ATORVASTATIN ENHANCED NITRIC OXIDE RELEASE AND REDUCED BLOOD PRESSURE, NITROXIDATIVE STRESS AND RANTES LEVELS IN HYPERTENSIVE RATS WITH DIABETES
2Elucida Research LLC, Beverly, MA, USA; 3Nanomedical Research Laboratory, Ohio University, Athens, OH, USA
INTRODUCTION
Clinical trials have shown that patients with diabetes benefit from treatment with HMG-CoA reductase inhibitors, including atorvastatin, even with normal low-density lipoprotein (LDL) levels (1). Up to 75% of adults with diabetes also have hypertension, and patients with hypertension alone often show evidence of insulin resistance. These risk factors lead to microvascular and macrovascular disorders linked to endothelial cell (EC) dysfunction and inflammation (2). Loss of EC function contributes to atherosclerosis and its clinical manifestations, including coronary artery disease (CAD) (3-5). EC dysfunction is observed in patients with the metabolic syndrome and evidenced by loss of nitric oxide (NO) release (6-8). Attenuated NO release contributes to platelet aggregation, leukocyte adhesion, and loss of normal vasodilation associated with hypertension and diabetes (2, 9, 10). Agents that restore vascular NO bioavailability may therefore have therapeutic value in the treatment and prevention of CAD (11).
Statins reduce the risk of CAD by lowering LDL levels, although other, lipid-independent mechanisms have also been reported (12-14). In patients with diabetes and no history of CAD, atorvastatin treatment was associated with a 42% reduction in major cardiovascular events, including a 61% reduction in stroke (1). Similar benefits with atorvastatin treatment have been observed in hypertensive patients with normal LDL levels (15). Also, atorvastatin decreases the production of proinflamatory cytokines and adhesion molecules in the process of wound healing (16). Atorvastatin has also been reported to exert anti-inflammatory and platelet inhibitory effects with reductions in the inflammatory chemokine RANTES (17). These direct effects on platelet function and inflammation may explain early benefits with atorvastatin in patients with acute coronary syndromes (18). Statins increase levels of eNOS and NO production, a regulator of platelet aggregation and other aspects of thrombus development (19, 20).
In this study, we tested the hypothesis that atorvastatin treatment can restore endothelial NO bioavailability while reducing RANTES, nitroxidative stress and blood pressure levels in a rat model of hypertension with induced diabetes. As a further control, we conducted these studies in SHR rats without diabetes. The results of the study indicate direct vascular endothelial effects with atorvastatin in an experimental model with multiple CV risk factors that were not predicted by changes in lipid or glucose levels.
MATERIALS AND METHODS
Materials
Male, stroke-prone, spontaneously hypertensive rats (SHR), aged 7–9 weeks and weighing 250 ± 20 g, were obtained from inbred colonies (Harlan Laboratories, Indianapolis, IN). Animals were maintained in an environment that provided free access to food (commercially standard pellets for rodents) and water during the study. Nicotinamide (NA), streptozotocin (STZ), L-arginine, sepiapterin reductase, apocynin, L-NAME, and the calcium ionophore A23187 (CaI) were purchased from Sigma-Aldrich (St. Louis, MO). Atorvastatin was provided by Pfizer Inc. (New York, NY).
Preparation of hypertensive animals with type 2 diabetes
All procedures, used in this study, were approved by the Ohio University Animal Care Committee. Experimental type 2 diabetes was induced in SHR rat using a NA-STZ regimen. NA (150 mg/kg bw) was dissolved in sterile saline and administered by intraperitoneal (i.p.) injection 15 minutes prior to treatment with STZ (prepared in sodium citrate buffer, pH 4.5, and administered i.p. at 65 mg/kg bw). The control group received sodium citrate buffer alone.
Before induction of diabetes, two weeks after NA-STZ injection, and at the end of the study (6 weeks), blood glucose levels were measured using an Accu-Check® Compact Plus glucometer. All glucose measurements were preceded by a 6–8 h fasting period. Only diabetic animals with blood glucose levels greater than 250 mg/dL were considered to have developed diabetes mellitus. Animals were then treated with atorvastatin at 20 mg/kg b.w./day (versus vehicle).
Nitric oxide and peroxynitrite nanosensors
Concurrent measurements of NO and ONOO– were performed with tandem electrochemical nanosensors combined into one working unit. Their design was based on previously developed and chemically modified carbon-fiber technology (21, 22). Each of thenanosensors was made by depositing a sensing material on the tip of a carbon fiber (length 4–5 µm, diameter 200–300 nm). The fibers were sealed with nonconductive epoxy and electrically connected to copper wires with conductive silver epoxy. Conductive films of polymeric nickel(II)tetrakis(3-methoxy-4-hydroxyphenyl)porphyrin and polymeric manganese(III)-[2,2]paracyclophenylporphyrin were used for the NO and ONOO– sensors, respectively.
The amperometric method (with a response time of 0.1 ms) provided a quantitative signal (current) that was directly proportional to changes (from basal levels) in NO or ONOO– concentration. Amperometric measurements were performed with a Gamry Reference 600TM dual potentiostat (Gamry Instruments, Warminster, PA). Basal NO or ONOO– levels were measured by differential pulse voltammetry in separate experiments.
Measurement of aortic nitric oxide and peroxynitrite
Rats were euthanized with sodium thiopental and aortic ring segments isolated and immobilized in an organ chamber containing Hank’s balanced salt solution (HBSS) at 37°C, pH 7.4. All measurements of NO and ONOO– were performed on intact endothelial cells. The module of NO and ONOO– nanosensors was positioned near the surface of individual endothelial cells using a remote-controlled micromanipulator (Sensapex, Finland) and a microscope fitted with a CCD camera (AmScope, Irvine, CA).
The sensors had a high reproducibility of measurement (± 8%) at a constant distance (5 ± 2 µm) from the surface of the endothelial cell. After establishing a background current, CaI was injected into the organ chamber using a micro-injector (World Precision Instruments, Sarasota, FL). Rapid changes in current (proportional to the concentrations of NO or ONOO–) were observed after the addition of CaI and were monitored continuously.
Measurement of glomerular nitric oxide and peroxynitrite
Immediately after euthanized animals as described above, the kidneys were removed, cut into thin sections, and transferred to an organ chamber containing HBSS (37°C, pH 7.4). The NO/ONOO– nanosensor module was positioned 5 ± 2 µm from the surface of a glomerular EC (cortical zone). All other aspects of NO and ONOO– measurement were performed as described for aortic endothelial cells above.
Blood pressure measurement
Blood pressure was measured with a CODA High-Throughput Non-invasive Tail Blood Pressure System (Kent Scientific Corporation, Torrington, CT). All animals were acclimated to this procedure for three days prior to measurement in order to minimize stress-induced variations in BP.
Measurement of insulin and RANTES
At various time points, preceded by a 6–8 h fasting period, blood was collected into MiniCollect K3 EDTA tubes (Greiner BioOne, Monroe, NC). Plasma was obtained by centrifugation (2.700×g for 10 min at 4°C) and stored at –80°C for further analysis. Plasma insulin was quantified using a rat insulin ELISA (Alpco Diagnostic, Salem, NH). The ELISA method was used to measure levels of RANTES (Millipore Corp., St. Charles, MO).
Statistical analyses
Data are reported as mean ± standard deviation (S.D.). The significance of differences between results from independent experimental conditions was tested using either the two-tailed, Student’s t-test (measurements of NO and ONOO– release, including ratio calculations, from SHR versus diabetic SHR rat aortic and glomerular ECs) or one-way analysis of variance with Student-Newman-Keuls multiple comparisons post hoc analysis (glucose and blood pressure measurements; drug effects on aortic and glomerular NO and ONOO– release from diabetic SHR rats). A value of P<0.05 was considered significant.
RESULTS
Effects of atorvastatin on mean blood pleasure, glucose and cholesterol levels in diabetic hypertensive animals
Glucose levels were significantly elevated in diabetic SHR animals as compared to non-diabetic SHR animals (Table 1). Diabetic SHR rats also had elevated levels of systolic, diastolic, and mean BP levels. Treatment with atorvastatin for five weeks reduced levels of systolic BP by 16%, diastolic BP by 24%, and mean BP by 21% (P<0.001) but had no significant effect on fasting glucose levels as compared to vehicle. Atorvastatin treatment had no effect on total cholesterol levels. In non-diabetic SHR animals, atorvastatin also reduced systolic BP levels by 21%, diastolic BP by 28%, and mean BP by 27% (P<0.001), with no significant effect on cholesterol levels.
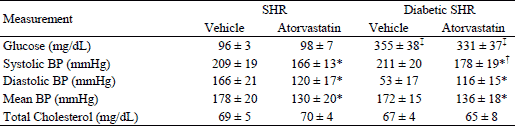
Nitric oxide and peroxynitrite release from spontaneously hypertensive rats versus diabetic spontaneously hypertensive rats in aortic and glomerular endothelial cells
Aortic endothelial or glomerular NO and ONOO– release was monitored and measured following maximal stimulation with a receptor-independent eNOS agonist, calcium ionophore (CaI). In the absence of an exogenous eNOS agonist, basal concentrations of NO (13 ± 2 nM) and ONOO– (4 ± 3 nM) were detected near the endothelial surface by nanosensors operating in differential pulse voltammetry mode. As shown in Fig. 1, NO and ONOO– levels increased significantly from baseline following stimulation with CaI, reaching a maximum after about one second and gradually decreasing during the next 2–6 s. Maximal NO release was measured at 267 ± 29 nM and 214 ± 20 nM in aortic endothelium for SHR and diabetic SHR rats, respectively (Fig. 2). Maximal ONOO– release reached 303 ± 27 nM for SHR rats and 350 ± 29 nM for diabetic SHR rats (an increase of 15%). We used the ratio of cytoprotective NO concentration to cytotoxic ONOO– concentration, [NO]/[ONOO–], as a marker to evaluate the level of eNOS uncoupling, endothelial dysfunction and nitroxidative stress. [NO]/[ONOO–] reflects the balance between bioavailable NO and the level of nitroxidative stress (ONOO–) produced mainly by eNOS after stimulation with CaI. In healthy, functional endothelium, [NO]/[ONOO–] is in the range of 3–5 (23). In the study presented here, [NO]/[ONOO–] was 0.88 ± 0.12 in aortic endothelium of SHR rats, indicating an uncoupling of eNOS and significant endothelial dysfunction. [NO]/[ONOO–] decreased further to 0.61 ± 0.08 in diabetic SHR (Fig. 2). This is about 30% decrease compared to SHR indication severe eNOS uncoupling and dysfunction of endothelial cells. In glomerular endothelial cells, the maximal NO and ONOO– concentrations were lower than that observed in aortic cells (Fig. 2). However, the pattern of relative changes was very similar between aortic and glomerular cells. [NO]/[ONOO–] was about 0.3 for glomerular cells and was about 50% lower than for aortic cells.
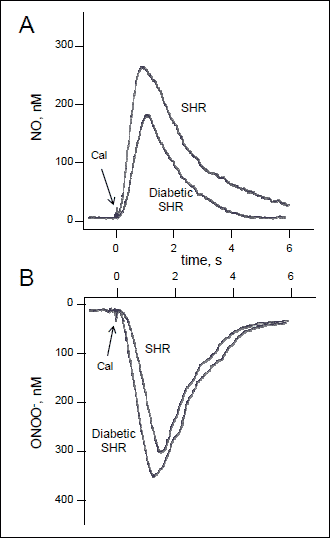 |
Fig. 1. Typical NO and ONOO– amperograms. The amperograms were recorded with nanosensors and show changes in NO concentration (A) and ONOO– concentration (B) released from aortic endothelial cells of SHR and diabetic SHR rats. The release of NO and ONOO– was simulated with calcium ionophore (CaI, 1 µM). |
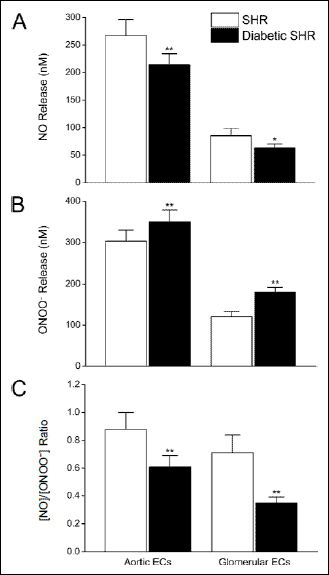 |
Fig. 2. Effects of hyperglycemia on of nitric oxide and peroxynitrite produced by aortic and glomerular endothelial cells isolated from SHR rats and diabetic SHR rats. Maximal NO and ONOO– concentrations, shown in panels (A) and (B), respectively, were measured from single endothelial cells immediately following stimulation with calcium ionophore. The ratio of maximal NO to ONOO– concentration (panel C) was calculated as the arithmetic quotient of simultaneous, separate NO and ONOO– measurements in situ. Values are mean ± S.D. (N = 10–15). *P<0.05 and ** P<0.001 versus cognate SHR group (Student-Newman-Keuls multiple comparisons test; overall ANOVA – Panel A: P<0.0001, F=281.06; Panel B: P <0.0001, F=331.43; Panel C: P<0.0001, F=50.550). |
Effects of atorvastatin on nitric oxide and peroxynitrite release from diabetic spontaneously hypertensive rat aortic endothelial cells
Atorvastatin treatment significantly increased the capacity of aortic endothelial cells to produce NO while simultaneously reducing ONOO– production, consistent with an increase in eNOS efficiency (coupling). Fig. 3 shows maximal NO and ONOO– release levels from aortic ECs isolated from diabetic SHR rats treated with atorvastatin for five weeks at 20 mg/kg/day. Atorvastatin treatment restored aortic endothelial function as compared to untreated, diabetic SHR animals, as evidenced by a 28% increase and 27% decrease in NO and ONOO– release, respectively. The [NO]/[ONOO–] increased by 80% (from about 0.6 to 1.1) with atorvastatin treatment, as compared to vehicle, in diabetic SHR animals (Fig. 3). This favorable effect on [NO]/[ONOO–] balance indicates that atorvastatin increased the bioavailability of NO in diabetic SHR rats. In SHR rats without diabetes, [NO]/[ONOO–] increased by 67% (from about 0.9 to 1.6) with atorvastatin treatment, as compared to vehicle.
 |
Fig. 3. Effects of atorvastatin on the release of nitric oxide and peroxynitrite from aortic endothelial cells isolated from diabetic and non-diabetic SHR rats. Maximal NO and ONOO–, shown in both panels A and B, respectively, were measured from single endothelial cells immediately following stimulation with calcium ionophore. The ratio of maximal concentration of NO to ONOO– (panel C) was calculated as the arithmetic quotient of separate, simultaneous measurement of NO and ONOO–, in situ. Values are mean ± S.D. (N = 10–14). * P<0.001 versus cognate vehicle-treated control; †P<0.001 versus SHR (Student-Newman-Keuls multiple comparisons test; overall ANOVA – Panel A: P<0.0001, F=61.710; Panel B: P<0.0001, F=52.327; Panel C: P<0.0001, F=80.548). |
Effects of atorvastatin on nitric oxide and peroxynitrite release from spontaneously hypertensive and diabetic spontaneously hypertensive rat glomerular endothelial cells
We also tested the effects of atorvastatin on the capacity of glomerular ECs to produce NO and ONOO–. As shown in Fig. 4, maximal NO release from glomerular ECs decreased from 85 ± 13 nM to 63 ± 7 nM for SHR versus diabetic SHR rats, respectively. We also observed a concomitant and pronounced increase in nitroxidative stress. ONOO– levels increased significantly from 120 ± 13 nM to 180 ± 12 nM for SHR and diabetic SHR rats, respectively (Fig. 4). The resultant [NO]/[ONOO–] in glomerular ECs decreased by 50% (from 0.7 to 0.3) in SHR versus diabetic SHR animals (Fig. 4).
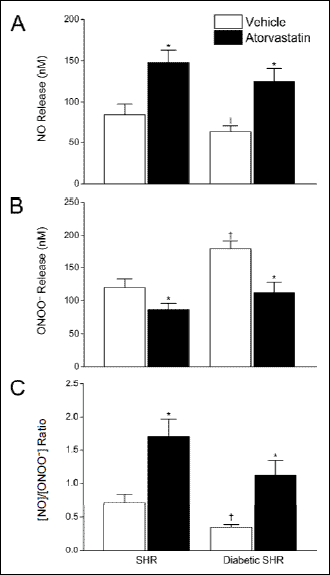 |
Fig. 4. Effects of atorvastatin on the release of NO and ONOO– from glomerular endothelial cells isolated from diabetic and non-diabetic SHR rats. Maximal NO and ONOO–, shown in panels (A) and (B), respectively, were measured from single endothelial cells immediately following stimulation with calcium ionophore. The ratio of maximal concentration of NO to ONOO– (panel C) was calculated as the arithmetic quotient of separate NO and ONOO– measurements. Values are mean ± S.D. (N = 10–15). *P<0.001 versus cognate vehicle-treated control; †P<0.001 versus SHR (Student-Newman-Keuls multiple comparisons test; overall ANOVA – Panel A: P<0.0001, F=87.791; Panel B: P<0.0001, F=125.02; Panel C: P<0.0001, F=102.37). |
Atorvastatin significantly increased the capacity of glomerular endothelial cells to produce NO while simultaneously reducing ONOO– production, consistent with an eNOS uncoupling effect. Atorvastatin increased NO release in ECs of diabetic SHR rats by 98% (63 ± 7 nM to 125 ± 16 nM) while decreasing ONOO– release by 38% (180 ± 12 nM to 112 ± 16 nM) as compared to vehicle alone (P<0.001). [NO]/[ONOO–] increased approximately three-fold with atorvastatin treatment (Fig. 4). Similar benefits on eNOS function were observed in non-diabetic SHR rats. In these animals, [NO]/[ONOO–] increased more than two-fold with atorvastatin treatment as compared to the effects of vehicle treatment alone (P<0.001).
eNOS uncoupling in diabetic spontaneously hypertensive rats
To elucidate the basis for enhanced NO bioavailability with atorvastatin, we compared its effects on the [NO]/[ONOO–] balance in glomerular ECs to other known modulators of eNOS function. We confirmed the essential role of eNOS activation in this process by first treating the cells with 300 µM NG-nitro-l-arginine methyl ester (L-NAME), which reduced NO and ONOO– production by about 80% (data not shown) but did not significantly increase [NO]/[ONOO–] (Fig. 5). Treatment with either L-arginine (3 mM), the metabolic precursor of NO, or sepiapterin (10 µM), a precursor of the eNOS cofactor, tetrahydrobiopterin, partially restored eNOS coupling and its function, as indicated by an increase in [NO]/[ONOO–] by 91% and 106%, respectively. Apocynin, an NADPH oxidase inhibitor, increased [NO]/[ONOO–] by 137%, confirming a significant role of NADPH oxidase to the overall formation of ONOO– and the diminished bioavailability of NO in glomerular ECs. By comparison, the atorvastatin increased [NO]/[ONOO–] by 220%, which was significantly greater (P<0.001) than the effects produced by any of the other eNOS modulators.
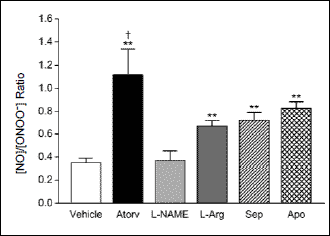 |
Fig. 5. Comparative effects of atorvastatin (Atorv) and the eNOS modulators, l-NAME, L-arginine (L-Arg), sepiapterin (Sep), and apocynin (Apo), on [NO]/[ONOO–] release from glomerular endothelial cells isolated from diabetic SHR rats. Values are mean ± S.D. (N = 5–11). **P<0.001 versus vehicle; †P <0.001 versus all other treatments (Student-Newman-Keuls multiple comparisons test; overall ANOVA: P<0.0001, F=46.841). |
Effects of atorvastatin on RANTES levels in diabetic hypertensive animals
Diabetic SHR rats also had elevated levels of the cytokine RANTES in circulating plasma. RANTES levels increased from 31.9 to 38.4 ng/mL in non-diabetic versus diabetic SHR rats, respectively (Fig. 6). Atorvastatin treatment reduced RANTES to 15.4 and 16.7 ng/mL (P<0.001) in non-diabetic and diabetic SHR rats, respectively.
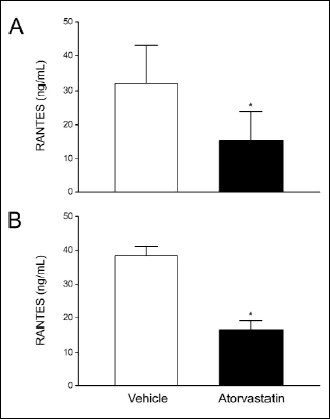 |
Fig. 6. Effects of atorvastatin on RANTES levels measured in non-diabetic (Panel A) and diabetic (Panel B) SHR study animals. Values are mean ± S.D. (N = 2–5). *P<0.05 (unpaired, two-tailed, Student’s t-test). |
DISCUSSION
In hypertensive rats, the induction of diabetes was associated with eNOS uncoupling and EC dysfunction as evidenced by reduced NO bioavailability along with higher RANTES, nitroxidative stress and blood glucose levels. Atorvastatin treatment partially, but significantly, restored eNOS coupling, EC function as evidenced by increased NO release in both aortic and glomerular ECs with concomitant reductions in ONOO– production. We previously introduced [NO]/[ONOO–] as a sensitive and accurate marker of eNOS uncoupling and endothelial dysfunction (24). NO release from endothelium is always accompanied by the release of ONOO–. The nanomedical approach was to use nanosensors for in situ monitoring of both NO and ONOO– concentrations, simultaneously, in small (picoliter) volumes of medium. In normal, functional endothelium, [NO]/[ONOO–] is in the range of 2–5, indicating a relatively high level of cytoprotective NO and a low level of cytotoxic ONOO–. With [NO]/[ONOO–] level below 2, gradual eNOS uncoupling and dysfunction of the endothelium is observed. In SHR rats, aortic [NO]/[ONOO–] is about 0.9, indicating significant eNOS uncoupling associated with dysfunction of endothelium. In diabetic SHR rats, the eNOS uncoupling and endothelial dysfunction are severe ([NO]/[ONOO–] (about 0.6). This negative effect is even more pronounced in glomerular ECs of diabetic SHR rats where [NO]/[ONOO–] is about 0.3. Treatment with atorvastatin significantly restored eNOS coupling and endothelial function SHR, as well as, diabetic SHR rats, in both aortic and glomerular eNOS- the most significant finding in this study. The improvement in [NO]/[ONOO–] balance and EC function with atorvastatin was associated with reduced blood pressure and RANTES levels despite no changes in glucose or cholesterol levels. Similar effects of atorvastatin on EC function, blood pressure and RANTES levels were observed in SHR rats without diabetes. Another important finding is that the effects of atorvastatin on the endothelial [NO]/[ONOO–] balance compared favorably to other modulators of eNOS coupling and function (L-NAME, L-arginine, sepiapterin) or an inhibitor of NADPH oxidase activity. This is an important observation which suggests that it would be much more efficient to restore [NO]/[ONOO–] balance in dysfunctional endothelium by restoring eNOS coupling than by shifting this balance with modulators of eNOS and/or NADPH oxidase.
In models of dyslipidemia, statins have been shown to improve NO synthesis through mechanisms unrelated to HMG-CoA reductase, including up-regulation of eNOS expression (19-20) and reduced O2– formation (25-26). Additionally, statins stimulate endothelial NO production by reducing plasma membrane caveolin levels (27). By interfering with cholesterol biosynthesis and lowering plasma membrane cholesterol levels, atorvastatin was shown to reduce the expression of caveolin-1 (27). Atorvastatin also promoted the agonist-induced association of eNOS and the molecular chaperone, Hsp90, resulting in eNOS activation (27). The active metabolites of atorvastatin have also been shown to have potent antioxidant properties under conditions of hyperglycemia that lead to reduced superoxide generation (28-29). In the current study, we observed improved eNOS function with atorvastatin in animals with hypertension and diabetes independently of changes in cholesterol levels.
Hypertension and diabetes are comorbid conditions that lead to microvascular and macrovascular changes linked to endothelial dysfunction. SHR rats are characterized by attenuated levels of functional endothelial NO, despite increased eNOS enzyme activity (30). The basis for this paradox is attributed to the excessive generation of O2–, which reacts rapidly with NO to form ONOO– (31). This hypothesis is supported by the observation of similar changes in eNOS expression and O2– generation in Sprague-Dawley rats made hypertensive by aortic banding (32). These findings are also consistent with other studies linking endothelial dysfunction in hypertension to excessive O2– production through eNOS uncoupling and NADPH oxidase activity (33). The induction of diabetes in the SHR rats further increases eNOS uncoupling as evidenced by an enhanced conversion of NO to ONOO– (34). The effects of atorvastatin on NO bioavailability were particularly evident in glomerular ECs. This may explain the renal benefits of treatment in patients with diabetes even with normal lipid levels, especially among those with evidence of microvascular complications (35).
The induction of diabetes further enhanced eNOS uncoupling in hypertensive rats as observed in this study. A relationship between NO bioavailability and insulin resistance has been described in transgenic mice that are eNOS-deficient and produced vascular abnormalities associated with insulin resistance, along with hyperinsulinemia (36). The relationship between low NO bioavailability and loss of insulin sensitivity was independent of blood pressure elevations in these animals. Insulin-induced uptake of glucose into skeletal muscle was also reversibly blocked by infusion of a specific NOS inhibitor, L-NMMA (37-39). Similarly, the endogenous eNOS inhibitor, asymmetrical dimethylarginine (ADMA), reduced insulin sensitivity in a transgenic animal model (40). Agents that improve NO bioavailability with diabetes may have an important therapeutic role in restoring insulin sensitivity.
In SHR rats, with or without diabetes, atorvastatin treatment reduced elevated levels of RANTES by more than 50%. This is consistent with a direct anti-inflammatory action as well as an ability to interfere with platelet activation. The chemokine RANTES is produced by a variety of leukocytes and platelets where it mediates platelet activation, platelet-leukocyte interaction, as well as attraction and homing of leukocytes in vascular injury and platelet deposition (41-42). Levels of RANTES also influence the progression of atherosclerosis by promoting monocyte MCP-1 production, macrophage accumulation, and neointimal growth (42-43). RANTES contributes to lesion progression by inducing monocyte survival and differentiation into macrophages (43). Additionally, RANTES contributes to smooth muscle cell proliferation which influences progression to the fibrous plaque (44). RANTES receptor antagonists inhibit the infiltration of monocytes and limit atherosclerotic plaque formation in proatherogenic mice models (46-48). In a clinical study that included 56 patients with CAD, high-dose atorvastatin reduced RANTES levels in a manner that could not be reproduced by a lower dose, when used in combination with ezetimibe, despite comparable reductions in overall LDL levels (17). Thus, these findings support a non-LDL related effect with atorvastatin by reducing a key mediator of both inflammation and platelet aggregation.
In conclusion, the results of this study provide clear and significant insight into the relationship between NO bioavailability, nitroxidative stress (ONOO–) and cardiovascular risk factors such as diabetes and hypertension. The SHR animals, with induced diabetes, demonstrated statistically significant reductions in endothelial-dependent NO release concomitant with increases in ONOO– concentration and significant [NO[/[ONOO–] imbalance, along with elevated nitroxidative stress, RANTES and blood pressure levels. Treatment with atorvastatin improved vascular function as evidenced by increased NO bioavailability in both aortic and glomerular ECs. This effect is primarily attributed to changes in eNOS coupling mechanisms and also with direct free radical scavenging properties of atorvastatin. Atorvastatin treatment was also associated with reductions in RANTES, an important mediator of platelet activity, in hypertensive rats with and without diabetes. These findings may also provide insight into the effects of atorvastatin on atherothrombotic risk in patients with otherwise normal baseline LDL levels.
Acknowledgments: Special thanks to Collin Arocho for his technical assistance in the preparation of this manuscript. This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR-014575-01 from the National Center for Research Resources, National Institutes of Health.
Conflict of interests: This study was supported, in part, by an investigator-initiated research grant (RPM) from Pfizer Inc., whom also provided the drug used in this study.
REFERENCES
- Colhoun HM, Betteridge DJ, Durrington PN, et al. Primary prevention of cardiovascular disease with atorvastatin in type 2 diabetes in the Collaborative Atorvastatin Diabetes Study (CARDS): Multicentre randomised placebo-controlled trial. Lancet 2004; 364: 685-696.
- Panza JA, Quyyumi AA, Brush JE, Epstein SE. Abnormal endothelium-dependent vascular relaxation in patients with essential hypertension. N Engl J Med 1990; 323: 22-27.
- Harrison DG, Freiman PC, Armstrong ML, Marcus ML, Heistad DD. Alterations of vascular reactivity in atherosclerosis. Circ Res 1987; 61: 74-80.
- Liao JK. Endothelium and acute coronary syndromes. Clin Chem 1998; 44: 1799-1808.
- Oemar BS, Tschudi MR, Godoy N, Brovkovich V, Malinski T, Luscher TF. Reduced endothelial nitric oxide synthase expression and production in human atherosclerosis. Circulation 1998; 97: 2494-2498.
- Kurioka S, Koshimura K, Murakami Y, Nishiki M, Kato Y. Reverse correlation between urine nitric oxide metabolites and insulin resistance in patients with type 2 diabetes mellitus. Endocr J 2000; 47: 77-81.
- Node K, Kitakaze M, Yoshikawa H, Kosaka H, Hori M. Reduced plasma concentrations of nitrogen oxide in individuals with essential hypertension. Hypertension 1997; 30: 405-408.
- Grattagliano I, Palmieri VO, Portincasa P, Moschetta A, Palasciano G. Oxidative stress-induced risk factors associated with the metabolic syndrome: a unifying hypothesis. J Nutr Biochem 2008; 19: 491-504.
- Paniagua OA, Bryant MB, Panza JA. Role of endothelial nitric oxide in shear stress-induced vasodilation of human microvasculature: diminished activity in hypertensive and hypercholesterolemic patients. Circulation 2001; 103: 1752-1758.
- Mason RP, Jacob RF, Kubant R, et al. Effect of enhanced glycemic control with saxagliptin on endothelial nitric oxide release and CD40 levels in obese rats. J Atheroscler Thromb 2011; 18: 774-783.
- Mason RP, Cockcroft JR. Targeting nitric oxide with drug therapy. J Clin Hypertens 2006; 8 (12 Suppl. 4): 40-52.
- Cosentino F, Bonetti S, Rehorik R, et al. Nitric-oxide-mediated relaxations in salt-induced hypertension: effect of chronic beta1-selective receptor blocker. J Hypertens 2002; 20: 421-428.
- Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol 2003; 23: 168-175.
- Mason RP, Walter MF, Day CA, Jacob RF. Intermolecular differences for HMG-CoA reductase inhibitors contribute to distinct pharmacologic and pleiotropic actions. Am J Cardiol 2005; 96 (5A): 11F-23F.
- Sever PS, Dahlof B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet 2003; 361: 1149-1158.
- Korybalska K, Kawka E, Breborowicz A, Witowski J. Atorvastatin does not impair endothelial cell wound healing in an in vitro model of vascular injury. J Physiol Pharmacol 2012; 63: 389-395.
- Piorkowski M, Fischer S, Stellbaum C, et al. Treatment with ezetimibe plus low-dose atorvastatin compared with higher-dose atorvastatin alone: is sufficient cholesterol-lowering enough to inhibit platelets? J Am Coll Cardiol 2007; 49: 1035-1042.
- Patti G, Pasceri V, Colonna G, et al. Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: results of the ARMYDA-ACS randomized trial. J Am Coll Cardiol 2007; 49: 1272-1278.
- Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998; 97: 1129-1135.
- Mason RP, Kubant R, Heeba G, et al. Synergistic effect of amlodipine and atorvastatin in reversing LDL-induced endothelial dysfunction. Pharm Res 2008; 25: 1798-1806.
- Lvovich V, Scheeline A. Amperometric sensors for simultaneous superoxide and hydrogen peroxide detection. Anal Chem 1997; 69: 454-462.
- Malinski T, Taha Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature 1992; 358: 676-678.
- Funovic P, Korda M, Kubant R, et al. Effect of beta-blockers on endothelial function during biological aging: a nanotechnological approach. J Cardiovasc Pharmacol 2008; 51: 208-215.
- Mason RP, Kubant R, Jacob RF, Walter MF, Boychuk B, Malinski T. Effect of nebivolol on endothelial nitric oxide and peroxynitrite release in hypertensive animals: Role of antioxidant activity. J Cardiovasc Pharmacol 2006; 48: 862-869.
- Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol 2000; 20: 61-69.
- Wassmann S, Laufs U, Muller K, et al. Cellular antioxidant effects of atorvastatin in vitro and in vivo. Arterioscler Thromb Vasc Biol 2002; 22: 300-305.
- Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylgluataryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation 2001; 103: 113-118.
- Mason RP, Walter MF, Day CA, Jacob RF. Active metabolite of atorvastatin inhibits membrane cholesterol domain formation by an antioxidant mechanism. J Biol Chem 2006; 281: 9337-9345.
- Self-Medlin Y, Byun J, Jacob RF, Mizuno Y, Mason RP. Glucose promotes membrane cholesterol crystalline domain formation by lipid peroxidation. Biochim Biophys Acta 2009; 1788: 1398-1403.
- McIntyre M, Hamilton CA, Rees DD, Reid JL, Dominiczak AF. Sex differences in the abundance of endothelial nitric oxide in a model of genetic hypertension. Hypertension 1997; 30: 1517-1524.
- Kerr S, Brosnan MJ, McIntyre M, Reid JL, Dominiczak AF, Hamilton CA. Superoxide anion production is increased in a model of genetic hypertension role of the endothelium. Hypertension 1999; 33: 1353-1358.
- Bouloumie A, Bauersachs J, Ling W, et al. Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension 1997; 30: 934-941.
- Landmesser U, Dikalov S, Price SR, et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest 2003; 111: 1201-1209.
- Mason RP, Kubant R, Jacob RF, et al. Loss of arterial and renal nitric oxide bioavailability in hypertensive rats with diabetes: effect of beta-blockers. Am J Hypertens 2009; 22: 1160-1166.
- Ballantyne CM, Bays HE, Kastelein JJ, et al. Efficacy and safety of eicosapentaenoic acid ethyl ester (AMR101) therapy in statin-treated patients with persistent high triglycerides (from the ANCHOR study). Am J Cardiol 2012; 110: 984-992.
- Duplain H, Burcelin R, Sartori C, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001; 104: 342-345.
- Baron AD, Tarshoby M, Hook G, et al. Interaction between insulin sensitivity and muscle perfusion on glucose uptake in human skeletal muscle: evidence for capillary recruitment. Diabetes 2000; 49: 768-774.
- Sasahara M, Raines EW, Chait A, et al. Inhibition of hypercholesterolemia-induced atherosclerosis in the nonhuman primate by probucol. I. Is the extent of atherosclerosis related to resistance of LDL to oxidation? J Clin Invest 1994; 94: 155-164.
- Adamcova M, Ruzickova S, Simko F. Multiplexed immunoassays for simultaneous quantification of cardiovascular biomarkers in the model of H(G)-nitro-L-arginine methylester (L-NAME) hypertensive rat. J Physiol Pharmacol 2013; 64: 211-217.
- Sydow K, Mondon CE, Schrader J, Konishi H, Cooke JP. Dimethylarginine dimethylaminohydrolase overexpression enhances insulin sensitivity. Arterioscler Thromb Vasc Biol 2008; 28: 692-697.
- Gear AR, Camerini D. Platelet chemokines and chemokine receptors: linking hemostasis, inflammation, and host defense. Microcirculation 2003; 10: 335-350.
- Huo Y, Schober A, Forlow SB, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med 2003; 9: 61-67.
- Prescott SM, McIntyre TM, Zimmerman GA, Stafforini DM. Sol Sherry lecture in thrombosis: molecular events in acute inflammation. Arterioscler Thromb Vasc Biol 2002; 22: 727-733.
- Scheuerer B, Ernst M, Durrbaum-Landmann I, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 2000; 95: 1158-1166.
- von Hundelshausen P, Weber KS, Huo Y, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 2001; 103: 1772-1777.
- Braunersreuther V, Steffens S, Arnaud C, et al. A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler Thromb Vasc Biol 2008; 28: 1090-1096.
- Schober A, Manka D, von Hundelshausen P, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 2002; 106: 1523-1529.
- Veillard NR, Kwak B, Pelli G, et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res 2004; 94: 253-261.
A c c e p t e d : December 12, 2014