ANTI-INFLAMMATORY EFFECTS OF ATORVASTATIN TREATMENT IN CHRONIC OBSTRUCTIVE PULMONARY DISEASE. A CONTROLLED PILOT STUDY
INTRODUCTION
Statins are potent inhibitors of cholesterol biosynthesis and have been shown to decrease mortality from cardiovascular disease(1, 2). In addition to their lipid lowering properties by inhibiting 3-hydroxy-3-methylgluteryl coenzyme A (HMG-CoA) reductase, statins also possess a wide range of pleiotropic effects, including anti-inflammatory and anti-oxidant properties, modulating effects on endothelial function and modification of vascular wall structure (3-5).Thus statins have the potential as a treatment in chronic inflammatory conditions such as chronic obstructive pulmonary disease (COPD). Retrospective pharmaco-epidemiologic studies suggest that statins reduce hospitalizations from exacerbations, acute coronary events, and cardiovascular and respiratory mortality in patients with COPD (6-8). Moreover, there is evidence that statins may improve lung function in patients with COPD (9). Oxidative stress and airway obstruction, major features of asthma and COPD have been studied by the others (10-12). Recent animal studies have shown the anti-inflammatory effects of statins in the lungs (13-16). However, an anti-inflammatory effect of statins in the lungs of COPD patients has not been demonstrated. We therefore undertook a study with the aim of demonstrating the anti-inflammatory effects of statins in the lungs of COPD patients.
MATERIALS AND METHODS
Study design and treatment
This randomized, single blind, controlled, parallel group pilot study was conducted in Bialystok, Poland between December 2012 and March 2013 (NCT01748279). Following screening and a 4-week wash-out period to ensure stable standardized therapy over the period and to allow proper wash-out of not allowed concomitant medications, patients were randomized (2:1) using sequentially numbered containers to receive atorvastatin 40 mg once daily in the morning or matched placebo for 12 weeks. Patients remained blinded to treatment allocation throughout the study except in an emergency. Relief medication (salbutamol metered-dose inhaler 200 µg as needed) was provided for additional symptom control as needed (but was not used within the 6 hours prior to each visit). The study protocol was approved by the University of Bialystok Ethical Committee. Each subject gave written informed consent to participate in the study.
Patients
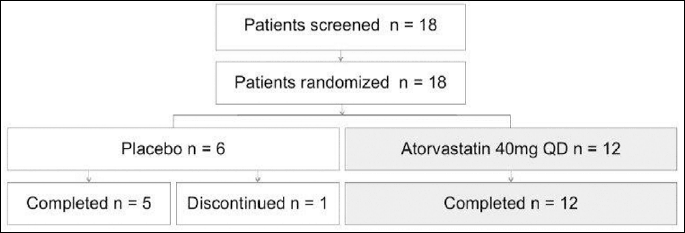
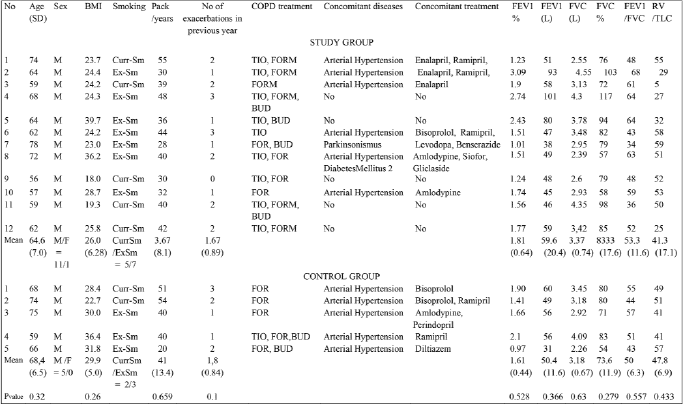
We enrolled eighteen statin naive COPD patients, current and ex-smokers. The diagnosis of COPD was based on the presence of clinical symptoms (chronic cough and/or exertional dyspnea), exposure to cigarette smoke (>10 pack years smoking) and a post bronchodilator FEV1/FVC ratio <0.7(17). Other lung conditions were excluded on history, clinical examination and chest X-ray. Seventeen patients completed the study (one control subject withdrew consent after randomization (Fig. 1). During four weeks of wash-out period ICS were withdrawn. Twelve patients (1 female and 11 males), mean age 64.6 years (range 56–78) were randomly allocated to receive atorvastatin 40 mg once daily for 3 months (treatment group) and 5 patients (all males), mean age 68.4 (range 59–75) received placebo (control group) as an add-on treatment to COPD maintenance therapy (Table 1). All patients received formoterol in a dose of 12 mcg twice daily by easyhaler and/or tiotropium bromide 18 mcg once daily by a handihaler as maintenance and salbutamol MDI 200 µg as a reliever treatment.
Study assessments
The primary endpoint was change in CD45+ cell expression measured by immunohistochemistry before and after 12 weeks of treatment. Secondary outcome measures were: change in health related quality of life assessed by St. George’s respiratory questionnaire, change in 6-minute walk distance (6-MWD) measured according to 6-MWD protocol, change in serum hs-C-reactive protein (hs-CRP), change in total cholesterol, LDL and HDL-cholesterol, triglycerides and intimal-medial thickness (IMT) measured in the common carotid artery (CCA) using a standard technique, and a change in gene expression in lung biopsy samples measured using microarrays before and after 12 weeks of therapy.
Lung function
Spirometry, lung volumes and diffusing capacity for carbon monoxide (DLCO) were performed in a body plethysmograph (Elite DL, Medgraphics, USA). The measurements were made using standard protocols (18).
Laboratory assessments
Blood samples were taken at screening (V0), baseline (V1) and after 3 months treatment. Twenty milliliters of blood was drawn from an antecubital vein using a 17-gauge cannula; a sample of whole blood was collected to measure full blood count and a further sample was collected, centrifuged within 2 hours of sampling and serum stored at –80°C until assayed. Total cholesterol, LDL and HDL-cholesterol and triglycerides were measured using a standard kit (Architect 8200, Abbott Laboratories, IL, USA). Serum creatine kinase isoenzyme (CK-MB) activity was measured by immuno-inhibition, using Enzyline CK-MB kits (BioMerieux, Lyon, France). Serum C-reactive protein (CRP) was measured using immunoturbidimetric Protiline® 103 CRP assay kits (bioMerieux, Lyon, France).
Sputum induction and processing
Sputum was induced before and after three months treatment as previously described (19).
Bronchoscopy and transbronchial lung biopsy (TBB)
Bronchoscopy and transbronchial lung biopsy (TBB) was performed under fluoroscopic guidance according to recommended guidelines (20-22). All subjects underwent bronchoscopy with TBB sampled from the right middle lobe in every patient before and after three months treatment with placebo or atorvastatin.
Immunohistochemistry
TBB samples were fixed in 4% phosphate-buffered formalin and after 24 h stored in 70% ethanol (POCH S.A., Gliwice, Poland) until further processing. Tissues were then dehydrated and paraffin embedded, according to standard histological techniques. The paraffin blocks were cut in slices (5 µm), which were mounted on silanized microscope slides (Sigma-Aldrich, Hamburg, Germany). For immunohistochemistry staining the slides were de-waxed in xylene and were hydrated in a graded series of ethanol to PBS. For antigen retrieval slices were heated in a microwave (650 W) in 400 ml of 10 mM citrate buffer (ph 6.0), twice for 5 min with a 5 min. pause. After 30 min. of cooling slices were rinsed with PBS (Sigma-Aldrich, Hamburg, Germany). A standard protocol of staining was then applied, using EnVision System HRP (DAB) (DAKO, Glostrup, Denmark) with specific sets of antibodies: mouse anti-human CD45 (Becton Dickinson, San Diego, CA, USA) in a 1:200 dilution. The results were analyzed on Bx-60 light microscope (Olympus Optical Co., Hamburg, Germany). Object recognition was performed using the MicroImage on the basis of integrated optical density (IOD - optical density × area of interest) of objects expressing the protein (brown colored) compared to the overall number of cells counted on the basis of IOD blue-related nuclei. Objects under 50 pixels in size were automatically eliminated and the remaining cells were counted. The results are expressed as the mean value of five randomly chosen slides before and after atorvastatin or placebo treatment for each COPD patient.
Gene expression profiling
Sample preparation
Samples obtained by TBB were immediately frozen in liquid nitrogen and stored at –80°C until analyzed. Total RNA was isolated, separately from twelve individual lung samples in patients pre and post atorvastatin treatment and from five individual samples from the placebo group using the NucleoSpin RNA II kit (Macherey-Nagel, Duren, Germany). The quantity and quality of each RNA sample was assessed using Nanodrop (NanoDrop Technologies, Houston, TX, USA) and Agilent 2100 Bioanalyzer (Agilent, Foster City, USA). The RNA Integrity Number (RIN) was above 8 for all samples. Due to insuficient RNA for individual assay, and also due to reduction of the influence of differences between the individuals, possible variation introduced by lung biopsy and tissue preparation, RNA samples were pooled. For the treatment group microarrays were hybridized with three pooled samples and each pool consisted of RNA isolated from four subsequent individuals (n=12). In the placebo group, microarrays were hybridized with three pooled samples and each pool consisted of RNA isolated from five individuals (n=5). Pooling proceeded with quantity and quality control of each RNA sample before and after using NanoDrop and Bioanalyzer. In the placebo group, total RNA samples from five individuals were pooled. Biotinylated cRNA was prepared using the Illumina RNA Amplification Kit (Ambion Inc., Austin, TX, USA) starting with 100 ng total RNA. Samples were purified with the RNeasy kit (Qiagen, Hilden, Germany).
Microarray, hybridization and fluorescent detection
Transcription profiling was performed by one color hybridization of the 47K microarray. Three independent biological microarray replicates were prepared for each experimental group. Each pool was separately converted to cRNA and hybridized to a single microarray. Overall six hybridizations in the treatment group (three before and three after atorvastatin ) and six hybridizations in the placebo group (three before and three after placebo) were performed. Hybridization to the Human HT-12 v4 Expression BeadChip (Illumina, Inc., San Diego USA), washing and scanning were performed using methodology that we have described previously (23-25).
Data normalization and selection of differentially expressed genes
Raw microarray data were processed with the Bead Array and LIMMA packages of the Bioconductor project (Bioconductor project; www.bioconductor.org). Empirical Bayes analysis was performed in order to identify differentially expressed genes based on our previous methodology (23-25). Genes with log fold-changes greater than 0.5 and an adjusted P-value less than 0.05 were considered to be significantly differentially expressed. The Benjamini and Hochberg method(26),for false discovery rate (FDR), was used to correct P-values.
Gene ontology functional clustering
Gene lists (GenBank accession numbers) from microarray results were submitted to the Database for Annotation, Visualization and Integrated Discovery DAVID 6.7 (DAVID; http://david.abcc.ncifcrf.gov/) functional annotation and clustering modules to detect the overrepresented functional gene categories and relationships between annotation categories. Analysis of the association of genes with physiological pathways was performed using the Kyoto Encyclopedia of Genes and Genomes database (KEGG; http://www.genome.jp/kegg/pathway.html). For details see Lisowski et al. (23-25).
Statistical analysis for immunohistochemistry and clinical data
Statistica 10.0 (StatSoft Inc. Tulsa, OK,. USA) statistical software was used in the statistical analysis. The Kolmogorov-Smirnov with Lilliefors significance correction and the Shapiro-Wilk tests were used to evaluate the normality of the distribution of the analyzed quantitative variables. Mann-Whitney U test was used to analyze non-parametric data between groups. The Wilcoxon Matched-Pairs Signed-Ranks Test was used for paired data before and after atorvastatin and placebo treatment within groups. A P value<0.05 was considered statistically significant.
RESULTS
Lung function and other clinical assessments
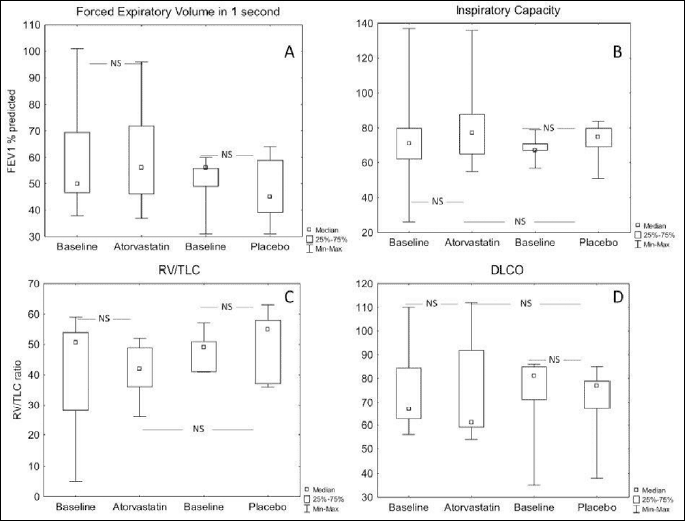
None of the measured parameters of lung function (FEV1, inspiratory capacity, lung volumes, or DLCO) changed after treatment with atorvastatin or placebo (P>0.05) (Fig. 2A-2D).
There was a significant increase in median 6MWD after treatment with atorvastatin (368 versus 375, P=0.002), with insignificant decrease in placebo group (350 versus 330, NS). The analysis of change with active versus change in placebo insignificantly favoured the atorvastatin group after 12-week treatment (375 versus 330, P=0.053) (Fig. 3A).
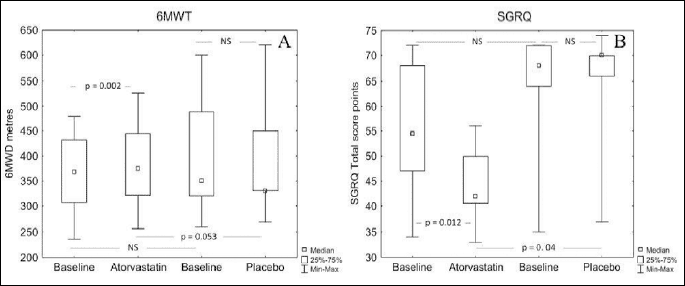
The total SGRQ score decreased after treatment with atorvastatin (54.5 versus 42, P=0.012), with no change in SGRQ in the placebo group (68 versus 70 points, NS) (Fig. 3B). The change with active treatment versus placebo favoured the atorvastatin (42 versus 70 points, P=0.04).
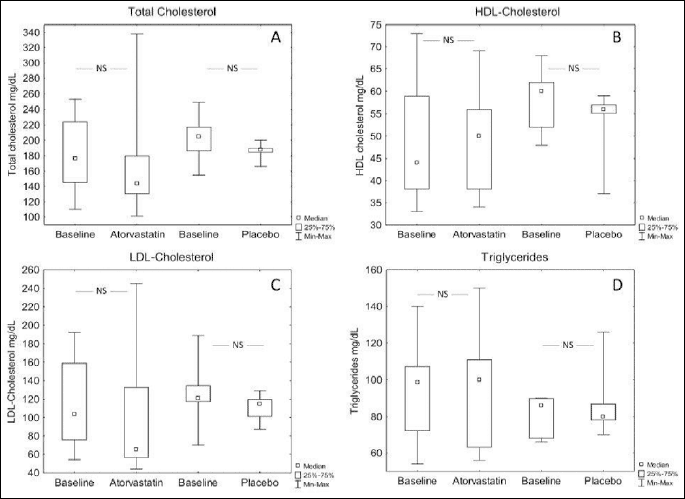
Twelve weeks treatment with atorvastatin or placebo did not change the blood lipid profile (Fig. 4).
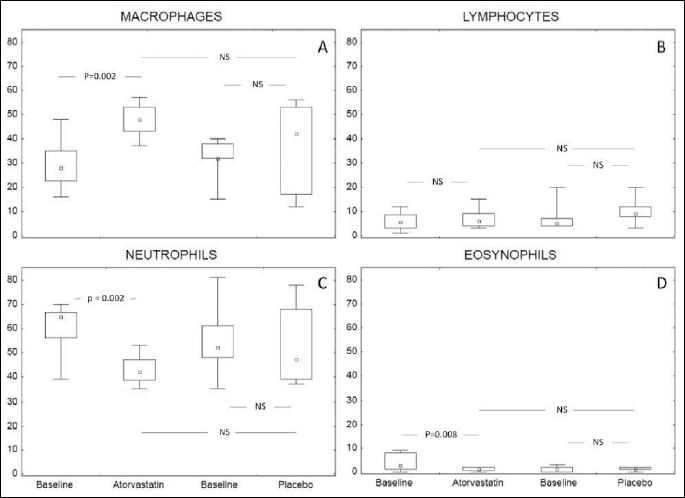
Induced sputum, laboratory assessment and immunohistochemistry
There was a significant decrease in median percentage neutrophil sand eosinophils in induced sputum after atorvastatin treatment (64% versus 42%, P=0.002 and 2.5 versus 1.0%, P=0.008, respectively) with no significant change in placebo group. There was a significant increase in median macrophage percentage after atorvastatin treatment (28% versus 48%, P=002), and insignificant increase in the placebo group (32% versus 42%, NS) (Fig. 5). The change with active versus placebo revealed no significant differencesin all induced cell subpopulations.
There was a significant decrease in median serum hs-CRP from 9.55 to 1.85 mg/l (P=0.002) after atorvastatin treatment with no change in the placebo group. The change with active versus placebo revealed no significant difference (median 1.85 versus 1.30 mg/l, respectively, NS) (Fig. 6).
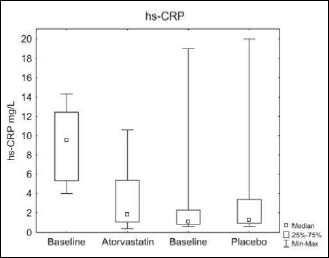 |
Fig. 6. Hs-CRP (A) before and after treatment with atorvastatin of placebo. |
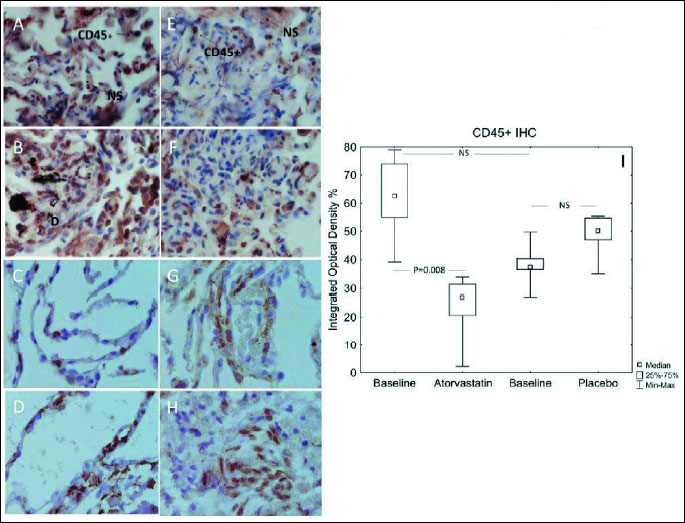
In lung biopsies (TBB) there was a significant decrease in inflammatory cells numbers (CD45+ cells expressed as IOD%) from a median IOD of 62.5 before to 27.0 after atorvastatin treatment (P=0.008). There was insignificant increase in median CD45+ IOD in the placebo group (37.29 versus 50.1, NS) (Fig. 7A-7I). The change in CD45+ cells comparing active versus placebo favoured the atorvastatin group (27.0 versus 50.1, IOD, respectively, P=0.002).
Transcriptome profiling
We assessed global mRNA expression to determine the impact of atorvastatin therapy on the transcriptome in lung tissue. To check whether the identified transcripts are expressed in lung tissue we used the Novartis Gene Expression Atlas database (http://www.biogps.gnf.org/), GeneCards Database (http://www.genecards.org/) screening, and ingenuity pathway analysis (IPA) of top canonical pathways (Fig. 8).
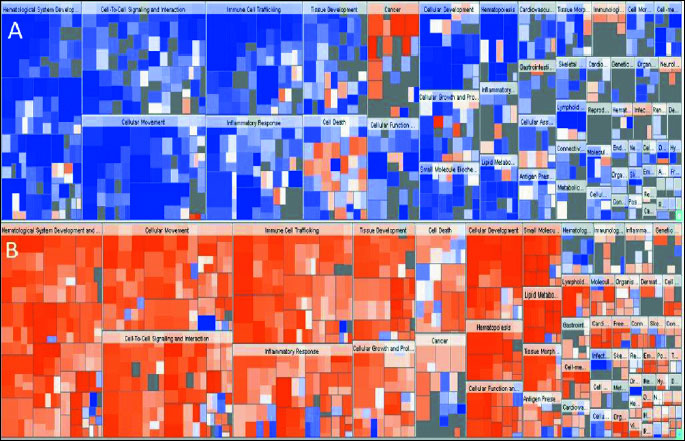
Heatmap shows differentially expressed genes, that are increased or decreased in the (A) atorvastatin dataset, and (B) placebo dataset. The color of the squares in the heatmap reflects the z-score. The color reflects the direction of change for the gene expression: orange -the z-score is positive. IPA predicts that the biological process is trending towards an increase. Z-scores ≥2 indicate that the function is statistically significantly increased. Blue - The z-score is negative. IPA predicts that the biological process is trending towards a decrease. Z-scores ≤ –2 indicate that the function is statistically significantly decreased. The intensity of the colors indicates the prediction strength. The size of the squares in the heatmap reflects the associated –log of the calculated P-value. Larger squares indicate more overlap between the genes that have changed their expression. The P-value is a measure of the likelihood that the association between a set of genes in the dataset and a related function is due to random association. The smaller the P-value (which means a larger –log of that value), the less likely, that the association is random and the more significant the association. In general, P-values <0.05 (–log=1.3) indicate a statistically significant, non-random association.
Transcriptome profiling in lung biopsies after 12-week treatment with atorvastatin revealed more than six hundred genes that met the criteria for differential expression (logFC>0.5, P<0.05). In comparison the lung transcriptomes following placebo revealed more than five hundred genes that met the criteria for differential expression (logFC>0.5, P<0.05). According to the Ingenuity® Knowledge Base the Differentially Expressed Genes (DEGs) are involved in inflammatory responses, tumorigenesis, cell movement and leukocyte migration. atorvastatin treatment resulted in downregulation of immunological response genes including genes related to inflammation of the respiratory system, tumorigenesis, immune cell trafficking and hematological system function which was not shown following placebo. Among the DEGs in the atorvastatin treated group we observed downregulation of key genes involved in the immune response, inflammatory processes and leukocyte/lymphocyte activation including selectin P (SELP), integrin alpha M (CD11B), intracellular adhesion molecule 1 (ICAM1), endothelin 1 (ET1), and endothelin receptor type A (ETAR).
Functional clustering of DEGsin atorvastatin and placebo treated individuals revealed several functional groups of genes. Altogether, 8 clusters were found to be downregulated and 4 clusters were found to be upregulated in the atorvastatin treatment group (Table 2 and 3). In the placebo group 1 cluster was downregulated, and 4 clusters were upregulated (Tables 4 and 5). Specific clusters for atorvastatin treatment revealed downregulation of genes involved in signal transduction, immune effector processes, or activation, regulation and differentiation of leukocytes and lymphocytes. Upregulated genes after atorvastatin treatment were those involved in epigenetic processes of nucleosomes and chromatin assembly, nucleosome organization and DNA packaging, negative regulation of T cell, B cell, leukocyte activation and differentiation. Specific clusters of placebo group contain downregulated genes involved in cell adhesion while upregulated genes are involved in immune system activation and response. Tables 2-5 summarize significantly enriched functional clusters for atorvastatin and placebo treatment comparisons with corresponding P-values and up- and downregulated DEGs.
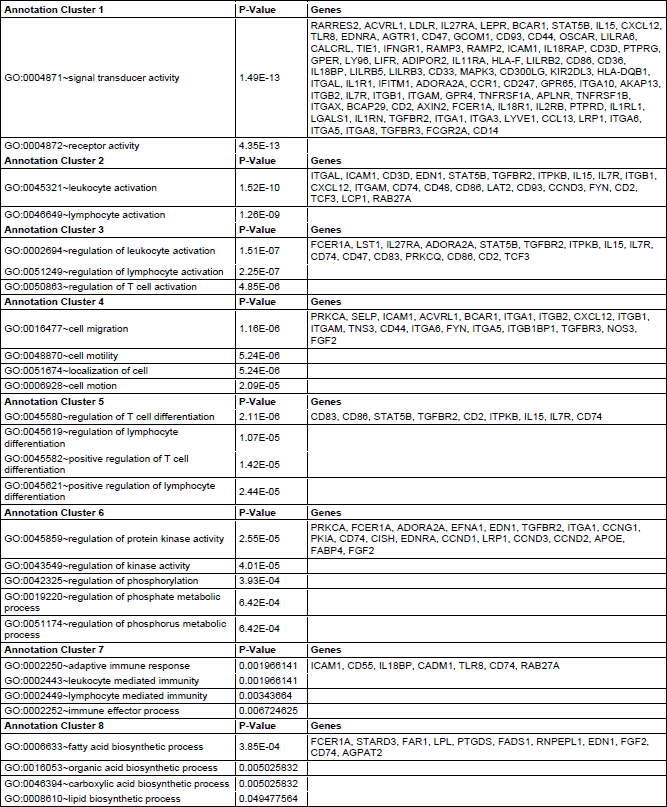
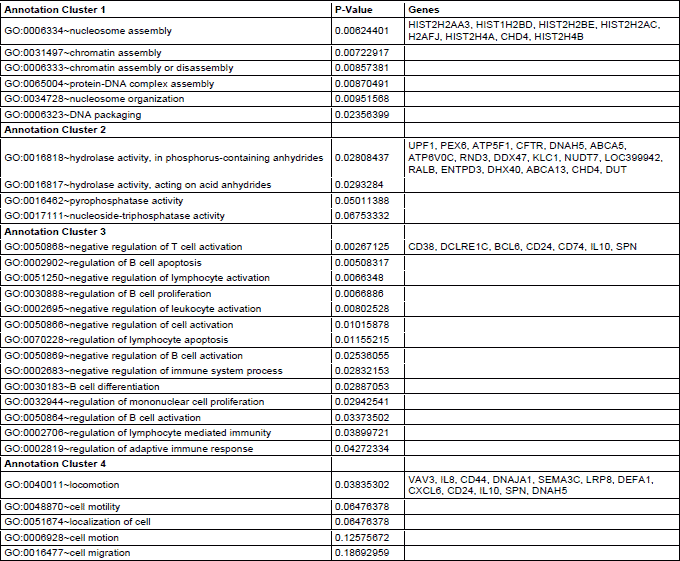
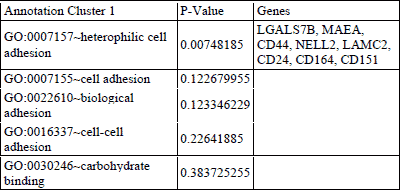
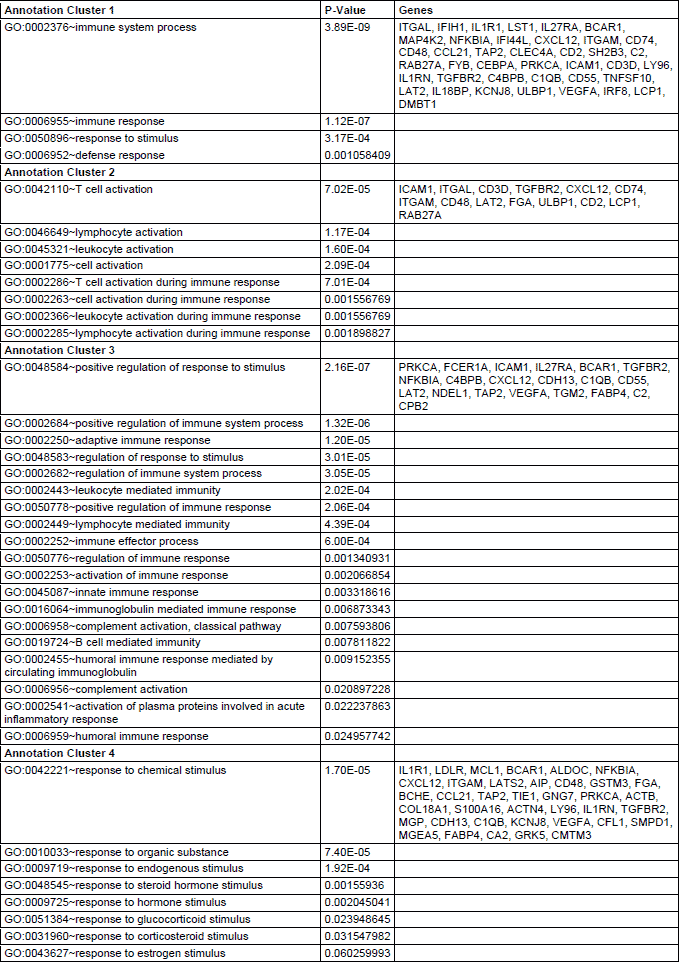
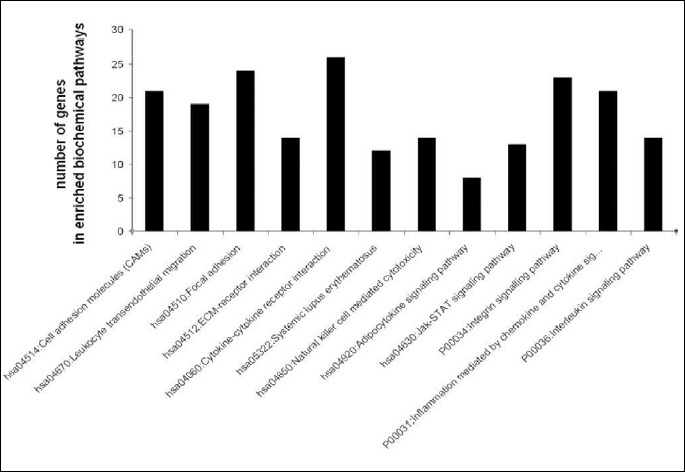
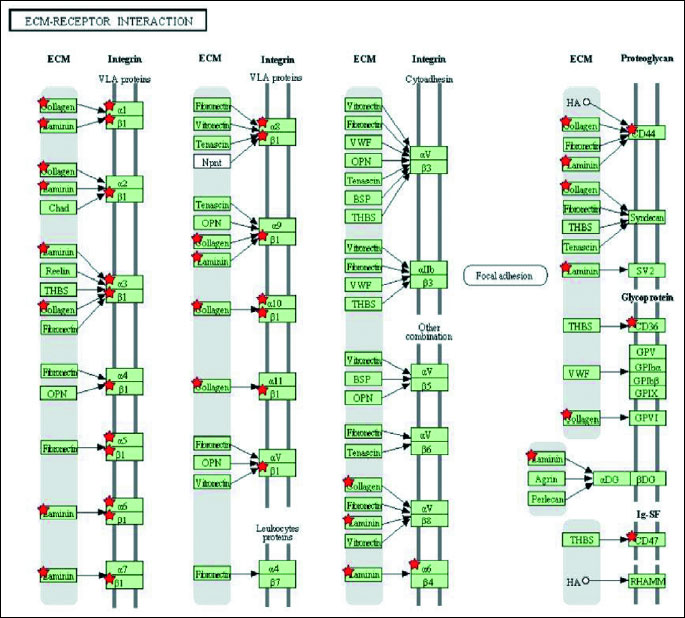
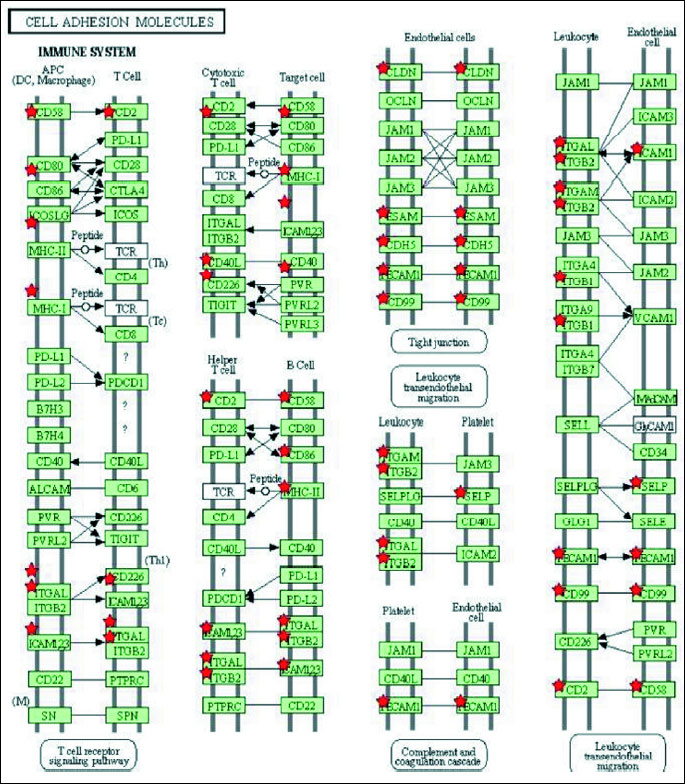
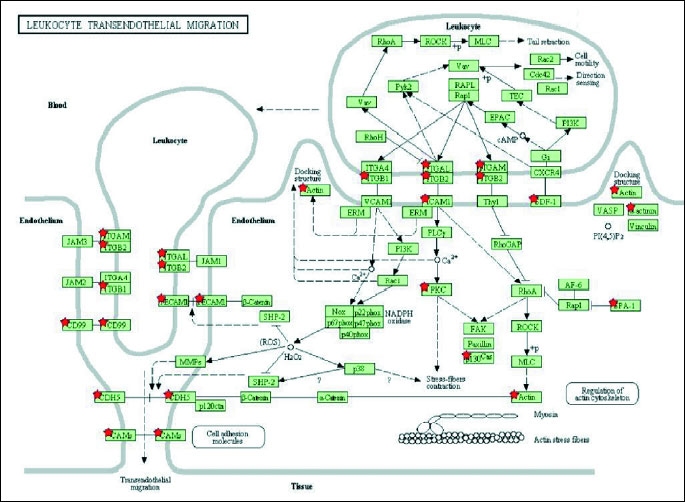
To determine significantly over-represented pathways in the lists of DEGs we searched the Gene Ontology (GO) and KEGG biochemical pathways databases. The most significantly enriched pathways associated with downregulated genes in the atorvastatin group were the cell adhesion molecules (CAMs), those related to leukocyte transendothelial migration, focal adhesion, ECM-receptor interaction, cytokine receptor interaction, adipocytokine signalling pathway, integrin signalling, inflammation mediated by chemokine and cytokine signalling and interleukin signalling; while upregulated genes were involved in pathways associated with drug metabolism. In the placebo group we observed upregulation of pathways involved in focal adhesion, leukocyte migration, complement cascades and ECM- receptor interaction. No pathways could be detected for the group of downregulated genes after placebo treatment. A complete list of identified biochemical pathways with corresponding p-values and associated genes are provided in Table 6. This finding is consistent with a search of the Gene Ontology (GO) database using the annotations for the DEGs, which reveals an enrichment in terms related to immune system functioning in the atorvastatin group. Figs. 9 and 10 show GO biological process and molecular function, Fig. 11 shows biochemical pathway terms associated with DEGs in atorvastatin group. Figs. 12, 13, and 14 represent examples of identified KEGG biochemical pathways in atorvastatin group with the DEGs from the microarray localized on the pathway. No side effects of the treatment were reported during the study period.
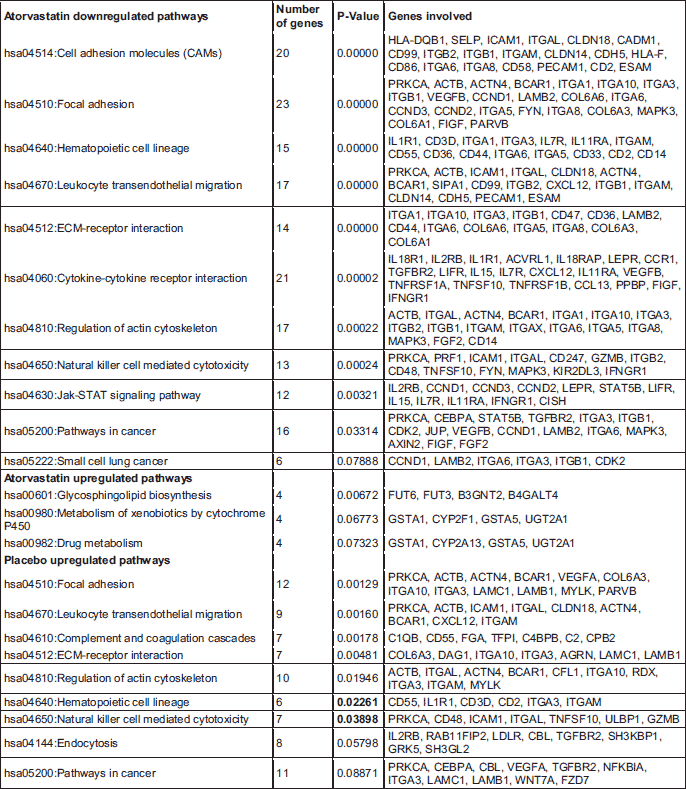
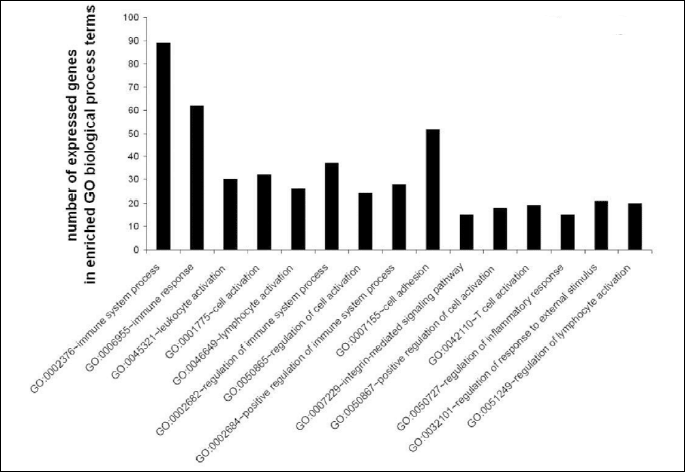
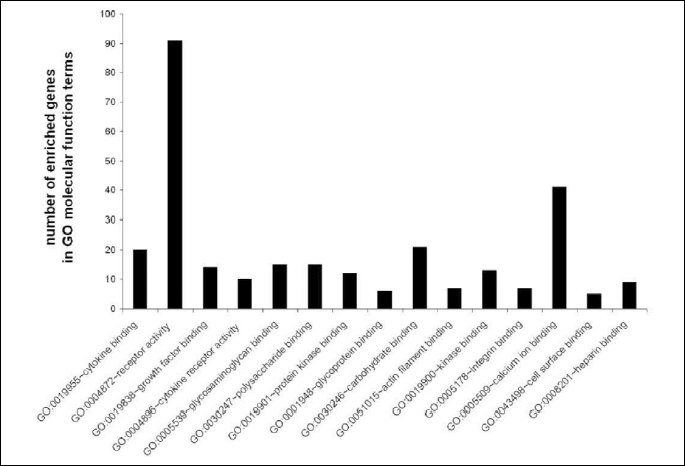
DISCUSSION
There is considerable interest in the use of statins, as a potential treatment in COPD, however the role of statins in treating COPD is still unknown (27, 28). Retrospective observational studies indicate that statin therapy may have beneficial effects on several clinical outcomes in COPD patients, including the number of COPD exacerbations, pulmonary function, exercise capacity, mortality from COPD, and all-cause mortality (9, 29). The short period of treatment in our study did not allow us to test most of these clinical outcomes. However we found a statistically and clinically i.e. 4 or more points drop, decrease in SGRQ and a trend to an increase in 6-MWD without required 25 m increase of clinically significant improvement with no significant changes in pulmonary function after treatment with atorvastatin. Further research on larger groups and the effects of longer term of intervention on clinical outcomes are needed. The impact of atorvastatin on lung function in asthma patients has been studied by Braganza et al. who showed, that short-term treatment with atorvastatin in mild asthmatic smokers did not alter lung function, but improved asthma quality of life (30).
Emerging evidence indicates important consequences of the systemic effects of COPD, particularly adverse cardiovascular effects, and has led to consideration of potential therapies directed at these systemic effects (27). In our study we found that statin treatment produced a reduction in systemic inflammation as shown by a significant decrease in serum hs-CRP after atorvastatin treatment. To the best of our knowledge this is the first confirmation of the anti-inflammatory effects of statins in the lungs in COPD patients. An anti-inflammatory mechanisms of statins and its impact on lungs has recently been studied in in vitro experiments. Marin et al. showed that simvastatin exerts anti-inflammatory effect via inhibition of the mevalonic acid cascade in alveolar macrophages (31). In a prospective study statin treatment in patients with COPD produced a reduction in pulmonary arterial pressures (32). In another study, 3-months treatment with simvastatin did not reduce circulating inflammatory markers in patients with COPD (33). In an experimental study Lee et al. showed in smoke exposed rats that simvastatin inhibited not only lung parenchymal destruction and development of pulmonary hypertension, but also peribronchial and perivascular infiltration with inflammatory cells and induction of matrix metalloproteinase-9 activity in lungs (34). Pretreatment with simvastatin prior to and continued throughout smoke exposure reduced the total influx of leukocytes, neutrophils and macrophages into the lung and airways (16). In our study we observed significant decrease in CD45+ cell number after atorvastatin treatment. Little is known regarding signaling pathway which may be involved. Recent data suggest ERK5 transcriptional activation (35), inhibition of farnesyltransferase and CXC chemokines formation leading to reduction of neutrophil recruitment (36).
To date only a limited number of high-throughput gene expression studies have been conducted on lung tissue samples from individuals with COPD. Gene expression profiling studies found distinct gene expression patterns in lung tissue of patients with severe COPD compared with normal lung. These studies showed differential expression in pathways of oxidative stress, apoptosis, angiogenesis, proteolysis, extracellular matrix, metalloproteinases, infiammatory markers, DNA binding, and regulation of transcription and transcription factors in COPD patients compared with control subjects (37-41). Moreover, Wang et al. observed downregulation of genes participating in anti-infiammatory responsesin COPD patients (40). According to these studies genes involved in tissue remodeling and repair are differentially regulated in the lungs of smokers and may be potential therapeutic targets. In COPD patients who received atorvastatin treatment we report differences in transcriptomic profiles before and after treatment compared with placebo treatment. Atorvastatin therapy resulted in downregulation of several groups of genes involved in immune signalling, leukocyte activation, or immune response, and according to the KEGG pathway classification - downregulation of cell adhesion, focal adhesion, leukocyte transendothelial migration, ECM and cytokine receptors interaction, and NK cell mediated cytotoxicity biochemical pathways. Moreover we observed downregulation of Jak-STAT signalling and several genes involved in pathway related to lung cancer. Interestingly, the placebo group was characterized by upregulation of several groups of genes involved in immune system processes and upregulation of pathways related to cancer, focal adhesion, leukocyte migration, ECM receptor interaction or complement cascades which indicates, in general, the opposite changes in gene expression comparing transcriptomes after placebo and statin treatment. However we did not observed regulation of COPD candidate genes suggested by previous microarray studies, such as SERPINE2 (serpin peptidase inhibitor) or clade E (nexin, plasminogen activator inhibitor type 1) (38, 42), but EGR1 (early growth response 1) was upregulated in atorvastatin treated group with no differences in the placebo group.
In studies conducted by others, several genes were found to be differentially expressed comparing lung tissue from subjects with severe or little to no emphysema. These included genes involved in the extracellular matrix (upregulated in patients with emphysema), immune and cell signalling (downregulated in patients with emphysema), as well as those involved in stress-related processes including oxidoreductases, isomerases, and genes involved in complement activity (37, 43-45). In our study, among genes downregulated after atorvastatin treatment we found downregulation of a key genes involved in the immune response such as selectin P (SELP), granule membrane protein 140 kDa, antigen (CD62), integrin alpha M (complement component 3 receptor 3 subunit, intercellular adhesion molecule 1(ICAM1), intercellular adhesion molecule 1, (ET1), and endothelin receptor type A (ETAR). In contrast, in the placebo group we found upregulation of several genes involved in pathway related to complement and coagulation cascades.
Another important observation was downregulation of pathways related to cancer following statin treatment. COPD and lung cancer occur commonly together and COPD could be a confounding factor in the genetics of lung cancer susceptibility (46). In the atorvastatin treated group 16 downregulated genes were classified in cancer pathways, while in the placebo group we observed upregulation of eleven genes involved in cancer indicating opposite regulation of cancer related gene expression in the atorvastatin and placebo groups.
Conclusions
The main finding of our study is the demonstration of an anti-inflammatory effect of statins in the lungs of patients with COPD associated with changes in the pattern of gene expression in lung tissue after atorvastatin treatment compared to placebo treated patients. These data support the use of atorvastatin as an anti-inflammatory drug in COPD.
Limitations
Gene expression is routinely quantified by measuring mRNA levels. However, it is not certain how closely levels of mRNA relate to levels of their corresponding proteins.
Our study does not elucidate the mechanism for the anti-inflammatory effect of statins in the lungs of COPD patients. The study is in small numbers and the measurements of cellular inflammation are limited.
Authors contribution: R.M.M. conceived of the study, and participated in its design and coordination, carried out bronchoscopy and TBB, analysis and interpretation of the results and drafting of the manuscript. PL participated in study design and carried out the molecular genetic studies, contributed to the analysis and interpretation of the results, and revision of the manuscript. AT participated in study design, carried out cardiology clinical studies and helped to draft the manuscript. JB participated in study design and carried out IHC studies and helped to draft the manuscript. PZT carried out IHC studies and helped to draft the manuscript. LM performed 6MWT and SGRQ and helped to draft the manuscript. RM performed statistical analysis and helped to draft the manuscript. AL carried out cardiology clinical studies and helped to draft the manuscript. PB performed lung function statistical analysis and helped to draft the manuscript. BS contributed to the analysis and interpretation of the results, and revision of the manuscript. AMD contributed to the analysis and interpretation of the results, and revision of the manuscript. EC contributed to the analysis and interpretation of the results, and revision of the manuscript. WJM contributed to the analysis and interpretation of the results, and revision of the manuscript. WMN contributed to the study idea and design, analysis and interpretation of the results, and revision of the manuscript. All authors read and approved the final manuscript.
Acknowledgements: The study was supported by National Science Centre (NCN) project No: N N402 593440.
Conflict of interest: None declared.
REFERENCES
- Kjekshus J, Pedersen TR. Reducing the risk of coronary events: evidence from the Scandinavian Simvastatin Survival Study (4S). Am J Cardiol 1995; 76: 64C-68C.
- Gordon DJ, Rifkind BM. 3-Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors: a new class of cholesterol-lowering agents. Ann Intern Med. 1987; 107: 759-761.
- Faggiotto A, Paoletti R. State-of-the-art lecture. Statins and blockers of the renin-angiotensin system: vascular protection beyond their primary mode of action. Hypertension 1999; 34: 987-996.
- Farmer JA. Pleiotropic effects of statins. Curr Atheroscler Rep 2000; 2: 208-217.
- Korybalska K, Kawka E, Breborowicz A, Witowski J. Atorvastatin does not impair endothelial cell wound healing in an in vitro model of vascular injury. J Physiol Pharmacol 2012; 63: 389-395.
- Briel M, Studer M, Glass TR, Bucher HC. Effects of statins on stroke prevention in patients with and without coronary heart disease: a meta-analysis of randomized controlled trials. Am J Med 2004; 117: 596-606.
- Briel M, Schwartz GG, Thompson PL, et al. Effects of early treatment with statins on short-term clinical outcomes in acute coronary syndromes: a meta-analysis of randomized controlled trials. JAMA 2006; 295: 2046-2056.
- Gurm HS, Hoogwerf B. The Heart Protection Study: high-risk patients benefit from statins, regardless of LDL-C level. Cleve Clin J Med 2003;70:991-997.
- Janda S, Park K, FitzGerald JM, Etminan M, Swiston J. Statins in COPD: a systematic review. Chest 2009; 136: 734-743.
- van Eijl S, Mortaz E, Versluis C, Nijkamp FP, Folkerts G, Bloksma N. A low vitamin A status increases the susceptibility to cigarette smoke-induced lung emphysema in C57BL/6J mice. J Physiol Pharmacol 2011; 62: 175-182.
- Mikolka P, Mokra D, Drgova A, Petras M, Mokry J. Dimethyl sulfoxide in a 10% concentration has no effect on oxidation stress induced by ovalbumin-sensitization in a guinea-pig model of allergic asthma. J Physiol Pharmacol 2012; 63: 179-186.
- Sharman JE, Johns DP, Marrone J, Walls J, Wood-Baker R, Walters EH. Cardiovascular effects of methacholine-induced airway obstruction in man. J Physiol Pharmacol 2014; 65: 401-407.
- Chen YJ, Chen P, Wang HX, et al. Simvastatin attenuates acrolein-induced mucin production in rats: involvement of the Ras/extracellular signal-regulated kinase pathway. Int Immunopharmacol 2010; 10: 685-693.
- Arimura K, Aoshiba K, Tsuji T, Tamaoki J. Chronic low-grade systemic inflammation causes DNA damage in the lungs of mice. Lung 2012; 190: 613-620.
- Miyata R, Bai N, Vincent R, Sin DD, Van Eeden SF. Statins reduce ambient particulate matter-induced lung inflammation by promoting the clearance of particulate matter, < 10 µm from lung tissues. Chest 2013; 143: 452-460.
- Davis BB, Zeki AA, Bratt JM, Wang L, Filosto S, Walby WF, et al. Simvastatin inhibits smoke-induced airway epithelial injury: implications for COPD therapy. Eur Respiratory J 2013; 42: 350-361.
- Celli BR, MacNee W, ATS/ERS Task Force. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J 2004; 23: 932-946.
- Miller MR, Crapo R, Hankinson J, et al. General considerations for lung function testing. Eur Respir J 2005; 26: 153-161.
- Holownia A, Mroz R, Noparlik J, Chyczewska E, Braszko J. Expression of CREB-binding protein and peroxisome proliferator-activated receptor gamma during formoterol or formoterol and corticosteroid therapy of chronic obstructive pulmonary disease. J Physiol Pharmacol 2008; 59 (Suppl. 6): 303-309.
- Guidelines for fiberoptic bronchoscopy in adults. American Thoracic Society. Medical Section of the American Lung Association. Am Rev Respir Dis 1987; 136: 1066.
- British Thoracic Society Bronchoscopy Guidelines Committee, a Subcommittee of Standards of Care Committee of British Thoracic Society. British Thoracic Society guidelines on diagnostic flexible bronchoscopy. Thorax 2001; 56 (Suppl. 1): i1-i21.
- Ernst A, Silvestri GA, Johnstone D, American College of Chest Physicians. Interventional pulmonary procedures: Guidelines from the American College of Chest Physicians. Chest 2003; 123: 1693-1717.
- Lisowski P, Stankiewicz AM, Goscik J, et al. Effects of chronic stress on prefrontal cortex transcriptome in micedisplaying different genetic backgrounds. J Mol Neurosci 2013; 50: 33-57.
- Lisowski P, Stankiewicz AM, Goscik J, Wieczorek M, Zwierzchowski L, Swiergiel AH. Selection for stress-induced analgesia affects the mouse hippocampal transcriptome. J Mol Neurosci 2012; 47: 101-112.
- Lisowski P, Juszczak GR, Goscik J, Wieczorek M, Zwierzchowski L, Swiergiel AH. Effect of chronic mild stress on hippocampal transcriptome in mice selected for high and low stress-induced analgesia and displaying different emotional behaviors. Eur Neuropsychopharmacol 2011; 21: 45-62.
- Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med 1990; 9: 811-818.
- Cazzola M, Matera M, Rogliani P, Page C. Treating systemic effects of COPD. Trends Pharmacol Sci 2007; 28: 544-550.
- Young RP, Hopkins R, Eaton TE. Pharmacological actions of statins: potential utility in COPD. Eur Respir Rev 2009; 18: 222-232.
- Lawes CM, Thornley S, Young R, et al. Statin use in COPD patients is associated with a reduction in mortality: a national cohort study. Prim Care Respir 2012; 12: 35-40.
- Braganza G, Chaudhuri R, McSharry C, et al. Effects of short-term treatment with atorvastatin in smokers with asthma - a randomized controlled trial. BMC Pulm Med 2011; 11: 16.
- Marin L, Colombo P, Bebawy M, Young PM, Traini D. Chronic obstructive pulmonary disease: patho-physiology, current methods of treatment and the potential for simvastatin in disease management. Expert Opin Drug Deliv 2011; 8: 1205-1220.
- Reed RM, Iacono A, DeFilippis A, et al. Statin therapy is associated with decreased pulmonary vascular pressures in severe COPD. COPD 2011; 8: 96-102.
- Kaczmarek P, Sladek K, Skucha W, et al. The influence of simvastatin on selected inflammatory markers in patients with chronic obstructive pulmonary disease. Pol Arch Med Wewn 2010; 120: 11-17.
- Lee JH, Lee DS, Kim EK, et al. Simvastatin inhibits cigarette smoking-induced emphysema and pulmonary hypertension in rat lungs. Am J Respir Crit Care Med 2005; 172: 987-993.
- Le NT, Takei Y, Izawa-Ishizawa Y, et al. Identification of activators of ERK5 transcriptional activity by high-throughput screening and the role of endothelial ERK5 in vasoprotective effects induced by statins and antimalarial agents. J Immunol 2014; 193: 3803-3815.
- Zhang S, Rahman M, Zhang S, Jeppsson B, Herwald H, Thorlacius H. Streptococcal m1 protein triggers farnesyltransferase-dependent formation of CXC chemokines in alveolar macrophages and neutrophil infiltration of the lungs. Infect Immun 2012; 80: 3952-3959.
- Golpon HA, Coldren CD, Zamora MR, et al. Emphysema lung tissue gene expression profiling. Am J Respir Cell Mol Biol 2004; 31: 595-600.
- Ning W, Lee J, Kaminski N, et al. Comprehensive analysis of gene expression on GOLD-2 versus GOLD-0 smokers reveals novel genes important in the pathogenesis of COPD. Proc Am Thorac Soc 2006; 3: 466.
- Zeskind JE, Lenburg ME, Spira A. Translating the COPD transcriptome: insights into pathogenesis and tools for clinical management. Proc Am Thorac Soc 2008; 5: 834-841.
- Wang IM, Stepaniants S, Boie Y, et al. Gene expression profiling in patients with chronic obstructive pulmonary disease and lung cancer. Am J Respir Crit Care Med 2008; 177: 402-411.
- Bhattacharya S, Tyagi S, Srisuma S, et al. Peripheral blood gene expression profiles in COPD subjects. J Clin Bioinforma 2011; 1: 12. doi: 10.1186/2043-9113-1-12.
- Hersh CP, DeMeo DL, Raby BA, et al. Genetic linkage and association analysis of COPD-related traits on chromosome 8p. COPD 2006; 3: 189-194.
- Spira A, Beane J, Pinto-Plata V, et al. Gene expression profiling of human lung tissue from smokers with severe emphysema. Am JRespir Cell Mol Biol 2004; 31: 601-610.
- Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol 2002; 3: 932-943.
- Silverman EK. Genetic epidemiology of COPD. Chest 2002; 121 (Suppl. 3): 1S-6S.
- Young RP, Hopkins RJ, Hay BA, Epton MJ, Black PN, Gamble GD. Lung cancer gene associated with COPD: triple whammy or possible confounding effect? Eur Respir J 2008; 32: 1158-1164.
A c c e p t e d : November 15, 2014