Recently, it was shown that the renin-angiotensin system (RAS) is a crucial component of atherosclerotic process and angiotensin II (AT II) promotes atherogenesis (5). On the other hand, angiotensin-(1-7) [AT-(1-7)] opposes AT II action (6).
Here, using apolipoprotein E (apoE)-knockout mice model of atherosclerosis we investigate, whether angiotensin-(1-7) receptor agonist: AVE 0991 (Fig. 2A), is able to ameliorate progression of atherosclerosis. Since our preliminary results, (concerning only morphological comparison between AVE 0991 and control - treated atherogenic mice) seemed plausible (7), we expanded the study on investigation of molecular mechanisms of AT-(1-7) action.
Animals
Fifty female apoE-knockout mice on the C57BL/6J background were obtained from Taconic (Ejby, Denmark) (8). Mice were maintained on 12-h dark/12-h light cycles in air-conditioned rooms (22.5±0.5°C, 50±5% humidity) and access to diet and water ad libitum. All animal procedures were approved by the Jagiellonian University Ethical Committee on Animal Experiments (approval nr Zi/496/2009).
Protocol and atherosclerosis studies
At the age of 8 weeks mice were put on chow diet made by Ssniff (Soest, Germany) for 4 months. Four experimental groups (in each n=10) received the same diet as a control group, mixed with: AVE 0991 (kind gift from Sanofi-Aventis Deutschland GmbH, Frankfurt am Main, Germany) at a dose 0.58 µmol per kg of body weight per day or perindopril (Sigma-Aldrich Poland, Poznan) at a dose 0.4 mg per kg of body weight per day, as well as tiorphan (Sigma-Aldrich Poland, Poznan) at a dose 2.5 mg per kg of body weight per day. In contrast, A-779 [(D-alanin7)-angiotensin-(1-7)] (Bachem AG, Bubendorf, Switzerland) - AT-(1-7) antagonist, was given three times weekly intraperitoneally at a dose of 3.3 mg/kg body weight to the last group.
At the age of 6 months 1000 UI of fraxiparine (Sanofi-Synthelabo, France) was injected i.p. and after 10 min mice were sacrificed in chamber filled with carbon dioxide. The blood was collected from the right ventricle. Plasma was separated by centrifugation at 1000 g at 4°C for 10 min and stored in -80°C. Then, right atrium was incised and the heart was perfused with PBS through the apex of the left ventricle at a constant pressure of 100 mm Hg. Next, the heart and the whole aorta were dissected.
Total cholesterol and triglycerides were assayed using commercially available kits (Roche Molecular Biochemical, USA).
The heart and ascending aorta were embedded in OCT compound (CellPath, UK) and snap-frozen. Ten micrometer-thick cryosections were cut from the aortic root using a standardized protocol (9, 10).
Serial sections were cut from the proximal 1 mm of the aortic root. Eight adjacent sections were collected at 100 µm intervals starting at a 100 µm distance from the appearance of the aortic valves. Sections were thaw-mounted on poly-L-lysine coated slides and air dried. After fixation in 4% paraformaldehyde (pH=7), sections were stained with Meyer's hematoxylin and oil red-O (Sigma-Aldrich, USA). Oil red O-stained sections were examined under Olympus BX50 (Olympus, Tokyo, Japan) microscope and used for quantitative evaluation. Images of the aorta were recorded using Olympus Camedia 5050 digital camera and stored as TIFF files of resolution 1024 768 pixels. Total area of the lesion was measured semiautomatically in each slide using LSM Image Browser 3 software (Zeiss, Jena, Germany). For each animal a mean lesion area was calculated from eight sections, reflecting the cross-section area covered by atherosclerosis.
The aorta from arch to bifurcation was fixed in 4% formaldehyde, opened longitudinally, pinned onto black wax plates and stained with Sudan IV (Sigma-Aldrich, St. Louis, MO, USA). Aortic lesion area and total aortic area were calculated using LSM Image Browser software (11, 12).
For indirect immunohistochemistry acetone-fixed sections of ascending aorta were used. Sections were preincubated overnight with 5% of non-immunogenic goat serum with 2% of fat-free milk to block nonspecific binding of antibodies. Incubations with primary antibodies were performed overnight at room temperature in wet chambers in the following combination of sera: Cy3-conjugated anti a-smooth-muscle actin (SMA) (Sigma-Aldrich, St. Louis, MO, USA) (dilution 1:600) and rat anti-mouse CD68 (Serotec, Oxford, UK) (dilution 1:800). Goat anti-rat IgG biotinylated antibodies followed by DTAF-conjugated streptavidin (Jackson IR, West Grove, PA), were applied to visualize rat antibodies. Sections were examined using an epifluorescence Olympus BX50 microscope equipped with appropriate filter cubes to show Cy3 (red) and DTAF (green) fluorescence. Images were registered with a Camedia 5050 digital camera.
In each section, total area with CD68-immunopositive macrophages as well as
Plasma levels of interleukin-6 (IL-6), interleukin-12 (IL-12) and serum amyloid A (SAA) (all from R&D Systems, Minneapolis, MN, USA) as well as macrophage chemotactic protein-1 (MCP-1) (BioSource, Camarillo, CA, USA) were measured using ELISA test.
Real time RT-PCR
Total RNA isolation and cDNA synthesis
Total RNA was isolated from the homogenized mouse aortas using QIAzol® Lysis Reagent (QIAGEN, USA) according to manufacturer's instructions. Extracted RNA was dissolved in 15 µl of nuclease-free water (Fermentas, Canada). The RNA concentration of each sample was measured at a wavelength of 260 nm (A260) in an EPOCH microplate spectrophotometer (BioTek Instruments Inc., USA). The purity of extracted total RNA was determined by the A260/A280 ratio. The integrity of RNA samples was confirmed by denaturing agarose gel electrophoresis.
cDNA was synthesized by the reverse transcription of 950 ng of total RNA from each sample using 0.5 µl oligo(dT)18 primer (0.5 µg/µl) (Fermentas, Canada) and 0.4 µl M-MuLV reverse transcriptase (200 U/µl) (Fermentas, Canada) in a total volume of 10 µl [including also: 2 µl 5×reaction buffer for M-MuLV RT (containing 250 mM Tris -HCl pH 8.3 at 25°C, 250 mM KCl, 20 mM MgCl2, 50 mM DTT), 0.5 µl dNTPs mixture (10 mM), 0.3 µl RiboLock RNAse Inhibitor (40 U/µl) and nuclease-free water (Fermentas, Canada)]. The reaction was performed in T3 Thermocycler (Biometra, Germany) at 42°C during 1 h, and the enzyme was then denatured at 95 C for 5 min. The cDNA was diluted ten-fold prior to PCR amplification.
Quantitative real time PCR
Relative gene expression quantitation for p22phox in aortas with ß-actin as internal reference gene, was carried out using 7900HT fast real-rime PCR System (Applied Biosystems; USA) in triplicate. Primers were designed with the Primer BLAST software (http://www.ncbi.nlm.nih.gov/tools/primer-blast), taking into account primer dimer, self-priming formation, and primer melting temperature (Table 1).
| Table 1. Primer sequences used for quantitative real-time polymerase chain reaction. |
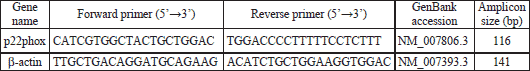 |
All reactions were performed using the SYBR® Green JumpStart Taq ReadyMix for quantitative PCR (Sigma, USA) in a total volume of 15 µl. Each reaction mixture contained 7.5 µl 2x SYBR Green JumpStart Taq Ready Mix (including in final concentration: 1.25 units Taq DNA polymerase, 10 mM Tris-HCl, 50 mM KCl, 3.5 mM MgCl2, 0.2 mm dNTP, and SYBR Green I), 0.75 µl 0.5 µM each primer (Table 1), and 2 µl diluted cDNA.
After 10 minutes at 95°C for activation of the Taq polymerase, the amplification program consisting of the following parameters: 30 s at 95°C, 60 s at 63°C, and 45 s at 72°C was performed for 40 cycles. The amplification was followed by a melting curve analysis to control the PCR products. As negative controls water instead of cDNA was run with every PCR experiment. To verify the accuracy of the amplification, PCR products were further analyzed by gel electrophoresis. Analysis of the data was performed using RQ Manager 1.2.1 software (Applied Biosystems, USA). The data were evaluated by the 2-CT
Flow cytometry
Cells
Spleens were cut into small pieces and gently rubbed through a 40 µm nylon cell strainers (BD Biosciencies, USA) into RPMI-1460 (Invitrogen, USA), containing 10% FBS (Invitrogen, USA). Then cells were washed twice with RPMI-1640 containing 1% FBS, suspended in RPMI-1640 containing 10% FBS, and counted (16).
Flow cytometry staining
Cells were washed with cold PBS containing 2% FCS and 0.05% sodium azide (FACS buffer) and incubated 40 min on ice (in the dark) with following monoclonal antibodies: FITC-conjugated rat anti-mouse CD4, APC-conjugated rat anti-mouse CD11c, PE-conjugated rat anti-mouse CD86, FITC-conjugated rat anti-mouse CD80, biotin-conjugated rat anti-mouse CD69, biotin-conjugated rat anti-mouse CD40 (all from BD Pharmingen), APC-conjugated anti-mouse F4/80 (from eBiosciences). In case of biotin conjugated antibodies incubation with PE-conjugated streptavidin (BD Pharmingen) followed for 30 min, on ice, in the dark. Unspecific binging of antibodies was blocked with anti-mouse CD16/32 monoclonal antibody (BD Pharmingen). Cells were washed, suspended in cold PBS containing 2% FCS and 0.05% sodium azide and analyzed on a FACSCanto II flow cytometer (BD, USA) using FACSDiva software (17).
Statistical analyses
Results are expressed as mean ±S.E.M. The nonparametric Mann-Whitney U test was used for statistical analysis of the data. P<0.05 was considered as statistically significant.
Lipid levels
None of the drugs changed the level of cholesterol and triglycerides in blood, as compared to the control group (Table 2).
| Table 2. The level
of total cholesterol and triglycerides in mice plasma (n=10 in each group). NS - lack of statistical significance in comparison with control |
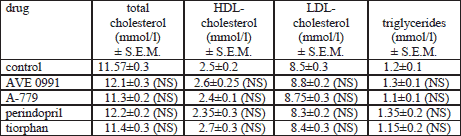 |
Comparison of atherosclerosis development
Aortas differed in the degree of atherosclerosis between control group and experimental groups. Measured by "en face" method, the percentage of occupied by Sudan IV-stained surfaces were: 14.2±1.9% in control group, whereas in AVE 0991 treated group 7.25±1.3%, in perindopril group 2.6±0.5%, in tiorphan group 16.7±2.8%, and in A-779 group 19.2±0.6% (Figs. 1A, 1B). All the differences, except for tiorphan, were statistically significant.
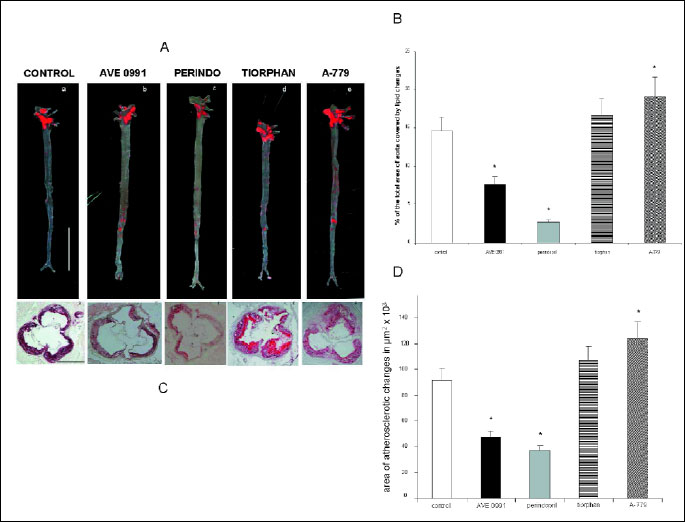 |
| Fig.
1. (A) Stained by Sudan IV aortas "en face"
from groups: control and drug-treated apoE-knockout mice. (a)- control,
(b)- AVE 0991, (c)- perindopril, (d)- tiorphan, (e)- A-779; Line on picture
"a" stands for 1 cm. (B) Area of lipid changes in aorta (Expressed as a percent of total area of the aorta) in apoE-knockout mice from control group and drug-treated groups (n=10 in each group). Results are expressed as a mean ±S.E.M. *p<0.05, comparing to the control group. (C) Representative microphotographs, showing oil red-O stained atherosclerotic changes in apoE - knockout mice, in control group and drug-treated groups (magnification×80). (a) control, (b) AVE 0991, (c) perindopril, (d) tiorphan, (e) A-779. Line on picture "a" stands for 500 µm. (D) Area of atherosclerotic changes in "aortic root", expressed in µm2, in oil red-O - stained 6-months-old apoE - knockout mice, from control and drug-treated groups (n=10 in each group). Results are expressed as a mean ± SEM. *p<0.05, comparing to the control group. |
"Cross-section" of aortic roots revealed the difference in atherosclerotic lesions. Measured in 8 consecutive sections mean surfaces areas ±S.E.M., occupied by oil red - O-stained changes were: 91.213±8.123 µm2 in control group versus 46.968±7.215 µm2 in AVE 0991-treated group, 37.107±2.824 µm2 in perindopril group, 107.599±9.735 µm2 in tiorphan group, and 124.201±10.373 µm2 in A-779 group (Fig. 1C, 1D). All the differences, except for tiorphan, were statistically significant.
Perindopril to a higher degree than AVE 0991 decreased atherosclerosis. Angiotensin converting enzyme inhibitors for a long time are well known very strong inhibitors of experimental atherosclerosis (18-23). It can be explained by the fact that the direct inhibition of AT II is stronger than "indirect" action of AT-(1-7), therefore the effect of perindopril is greater than AVE. Group receiving perindopril was a "positive control", since the low dose of this angiotensin converting enzyme inhibitor shows positive endothelial and anti-atherosclerotic action, at the same time not causing the fall of the blood pressure. Tiorphan neutral endopeptidase (NEP) inhibitor tends to increase atherosclerosis. NEP is the main enzyme converting AT I as well as AT-(1-9) into AT-(1-7). That is why inhibition of this enzyme blocks generation of AT-(1-7). Moreover, AVE peptide antagonist, A-779, statistically significantly increased atherogenesis, as compared to control.
Immunohistochemistry
Our results indicate that AVE 0991 may increase plaque stability by decreasing number of macrophages and increasing smooth muscle cells plaque content (Fig. 2B, Table 3).
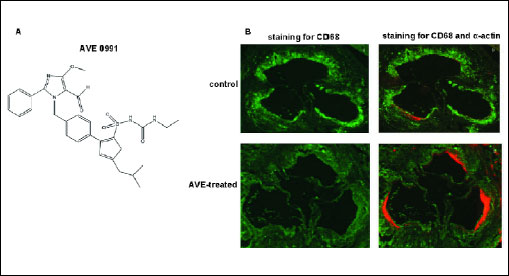 |
Fig. 2. (A) Chemical structure of AVE 0991. (B) Composition of atherosclerotic plaque in 6-months-old mice, treated by AVE 0991 (c and d), in comparison to control group (a and b). Representative microphotographs showing atherosclerotic changes in apoE-knockout mice in control group and in group treated by AVE 0991. Immunohistochemical staining for a-actin of smooth muscle cells (SMA) (in orange) and for macrophages marker CD68 (in green) (magnification 80). a and c - staining for CD68; b and d - connected staining for CD68 and a-actin. |
| Table 3. Composition of atherosclerotic plaque in 6-months-old mice, treated with AVE 0991 in comparison to the control group. |
 |
Inflammatory mediators in plasma
All inflammatory indicators: MCP-1, IL-6, IL-12 and SAA were diminished by AVE 0991 (as well as by perindopril) (Fig. 3).
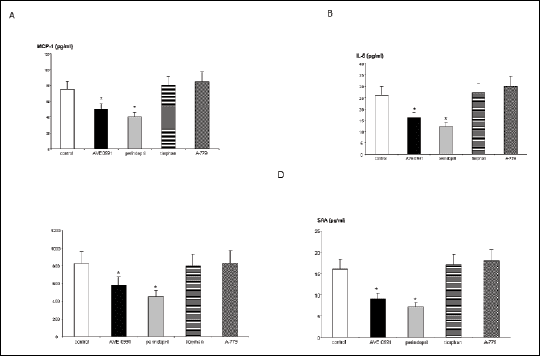 |
Fig. 3. The plasma level of MCP-1, IL-6, IL-12 and SAA. *p<0.05, comparing to the control group. |
Real time RT-PCR
Relative p22phox expression was significantly decreased in AVE-treated mice, whereas in A-779-treated mice it showed tendency to increase, as compared to control (Fig. 3). It means that AVE significantly inhibits NADPH oxidase expression. This enzyme is crucial in atherogenesis (24).
Flow cytometry
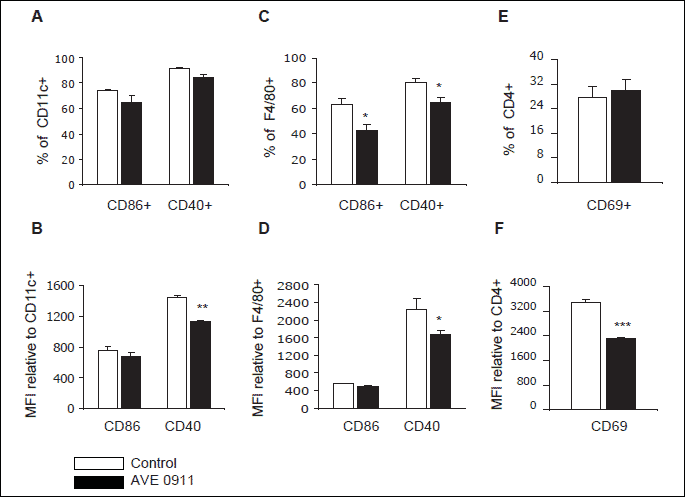
The population of spleen macrophages (F4/80+) expressing co-stimulatory molecule CD86 or CD40 were significantly reduced in number after AVE 0911 treatment (Fig. 4B). Similar tendency was observed in the population of spleen dendritic cells (CD11c+) (Fig. 4A). Moreover, the expression of these molecules, especially CD40, on both dendritic cells (Fig. 4B) and macrophages (Fig. 4D) was significantly decreased. CD86/CD80 molecules are indispensable in the process of antigen presentation to activate T cell. The study on CD80/CD86/low-density lipoprotein receptor - deficient mice showed that the ability of T cells to influence atherosclerosis depends on CD80/CD86 co-stimulation (25). Indeed, in mice treated with AVE 0991 the CD69 activation marker on spleen CD4+ T cells was greatly down-regulated (Fig. 4F) though the percentage of CD4+CD69+ cells was comparable to the control (Fig. 4E). The crucial role of CD40 molecule and its interaction with CD40L in pathogenesis of atherosclerosis has been also well documented (26). Therefore, our results suggest that AVE 0991 may ameliorate atherosclerosis via modulation of the immune cells activity/function.
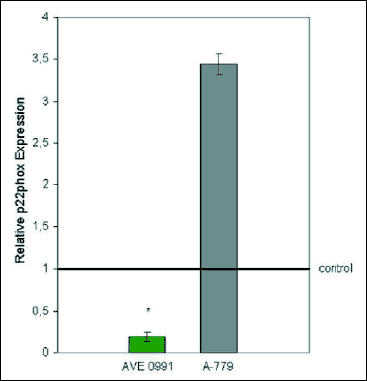 |
Fig. 4. Flow cytometric analysis of spleen dendritic cells (CD11c+gated), macrophages (F4/80+gated) and T cells (CD4+gated) from control (white bars) and AVE 0911 treated (black bars) mice. The percentage of CD40+ and CD86+ cells among dendritic cells (A) and macrophages (C) and the percentage of CD69+ cells among CD4+ T cells (E) is shown as a mean ± S.E.M. of three samples. The expression of CD40 and CD86 on dendritic cells (B) and macrophages (D) and the expression of CD69 on CD4+ T cells (F) is shown as a mean of median fluorescence (PE) intensity (MFI) ±S.E.M. of three samples. *p<0.05, **p<0.01, ***p<0.001 comparing to the control group. |
The renin-angiotensin system (RAS) is highly complicated hormonal system controlling cardiovascular system, kidney and adrenal glands, thus crucial for hydro-electrolyte balance and blood pressure regulation. AT II is the best described peptide of RAS. AT II increases activity of sympathetic nervous system, acts as a vasoconstrictor, increases aldosterone release and sodium retention. Additionally, AT II stimulates free radical production, plasminogen activator inhibitor-1 (PAI-1) release, tissue factor (TF) and adhesion molecules (VCAM-1) expression. Moreover, in blood vessels it stimulates smooth muscle cells proliferation and leukocyte adhesion. What is important, AT II inhibits nitric oxide synthase (NOS), thus diminishing all beneficial effects of nitric oxide (NO).
AT II acts by several potential mechanisms that may increase the atherogenic process (27). First, AT II may indirectly influence the atherogenic process via hemodynamic effects resulting from increased arterial blood pressure. Marked increases in arterial blood pressure have been demonstrated to augment the severity of experimental atherosclerosis. Second, AT II has been demonstrated to exert several direct effects relevant to the development of atherosclerosis including stimulation of monocyte recruitment, activation of macrophages, and enhanced oxidative stress, all of which have been linked to increase of the atherogenic process. These effects of AT II would occur independently of elevations in arterial blood pressure.
In presented experiment, except AVE 0991, we added: perindopril - as angiotensin converting enzyme inhibitor - the most powerful inhibitor of atherosclerosis in apoE-knockout mice (18-23). We also used tiorphan neutral endopeptidase inhibitor, which blocks the production of AT-(1-7) as well as A-779-direct inhibitor of AT-(1-7) receptor.
AT-(1-7) was discovered in 1988 (28). For a long time, it was considered only as a non-reactive product of RAS axis. The small interest in AT-(1-7) may have resulted also from the fact that initially, the specific enzymatic pathway of AT-(1-7) was unknown. The breakthrough was made already in 2000, when ACE homolog- ACE2 was described (29). Later it was discovered that AT-(1-7) acts via Mas oncogen as a metabotropic receptor, connected with G protein (30).
Therefore, knowledge about AT-(1-7) was gradually changing, leading to the current view that AT-(1-7) is an active, important component of RAS axis (31-33).
AT-(1-7) is an active peptide of RAS. It counteracts vasoconstriction by releasing nitric oxide and prostacyclin (34). Moreover, it opposites AT II mitogenic, arrhythmogenic and procoagulant activities (35). Water and sodium retention caused by AT II is inhibited by enhancing natriuresis and diuresis. On the other hand, AT-(1-7) independently of Mas-receptor increases bradykinin activity and antagonizes hypertrophic action of AT II. Recently, direct antiatherogenic and vasoprotective effect of AT-(1-7) in apoE-knockout mice model was discovered by Tesanovic et al. (36).
In 2002, non-peptide agonist of AT-(1-7) receptor: AVE 0991, has been described (37). In current study, we have shown that AVE 0991 inhibited atherogenesis in mouse model of atherosclerosis.
This observation is in agreement with the fact that AT-(1-7) is considered as "anti-AT II" factor (38). Since AT II is a potent proatherogenic agent, its functional antagonist should ameliorate atherogenesis (Fig. 5A).
 |
| Fig. 5. Relative p22phox expression in aortas from AVE 0991 and A-779-treated groups, in comparison to control. |
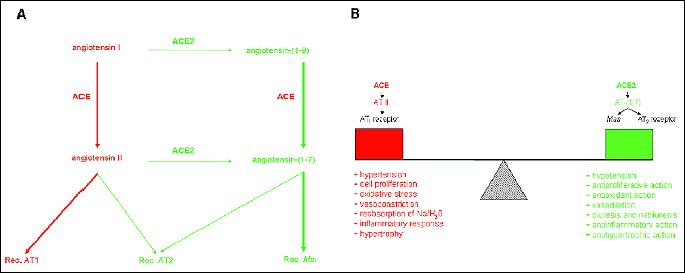 |
| Fig. 6. (A) Two "counteracting" systems in RAS axis. (B) Balance between ACE and ACE2 activity and its effect on development of cardiovascular diseases (modificated from Iwai & Horiuchi, 2009). |
What is a molecular explanation of AVE 0991 action? As we have shown in the current study, AVE 0991 inhibited NADPH oxidase expression, enzyme responsible for the production of reactive oxygen species, which are key players in the inflammatory process. Additionally, AVE 0991 reduced expression of co-stimulatory molecules CD40, CD86 and CD80 (not shown) on antigen presenting cells (macrophages and dendritic cells) and CD69 activation marker on CD4+ T lymphocytes.
Down-regulation of co-stimulatory molecules on antigen presenting cells was in line with reduced level of inflammatory factors (IL-6, IL-12, SAA, MCP-1) in plasma, suggesting that AVE 0911 may modulate the activity and function of innate immune cells (macrophages, dendritic cells) to suppress their involvement in the development of atherosclerosis. Although the CD69 activation marker was significantly down-regulated on CD4+ T cells, the role of this molecule in the atherosclerosis seems to be minor as shown in the study on CD69-/-apoE-/-mice (39).
Considering the presence of two kinds of ACE and two general mediators: AT II and AT-(1-7), recently, totally new view on RAS system has been proposed. In this concept, RAS system is divided into two contradictory systems, dependent on ACE/ACE2 balance: ACE-AT II-AT1 receptor vs. ACE2-AT-(1-7) - Mas receptor (Fig. 5B) (40).
AT-(1-7) is produced from AT I or AT II by ACE2. Since catalytical efficiency of ACE2 with AT II as substrate is about 400 times greater than with AT I, therefore it is the regulatory system of the ACE - AT II - receptor AT1 axis (41, 42).
During pharmacological blockade of AT1 receptors, greater amount of AT II may have a beneficial effect on the organism: by activation of type AT2 receptors or by changing into AT-(1-7) (43).
Angiotensin converting enzyme inhibitors increase plasma level of AT-(1-7) 10 times, which suggests that a part of beneficial action of these drugs might be mediated by AT-(1-7) (44). On the other hand, the expression of mRNA coding ACE2 increases 5 times during ACEI treatment or 3 times during angiotensin receptor blocker treatment (45).
Recently, it has been also proved that blockade of aldosterone inhibited ACE action and activated ACE2 action (46, 47). Therefore, drugs which influence RAS axis, perhaps not only inhibit the production and action of AT II, but also activate the "alternative", beneficial route which ends in AT-(1-7) production (Fig. 5B).
ACE2-AT-(1-7) - receptor Mas axis may become in the future an important therapeutic target in cardiovascular diseases as well as in metabolic diseases (48, 49). Therefore, a new therapeutic challenge has been established for pharmacology: creation of drugs which "stimulate" ACE2 - AT-(1-7) - Mas receptor axis. AVE 0991, nonpeptide agonist of receptor for AT-(1-7), might be one of such drugs.
Taking into account presented data, we can conclude that AVE 0991 inhibits atherogenesis. This gives promising perspectives for future clinical trials with this agent.
Introducing AVE 0991 as a drug for the treatment of atherosclerosis would be in line with the new postulate, that a new therapeutic agent should maintain the balance, shaken by activation of component: ACE - AT II - AT1 receptor, through the action, activating component: ACE2 - AT-(1-7) - receptor Mas receptor.
Acknowledgments: We thank Dr Jürgen Pünter from Sanofi - Aventis Deutschland GmbH, for providing us with AVE 0991. This article was supported by the grants from Polish Ministry of Science and Higher Education (MNiSW): N N401 548 340 and N N401 124 339.
Conflict of interests: None declared.
- Mendez-Fernandez YV, Stevenson BG, Diehl CJ, et al. The inhibitory
Fc
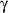 RIIb
modulates the inflammatory response and influences atherosclerosis in male
apoE(-/-) mice. Atherosclerosis 2011; 214: 73-80.
RIIb
modulates the inflammatory response and influences atherosclerosis in male
apoE(-/-) mice. Atherosclerosis 2011; 214: 73-80.
- Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol 2011; 12: 204-212.
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med 2005; 352: 1685-1695.
- Hansson GK, Libby P, Schonbeck U, Yan ZQ. Innate and adaptive immunity in the pathogenesis of atherosclerosis. Circ Res 2002; 91: 281-291.
- Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest 2000; 105: 1605-1612.
- Montecucco F, Pende A, Mach F. The renin-angiotensin system modulates inflammatory processes in atherosclerosis: evidence from basic research and clinical studies. Mediators Inflamm 2009; 2009: 752406.
- Toton-Zuranska J, Gajda M, Pyka-Fosciak G, et al. AVE 0991-angiotensin-(1-7) receptor agonist, inhibits atherogenesis in apoE-knockout mice. J Physiol Pharmacol 2010; 61: 181-183.
- Jawien J, Nastalek P, Korbut R. Mouse models of experimental atherosclerosis. J Physiol Pharmacol 2004; 55: 503-517.
- Jawien J, Csanyi G, Gajda M, et al. Ticlopidine attenuates progression of atherosclerosis in apolipoprotein E and low density lipoprotein receptor double knockout mice. Eur J Pharmacol 2007; 556: 129-135.
- Szczeklik A, Jawien J. Immunoglobulin E in acute phase response to surgical stress. Clin Exp Allergy 1996; 26: 303-307.
- Elhage R, Gourdy P, Jawien J, et al. The atheroprotective effect of 17ß-estradiol depends on complex interactions in adaptive immunity. Am J Pathol 2005; 167: 267-274.
- Jawien J, Gajda M, Wolkow PP, Zuranska J, Olszanecki R, Korbut R. The effect of montelukast on atherogenesis in apoE/LDLR - double knockout mice. J Physiol Pharmacol 2008; 59: 633-639.
- Kus K, Gajda M, Pyka-Fosciak G, et al. The effect of nebivolol on atherogenesis in apoE - knockout mice. J Physiol Pharmacol 2009; 60: 163-165.
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 22DDCT Method. Methods 2001; 25: 402-408.
- Zack M, Boyanovsky BB, Shridas P, et al. Group X secretory phospholipase A(2) augments angiotensin II-induced inflammatory responses and abdominal aortic aneurysm formation in apoE-deficient mice. Atherosclerosis 2011; 214: 58-64.
- McAfee JG, Subramanian G, Gagne G. Technique of leukocyte harvesting and labeling: problems and perspectives. Semin Nucl Med 1984; 14: 83-106.
- Chattopadhyay PK. Quantum dot technology in flow cytometry. Methods Cell Biol 2011; 102: 463-477.
- Arnal JF, Castano C, Maupas E, Mugniot A, et al. Omapatrilat, a dual angiotensin-converting enzyme and neutral endopeptidase inhibitor, prevents fatty streak deposit in apolipoprotein E-deficient mice. Atherosclerosis 2001; 155: 291-295.
- Candido R, Jandeleit-Dahm KA, Cao Z, et al. Prevention of accelerated atherosclerosis by angiotensin-converting enzyme inhibition in diabetic apolipoprotein E-deficient mice. Circulation 2002; 106: 246-253.
- Da Cunha V, Tham DM, Martin-McNulty B, et al. Enalapril attenuates angiotensin II-induced atherosclerosis and vascular inflammation. Atherosclerosis 2005; 178: 9-17.
- Hayek T, Attias J, Coleman R, et al. The angiotensin-converting enzyme inhibitor, fosinopril, and the angiotensin II receptor antagonist, losartan, inhibit LDL oxidation and attenuate atherosclerosis independent of lowering blood pressure in apolipoprotein E deficient mice. Cardiovasc Res 1999; 44: 579-587.
- Keidar S, Attias J, Coleman R, Wirth K, Scholkens B, Hayek T. Attenuation of atherosclerosis in apolipoprotein E-deficient mice by ramipril is dissociated from its antihypertensive effect and from potentiation of bradykinin. J Cardiovasc Pharmacol 2000; 35: 64-72.
- Knowles JW, Reddick RL, Jennette JC, Shesely EG, Smithies O, Maeda N. Enhanced atherosclerosis and kidney dysfunction in eNOS(-/-) apoE(-/-) mice are ameliorated by enalapril treatment. J Clin Invest 2000; 105: 451-458.
- Violi F, Basili S, Nigro C, Pignatelli P. Role of NADPH oxidase in atherosclerosis. Future Cardiol 2009; 5: 83-92.
- Buono C, Pang H, Uchida Y, Libby P, Sharpe AH, Lichtman AH. B7-1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation 2004; 109: 2009-2015.
- Lutgens E, Lievens D, Beckers L, Donners M, Daemen M. CD40 and its ligand in atherosclerosis. Trends Cardiovasc Med 2007; 17: 118-123.
- Daugherty A, Cassis L. Chronic angiotensin II infusion promotes atherogenesis in low density lipoprotein receptor -/- mice. Ann NY Acad Sci 1999; 892: 108-118.
- Santos RA, Brosnihan KB, Chappell MC, et al. Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension 1988; 11(2 Pt 2): I 153-157.
- Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 2000; 87: E1-E9.
- Santos RA, Simoes e Silva AC, Maric C, et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA 2003; 100: 8258-8263.
- Chappell MC. Emerging evidence for a functional angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS receptor axis: more than regulation of blood pressure? Hypertension 2007; 50: 596-599.
- Iusuf D, Henning RH, van Gilst WH, Roks AJ. Angiotensin-(1-7): pharmacological properties and pharmacotherapeutic perspectives. Eur J Pharmacol 2008; 585: 303-312.
- Santos RA, Campagnole-Santos MJ, Andrade SP. Angiotensin-(1-7): an update. Regul Pept 2000; 91: 45-62.
- Benter IF, Yousif MH, Cojocel C, Al-Maghrebi M, Diz DI. Angiotensin-(1-7) prevents diabetes-induced cardiovascular dysfunction. Am J Physiol Heart Circ Physiol 2007; 292: H666-H672.
- Kucharewicz I, Pawlak R, Matys T, Pawlak D, Buczko W. Antithrombotic effect of captopril and losartan is mediated by angiotensin-(1-7). Hypertension 2002; 40: 774-779.
- Tesanovic S, Vinh A, Gaspari TA, Casley D, Widdop RE. Vasoprotective and atheroprotective effects of angiotensin (1-7) in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2010; 30: 1606-1613.
- Wiemer G, Dobrucki LW, Louka FR, Malinski T, Heitsch H. AVE 0991, a nonpeptide mimic of the effects of angiotensin-(1-7) on the endothelium. Hypertension 2002; 40: 847-852.
- Benter IF, Yousif MH, Dhaunsi GS, Kaur J, Chappell MC, Diz DI. Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am J Nephrol 2008; 28: 25-33.
- Gomez M, Sanz-Gonzalez SM, Abu Nabah YN, Lamana A, Sanchez-Madrid F, Andres V. Atherosclerosis development in apolipoprotein E-null mice deficient for CD69. Cardiovasc Res 2009; 81: 197-205.
- Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-Mas receptor axis. Hypertens Res 2009; 32: 533-536.
- Castro CH, Santos RA, Ferreira AJ, Bader M, Alenina N, Almeida AP. Evidence for a functional interaction of the angiotensin-(1-7) receptor Mas with AT1 and AT2 receptors in the mouse heart. Hypertension 2005; 46: 937-942.
- Kramkowski K, Mogielnicki A, Leszczynska A, Buczko W. Angiotensin-(1-9), the product of angiotensin I conversion in platelets, enhances arterial thrombosis in rats. J Physiol Pharmacol 2010; 61: 317-324.
- Iyer SN, Ferrario CM, Chappell MC. Angiotensin-(1-7) contributes to the antihypertensive effects of blockade of the renin-angiotensin system. Hypertension 1998; 31: 356-361.
- Luque M, Martin P, Martell N, Fernandez C, Brosnihan KB, Ferrario CM. Effects of captopril related to increased levels of prostacyclin and angiotensin-(1-7) in essential hypertension. J Hypertens 1996; 14: 799-805.
- Ferrario CM, Jessup J, Chappell MC, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005; 111: 2605-2610.
- Keidar S, Gamliel-Lazarovich A, Kaplan M, et al. Mineralocorticoid receptor blocker increases angiotensin-converting enzyme 2 activity in congestive heart failure patients. Circ Res 2005; 97: 946-953.
- Szczepanska-Sadowska E, Cudnoch-Jedrzejewska A, Ufnal M, Zera T. Brain and cardiovascular diseases: common neurogenic background of cardiovascular, metabolic and inflammatory diseases. J Physiol Pharmacol 2010; 61: 509-521.
- Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin converting enzyme 2 and angiotensin-(1-7) in regulation of cardiovascular function. Am J Physiol 2005; 289: H2281-H2290.
- Raizada MK, Ferreira AJ. ACE2: a new target for cardiovascular disease therapeutics. J Cardiovasc Pharmacol 2007; 50: 112-119.