemical characterization of circulating parathyroid hormone-related protein in patients with humoral hypercalcaemia of cancer. N Engl J Med 1990; 322: 1106-1112.
- Burtis WJ, Fordero JP, Gaich G, Debessey M, Stewart AF. Preliminary characterization of circulating amino- and carboxy-terminal fragments of parathyroid hormone-related peptide in human hypercalcemia of malignancy. J Clin Endocrinol Metab 1992; 75: 1110-1114.
- Gagnon L, Jouishomme H, Whitfield JF, et al. Protein kinase C-activating domains of PTH-rP. J Bone Miner Res 1993; 8: 497-503.
- Fenton AJ, Kemp BE, Kent GN, et al. A carboxy-terminal peptide from the parathyroid hormone-related protein inhibits bone resorption by osteoclasts. Endocrinology 1991; 129: 3424-3426.
- Schluter K-D, Weber M, Piper HM. Effects of PTH-rP(107-111) and PTH-rP(7-34) on adult cardiomyocytes. J Mol Cell Cardiol 1997; 29: 3057-3065.
- Rouffet J, Coxam V, Gaumet N, Barlet JP. Preserved bone mass in ovariectomized rats treated with parathyroid-hormone-related peptide (1-34) and (107-111) fragments. Reprod Nutr Dev 1994; 34: 473-481.
- Baumgarten G, Kim S-E, Stapel H, et al. Myocardial injury modulates the innate immune system and primes for myocardial LPS responsiveness. Basic Res Cardiol 2006; 101: 427-435.
- Van Eickels M, Schreckenberg R, Doevendans PA, Meyer R, Grohe C, Schlüter K-D. The influence of oestrogen-deficiency and ACE inhibition on the progression of myocardial hypertrophy in spontaneously hypertensive rats. Eur J Heart Failure 2005; 7: 1079-1084.
- Wenzel S, Schorr K, Degenhardt H, et al. TGF-b1 downregulates PTHrP in coronary endothelial cells. J Mol Cell Cardiol 2001; 33: 1181-1190.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta C(T)). Methods 2001; 25: 402-408.
- Schlüter K-D, Katzer C, Frischkopf K, Wenzel S, Taimor G, Piper HM. Expression, release, and biological activity of PTHrP from coronary endothelial cells. Circ Res 2000; 61: 307-316.
- Almeida NAS. Cordeiro A, Machado DS, et al. Cionnexin 40 messenger ribonucleic acid is posititively regulated by thyroid hormone (TH) acting in cardiac atria via tha TH receptor. Endocrinology 2009; 150: 546-554.
- Bagwe S, Berwenfeld O, Vaidya D, Morley GE, Jalife J. Altered right atrial excitation and propagation in connexin 40 knockout mice. Circulation 2005; 112: 2245-2253.
- Qu J, Volpicelli FM, Garcia LI, et al. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ Res 2009; 104: 365-371 .
- Louhelainen M, Merasto S, Finckenberg P, et al. Effects of calcium sensitizer OR-1986 on cardiovascular mortality and myocardial remodelling in hypertensive Dahl/RAPP rats. J Physiol Pharmacol 2009; 60: 41-47.
- de Diego C, Pai KP, Chen F, et al. Electrophysiological consequences of acute regional ischemia/reperfusion in neonatal rat ventricular myocyte monolayers. Circulation 2008; 118: 2330-2337.
- Laird DW, Castillo M, Kasprzak L. Gap junction turnover, intracellular trafficking, and phosphorylation of connexin 43 in brefeldin A-treated rat mammary tumor cells. J Cell Biol 1995; 131: 1193-1203.
- Musil LS, Goodenough DA. Biochemical analysis of connexin 43 intracellular transport, phosphorylation, and assembly into gap junctional plaques. J Cell Biol 1991; 115: 1050-1057.
- Bowling N, Huang X-D, Sandusky GE, et al. Protein kinase C-a and -e modulate connexin 43 phosphorylation in human heart. J Mol Cell Cardiol 2001; 33: 789-798.
- Doble BW, Ping P, Kardami E. The e subtype of protein kinase C is required for cardiomyocyte connexin 43 phosphorylation. Circ Res 2000; 86: 293-301.
- Thomas SA, Schuessler RB, Berul CI, et al. Disparate effects of deficient expression of connexin 43 on atrial and ventricular conduction: Evidence for chamber-specific molecular determinants of conduction. Circulation 1998; 97: 686-691.
- Lidington D, Ouellette Y, Tyml K. Endotoxin increases intercellular resistance in microvascular endothelial cells by a tyrosine kinase pathway. J Cell Physiol 2000; 185: 117-125.
- Beauchamp P, Yamada KA, Baertschi AJ, et al. Relative contribution of connexion 40 and 43 to atrial impulse propagation in synthetic strands of neonatal and fetal murine cardiomyocytes. Circ Res 2006; 99: 1216-1224.
- Karagaratnam P, Rothery S, Patel P, Severs NJ, Peters NS. Relative expression of immunolocalized connexins -40 and -43 correlates with human atrial conduction properties. J Am Coll Cardiol 2002; 39: 116-123.
- Orloff JJ, Soifer NE, Fodero JP, Dann P, Burtis WJ. Accumulation of carboxy-terminal fragments of parathyroid hormone-related protein in renal failure. Kidney Int 1993; 43: 1371-1376.
- Meyer R, Schreckenberg R, Kretschmer F, et al. Parathyroid hormone-related protein (PTHrP) signal cascade modulates myocardial dysfunction in the pressure overloaded heart. Eur J Heart Failure 2007; 9: 1156-1162.
- Rybaczynska A, Jurska-Jasko A, Boblewski K, Lehmann A, Orlewska C. Blockade of calcium channels and AT1 receptors prevents the hypertensive effect of calcilytic NPS 2143 in rats. J Physiol Pharmacol 2010; 61: 163-170.