ANTI-INFLAMMATORY EFFECTS OF SIMVASTATIN IN NONSTEROIDAL
ANTI-INFLAMMATORY DRUGS-INDUCED SMALL BOWEL INJURY
2Gachon Medical Research Institute, Gachon University Gil Medical Center, Incheon, Republic of Korea
INTRODUCTION
Nonsteroidal anti-inflammatory drugs (NSAIDs) are used as anti-inflammatory agents for several diseases. However, NSAIDs have adverse effects on the stomach and small bowel, including the duodenum. NSAID-induced small bowel injury is generally ignored due to the difficulty in diagnosis. However, NSAIDs have been reported to cause damage to the gastric and small bowel mucosa with a relatively high frequency (1, 2). Gastrointestinal mucosal injury caused by NSAIDs can manifest as simple inflammatory mucosal changes, ulceration, bleeding, and even perforation (3, 4). Proton pump inhibitors have been used as the standard treatment for such gastric injuries. Moreover, other agents such as misoprostol, probiotics, lactoferrin, rebamipide, NSAID pro-drugs, and selective COX-2 inhibitors have been evaluated in terms of their ability to prevent or treat small bowel injury (5). However, no effective drug is available for the prevention or treatment of small bowel injury. NSAID-induced gastrointestinal injury occurs by various mechanisms: inhibition of COX-1 enzyme and gastro-protective prostaglandin synthesis, increased intestinal permeability, uncoupling of mitochondrial oxidative phosphorylation, and inflammation (6).
Statins are inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase and play important roles in cholesterol synthesis. For this reason, they are used as lipid-lowering agents. The efficacy of statins against other conditions such as osteoporosis (7), several type of cancer (8-10), and rheumatoid arthritis (11) have been evaluated. Anti-inflammatory and antioxidant effects of statins have also been reported (12-15). One mechanism of NSAID-induced gastrointestinal injury is oxidative stress-induced inflammation. Therefore, we evaluated the anti-inflammatory effect of statins against NSAID-induced gastrointestinal injury.
MATERIALS AND METHODS
Reagents and materials
TNF-α was purchased from Peprotech (Minneapolis, MN). Simvastatin was obtained from Sigma-Aldrich (St. Louis, MO). 2', 7'-dichlorofluorescein diacetate (DCFH-DA) and Alexa Fluor 488 goat anti-mouse antibody were purchased from Molecular Probes (Eugene, OR). Antibodies to phospho NF-κB p65 (Ser 536), NF-κB p65, Akt, phospho Akt (Ser 473), IkBa, and phospho-IkBa were purchased from Cell Signaling Technology (Beverly, MA). 8-Hydroxyguanosine antibody was purchased from Abcam. TNF-α antibody was purchased from Santa Cruz biotechnology.
Cell culture
Rat normal small bowel IEC-6 cells were purchased from the Korean Cell Lines Bank (KCLB, Seoul, Korea) and cultured in Dulbecco's modified Eagle's medium (GIBCO, NY) supplemented with 10% fetal bovine serum (GIBCO) and 1% antibiotic-antimycotic in a humidified 5% CO2 atmosphere. The cells were treated with TNF-α (50 ng/ml) or simvastatin (0.5 µM).
Real-time quantitative PCR
Total RNA was purified using an RNeasy Plus Mini Kit (Qiagen, Hilden, Germany), and cDNA was synthesized from 1 µg of total RNA using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems,USA). For relative quantitation, the reactions were tested using TaKaRa SYBR Premix Ex taq II (TaKaRa, Japan), and PCR processing was carried out in an iCycler (Bio-Rad, Hercules, CA). The sequences of primers for rat IL-6 were 5'-AAAGCCAGAGTCATTCAGAGC-3' (sense) and 5'-GAGCATTGGAAGTTGGGGTA-3' (antisense), and those for rat IL-8 were 5'-CAT TAA TAT TTA ACG ATG TGG ATG CGT TTC A-3' (sense) and 5'-GCC TAC CAT CTT TAA ACT GCA CAAT-3' (antisense). The sequences of primers for rat COX-1 were 5'-ATCGCCATGGAATTCAACCA-3' (sense) and 5'-GTGAGCCCACTTGGAAGGAA-3' (antisense), and those for rat COX-2 were 5'-TGATCGAAGACTACGTGCAACA-3' (sense) and 5'-AAAAGCAGCTCTGGGTCGAA-3' (antisense). The sequences of primers for GAPDH were 5'-ACT TTG TCA AGCTCA TTT CC-3' (sense) and 5'-TGC AGC GAA CTT TATTGA TG-3' (antisense). The copy number of the target gene was normalized to an endogenous reference, GAPDH. The fold change from normal samples was set at 1-fold, and the normalized fold change ratio was calculated.
Immunoblotting
Cells were lysed in cold RIPA lysis buffer (0.5 M Tris-HCl, pH 7.4, 1.5 M NaCl, 2.5% deoxycholic acid, 10% NP-40, 10 mM EDTA) containing protease and phosphatase inhibitors (GenDEPOT, CA, USA). Cell lysates were subjected to SDS-PAGE and transferred to a nitrocellulose membrane. After blocking with 8% skim milk or 5% bovine serum albumin, the membrane was probed with anti-phospho Akt, anti-phospho IkBa, and anti-phospho NF-κBα p65 antibodies, followed by incubation with a secondary antibody conjugated to horseradish peroxidase. The membranes were then stripped and reprobed with anti-Akt, anti-IκBα, anti-NF-κB p65, and anti-β-actin antibodies.
Reactive oxygen species assay
Intracellular ROS was assayed by measuring the fluorescence of DCF, which is the oxidized product of DCF-DA in stained cells. IEC-6 cells were stimulated with TNF-α or simvastatin for 15 min. Cells were then washed in PBS and incubated with 25 µM DCF-DA in the dark for 40 min. DCF fluorescence was detected by confocal microscopy.
Animal model
Animals were handled in an accredited animal facility in accordance with Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC International) guidelines under the facility named CACU (The Center of Animal Care and Use) of Gachon University Lee Gil Ya Cancer and Diabetes Institute after IRB approval (LCDI-2014-0051). Seven weeks old SPF male C57BL/6 mice (Orient Bio, Sungnam, Korea) were used for the experiments.
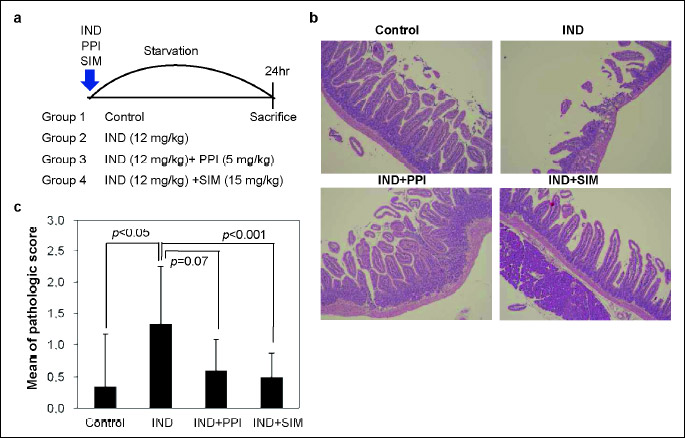
A total of 32 mice were divided into 4 groups (n = 8):
Group 1, a non-treated control group;
Group 2, indomethacin (12 mg/kg) induced damage group;
Group 3, indomethacin (12 mg/kg) with PPI (pantoprazole, 5 mg/kg);
Group 4, indomethacin (12 mg/kg) plus simvastatin (15 mg/kg).
All groups were starved for 4 hours before treatments, then animals were treated for 24 hours and sacrificed. All stomachs and small intestines of mice were removed and opened along the greater curvature and then washed with ice cold PBS solutions. Isolated proximal small intestines were subjected to H&E, immunohistochemistry, and immunofluorescencestaining.
Histological scoring
Histologic score for specimen was analyzed according to the previous report (16). Previous histologic score system for colitis evaluation was modified for analyzing the small bowel inflammation. Degree of lamina propria and submucosal mononuclear cellular infiltration, crypt hyperplasia, goblet cell depletion, and architectural distortion were used for histologic score.
Immunohistochemistry
Proximal small intestinal in paraffin-embedded blocks were sectioned and mounted on frost-free slides. Sections (3 – 10 µm) were deparaffinized in xylene and rehydrated through a graded ethanol series. Antigen retrieval was achieved by immersion of the sections in 10 mM sodium citrate buffer (pH 6.0), heating to 95°C for 10 min, and cooling in cold water for 30 min. Slides were treated with 3% hydrogen peroxidase for 10 min to block endogenous peroxidase, rinsed in phosphate-buffered saline (PBS) for 5 min, and then incubated with a mouse monoclonal antibody to 8-hydroxyguanosine (1:200, ab48508; Abcam, Cambridge, MA) at 4°C for 2 hours. After three PBS washes (5 min), sections were incubated with a secondary horseradish peroxidase-conjugated anti-rabbit/mouse antibody (Dako, Carpinteria, CA, USA) at room temperature for 20 min, followed by DAB + chromogen solution (Dako) for 5 min. Nuclei were counterstained with hematoxylin. Relatively normal mucosa in 4 slides of each IHC sample were observed at 20 × using microscope and images captured by an Olympus DP72 microscope (Olympus, Tokyo, Japan). Pictures were quantified by DP2-BSW (Olympus soft imaging solution GmbH, Germany).
Immunofluorescence
Immunofluorescence was used a TNF-α antibody (1:100, sc-52746, Santa Cruz biotechnology, Santa Cruz, CA) and secondary Alexa Fluor 488 antibody (1:500, A11001, molecular probes Inc., Eugene OR, USA). Nuclei were stained using Vectashield mounting medium for fluorescence with DAPI (H-1200, Vector Laboratories, Burlingame, CA). Images were performed using Zeiss LSM confocal microscope.
Statistical analysis
Histologic scores were analyzed with Mann Whitney test (SAS; Cary, NC). The paired Student's T-test was used to compare all other data. Statistical significance was defined as P < 0.05 for comparisons indicated. Data are reported as mean values ± standard error of the mean (S.E.), unless otherwise indicated.
RESULTS
Effects of simvastatin on TNF-α-induced inflammatory gene expression
Interleukin-6 (IL-6) and IL-8, inflammatory cytokines, contribute to inflammation caused by TNF-α. The effect of simvastatin on TNF-α-induced inflammation was investigated in IEC-6 cells, nontransformed small intestinal cells. IEC-6 cells were incubated in the presence of TNF-α for 24 hours. TNF-α treatment induced the expression of IL-6 and IL-8, but the effect was diminished by treatment with simvastatin (Fig. 1).
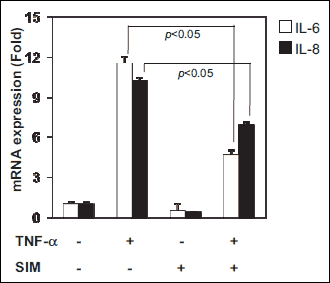 |
Fig. 1. Inhibition of IL-6 or IL-8 expression in 50 ng/ml TNF-α induced inflammation with 0.5 uM simvastatin in IEC-6 cell line. IEC-6 cells were treated with 50 ng/ml TNF-α and/or 0.5 uM simvastatin for 24 hours. mRNA expressions of IL-6 and IL-8 were determined by qRT-PCR. Asterisks indicate statistical significance (P < 0.05). |
Simvastatin inhibits TNF-α-induced phosphorylation of Akt
Activation of TNF-α can trigger the activation of Akt. We examined whether simvastatin modulates TNF-α-induced Akt phosphorylation and found that simvastatin treatment inhibited TNF-α-mediated Akt phosphorylation (Fig. 2a). TNF-α induced Ser-473 phosphorylation of Akt in control cells within 15 min, and the peak effect occurred at 45 min. However, TNF-α did not accelerate Akt phosphorylation in cells pretreated with 0.5 µM simvastatin. These results indicate that simvastatin inhibits TNF-α-induced Akt phosphorylation.
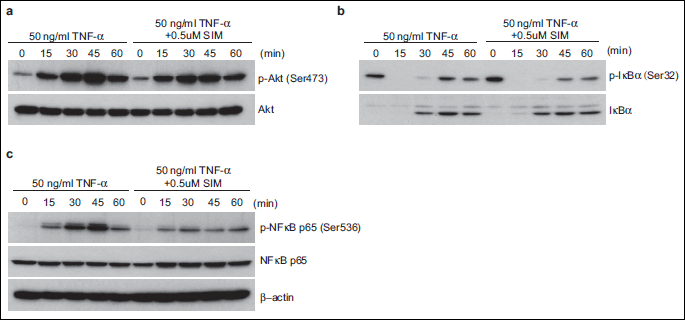
Simvastatin promotes TNF-α-mediated degradation of IkBa
Translocation of NF-κB to the nucleus is preceded by proteolytic degradation of IκBα (17). To determine whether the NF-κB-inhibitory activity of simvastatin is the result of IkBa degradation, we exposed IEC-6 cells pretreated with simvastatin to 50 ng/ml TNF-α for various times and examined the IkBa status in the cytoplasm by Western blotting. TNF-α induced IkBa degradation in control cells within 15 min, and the effect had disappeared at 45 min. Simvastatin maintained TNF-α-induced IκBα degradation (Fig. 2b). Under these conditions, simvastatin inhibited phosphorylation of IκBα by treatments of TNF-α. These results indicate that simvastatin directly activated TNF-α-induced IkBa degradation.
Simvastatin inhibits TNF-α-induced activation of p65
TNF-α induces the phosphorylation of NF-κB p65, which is required for its transcriptional activity (18). We next determined whether simvastatin affects TNF-α-induced NF-κB activation. IEC-6 cells were pretreated with 0.5 µM simvastatin and then stimulated with 50 ng/ml TNF-α. We examined whether simvastatin modulates TNF-α-induced Ser-536 phosphorylation of p65 through the Akt pathway. Western blot analysis showed that TNF-α induced the phosphorylation of p65, and the maximum effect occurred at 45 min (Fig. 2c). These results suggest that simvastatin inhibits TNF-α-mediated phosphorylation of NF-κB p65 via reducing of Akt phosphorylation.
Simvastatin inhibits TNF-α-induced cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) expression
To investigate the influence of simvastatin on TNF-α-induced COX-1 and COX-2 expression, we assessed their mRNA levels by qRT-PCR. Treatment with 50 ng/ml TNF-α induced overexpression of both COX-1 and COX-2 mRNA, while pretreatment with simvastatin reduced TNF-α-induced expression of these genes in IEC-6 cells (Fig. 3a). However, western blotting showed that the effect on COX-1 and COX-2 protein levels of treatment with the combination of TNF-α and simvastatin was similar to that of TNF-α alone (Fig. 3b). These results suggest that simvastatin inhibits the TNF-α-induced transcription of the COX-1 and COX-2 genes but does not affect COX-1 and COX-2 protein levels.
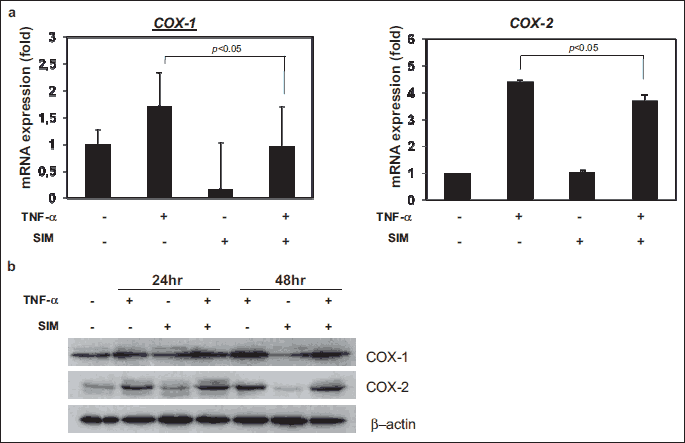
Simvastatin inhibits intracellular ROS generation in TNF-α-stimulated IEC-6 cells
TNF-α has been shown to bind to receptors and generate ROS in various cell lines (19). To investigate whether ROS production was induced during TNF-α-mediated inflammation, intracellular ROS levels were analyzed using the cell-permeable, oxidation-sensitive dye DCF-DA. ROS-mediated oxidation of this dye was detected by confocal laser-scanning microscopy. TNF-α-treated cells exhibited greater DCF-DA fluorescence intensity than untreated cells (Fig. 4). However, DCF-DA fluorescence intensity was decreased by treatment with simvastatin. These findings suggest that simvastatin reduces TNF-α-induced ROS production.
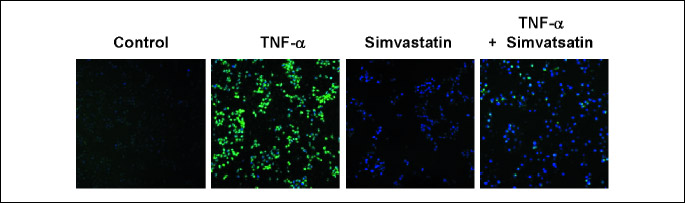
Effect of simvastatin in vivo
To investigate the anti-inflammatory effect of simvastatin (10 mg/kg), an indomethacin (12 mg/kg)-induced mouse model of inflammation was established (Fig. 5a).
Histopathological examination showed that indomethacin disrupted epithelial stratification and induced basal lamina degeneration and infiltration of inflammatory cells (Fig. 5b). The number of villi was significantly lower in mice after oral administration of indomethacin than control mice. In contrast, simvastatin pretreatment reduced the structural damage to the small intestine and increased the mean villus height compared with indomethacin treatment alone. The histological score for inflammation was evaluated for each group. Comparing to control group, indomethacin groups showed the statistically higher histological score. However, histological score for inflammation of duodenum was decreased after treatment with simvastatin (Fig. 5c).
Indomethacin-induced damage induced expression of TNF-α (20, 21). Immunofluorescence image of TNF-α show that simvastatin reduced indomethacin induced expression of TNF-α in small intestine (Fig. 6a).
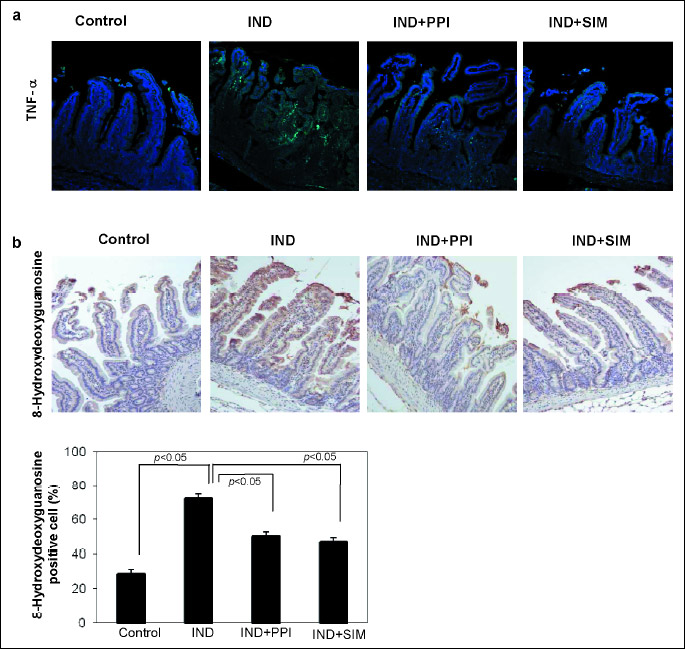
One of the key molecular mechanisms of NSAID-induced damage is the robust induction of ROS in the small intestine. 8-Hydroxydeoxyguanosine (8-OHdG) is a useful marker for assessing DNA damage due to ROS. Representative images of 8-OHdG staining of the small intestine are show in Fig. 6b. Treatment with indomethacin resulted in a higher level of 8-OHdG than in the control mucosal area. The numbers of 8-OHdG-positive cells were statistically smaller after treatment with the combination of indomethacin plus PPI or simvastatin than treatment with indomethacin alone.
DISCUSSION
NSAIDs can reduce inflammation and pain. However, in the presence of Helicobacter pylori infection, NSAIDs are one of two major causes of peptic ulcers (22, 23). Inhibition of COX1 and prostaglandin (24) increased membrane permeability (25), and generation of pro-inflammatory mediators are the three main mechanisms of NSAID-induced gastrointestinal injury (26). Many inflammatory mediators can be involved in response to gastrointestinal injury (27-29). Pro-inflammatory mediators, such as TNF-α, can release oxygen-derived free radicals in damaged microvessels, which can damage or destroy the mucosa by lipid peroxidation (30).
This study investigated the effect of simvastatin on gastro-intestinal inflammation by assessing TNF-α-induced expression of NF-κB-mediated genes, COX expression, ROS production, and NSAID-induced inflammation in the mouse small intestine.
We found that simvastatin inhibited the expression of inflammatory genes induced by TNF-α (Fig. 1). This effect of simvastatin correlated with down-regulation of NF-κB-mediated gene expression, which was regulated by TNF-α. Simvastatin suppressed constitutive, but not inducible, NF-κB expression. Suppression of NF-κB activation by simvastatin was due to inhibition of TNF-α-induced NF-κB phosphorylation.
We report here for the first time that the anti-inflammatory effect of simvastatin was due to inhibition of TNF-α-induced inflammation in the small intestine. Simvastatin specifically reduced TNF-α-induced Akt phosphorylation at Ser373. However, it had no effect on Akt phosphorylation at Thr308 (data not shown). Therefore, simvastatin affects a specific site of Akt or upstream protein kinases that phosphorylates Akt at the Ser373 residue.
Moreover, TNF-α-induced IκBα degradation was increased by simvastatin. Simvastatin did not affect phosphorylation of IκBα at Ser 32. However, simvastatin treatment increased the degradation of IkBa, which in turn inhibited phosphorylation of NF-κB p65.
Simvastatin inhibited TNF-α-induced transcription of the COX-1 and COX-2 genes. However, simvastatin did not affect total COX-1 and COX-2 protein levels. This result suggests that simvastatin does not influence gastrointestinal mucosal protection regulated by prostaglandins.
We also found that simvastatin reduced TNF-α-induced generation of ROS. Akt activation can be increased by ROS. Simvastatin reduced TNF-α-mediated activation of Akt and NF-κB via reducing ROS production. Thus, simvastatin inhibited TNF-α-induced inflammation in the small intestine. Increased level of ROS by indomethacin enhanced intestinal damage (31). IHC staining for 8-OHdG in mice small intestine exposure to indomethacin suggested that simvastatin also functions as an ROS scavenger. Detection of a high level of 8-OHdG is suggestive of ROS-induced DNA damage. 8-OHdG is also an indicator of recovery of DNA damage (32). The reduced 8-OHdG level following treatment of simvastatin suggested amelioration of indomethacin-induced ROS production.
The structure of the mouse small intestine was disrupted by indomethacin-induced inflammation, as determined by histology (Fig. 5). However, simvastatin treatment reduced this indomethacin-induced loss of small intestinal structural integrity. Therefore, simvastatin protects the small intestine by reducing ROS production in indomethacin-mediated inflammation. Indeed, simvastatin has been reported to protect against damage to the small intestine in a murine model of hyper-acute Th1-type ileitis with Toxoplasma gondii by inactivating Th1-type immune responses (13).
No definitive effective drug is available for the treatment of small bowel injury. Satoh et al. reported that a diet without or with small amounts of dietary fiber could decrease gastrointestinal inflammation associated with the use of NSAIDs (33). Statin has therapeutic effects on gastrointestinal damage. Anti-inflammatory effect of HMG-CoA reductase inhibitors, fluvastatin, pravastatin and atorvastatin on ulcer formation in 5-bromo-2-(4-fluorophenyl)-3-(4-methylsulfonylphenyl) thiophene (BFMeT)-treated rats (34). Simvastatin reduced the risk of progression from inflammatory bowel disease (35) to colorectal cancer (36, 37). Simvastatin reduced lesion score of the colitis, GSH level, and myeloperoxidase (38) activity on trinitrobenzene sulfonic acid (TNBS)-induced colonic inflammation in rats (39). Antiulcer effect of simvastatin decreased the formation of gastric lesions. Simvastatin altered expression of antioxidant proteins and enzymatic activity of antioxidant proteins on indomethacin- and ethanol-induced gastric ulcer in rats (40, 41). Simvastatin decreased gastric TNF-α and gastric ulcer area in diabetic rats (42).
Anti-inflammatory effect of statin was dependent on their antioxidant role and unrelated to its cholesterol-lowering activity. Previous studies suggested that simvastatin has an antioxidant and anti-inflammatory effect regardless of cholesterol-lowering action. Mason et al. reported that atorvastatin could reduce blood pressure, nitroxidative stress and rantes level in hypertensive rats with diabetes by enhancing nitric oxide (NO) release (43). In normocholesterolemic rats, simvastatin reduced leukocyte-endothelial cell interaction and expression of P-selectin, as an endothelial cell adhesion molecule, on the mesenteric venular endothelium after superfusion with either N(G)-nitro-L-arginine methyl ester (L-NAME) or thrombin (44). A study of Huang et al. showed that simvastatin suppressed H2O2 mediated oxidative stress through decreased protein level of Nox4 in murine osteoblastic cells (45).
In summary, we investigated simvastatin-mediated inhibition of TNF-α-induced inflammation and found that it acts as an ROS scavenger. Our results also showed that simvastatin suppresses TNF-α-mediated inflammation through inhibition of ROS-induced Akt signaling pathways. Thus, the simvastatin suppressed the inflammatory related production of intracellular ROS levels and reduced inflammatory reaction in indomethacin-induced small bowel injury of mice. This phenomenon may have contributed to protection against NSAID-mediated injury in gastrointestinal tract.
Abbreviations: 8OHdG, 8-hydroxy-2'-deoxyguanosine; COX, cyclooxygenase; DCFH-DA, 2', 7'-dichlorofluorescein diacetate; HMG-CoA reductase, 3-hydroxy-3methylglutaryl coenzyme A reductase; IL, interleukin; L-NAME, N(G)-nitro-L-arginine methyl ester; NF-κB, nuclear factor-kB; NSAIDs, nonsteroidal anti-inflammatory drugs; ROS, reactive oxygen species; SPF, specific-pathogen-free; TNBS, trinitrobenzene sulfonic acid, TNF-α, tumor necrosis factor alpha.
Eun-Kyung Kim and Jae Hee Cho equally performed the majority of the work including collecting, analyzing, and interpreting the data and writing the manuscript. A Reum Jeong, Eui Joo Kim, and Dong Kyun Park contribute to designing experiments of mouse model. Kwang An Kwon, Jun-Won Chung, and Kyoung Oh Kim edited the report. Yun Soo Kim, Ju Hyun Kim, and Yoon Jae Kim designed and coordinated the study.
Acknowledgments: This study was funded by the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (2014R1A1A1A05008202) and Gachon University Gil Medical Center (Grant number: 2016 - 15).
Conflict of interests: None declared.
REFERENCES
- Maiden L, Thjodleifsson B, Theodors A, Gonzalez J, Bjarnason I. A quantitative analysis of NSAID-induced small bowel pathology by capsule enteroscopy. Gastroenterology 2005; 128: 1172-1178.
- Graham DY, Opekun AR, Willingham FF, Qureshi WA. Visible small-intestinal mucosal injury in chronic NSAID users. Clin Gastroenterol Hepatol 2005; 3: 55-59.
- Bjarnason I, Hayllar J, MacPherson AJ, Russell AS. Side effects of nonsteroidal anti-inflammatory drugs on the small and large intestine in humans. Gastroenterology 1993; 104: 1832-1847.
- Dorais J, Gregoire G, LeLorier J. Gastrointestinal damage associated with nonsteroidal antiinflammatory drugs. N Engl J Med 1992; 327: 1882-1883.
- Scheiman JM. Prevention of damage induced by aspirin in the GI tract. Best Pract Res Clin Gastroenterol 2012; 26: 153-162.
- Matsui H, Shimokawa O, Kaneko T, Nagano Y, Rai K, Hyodo I. The pathophysiology of non-steroidal anti-inflammatory drug (NSAID)-induced mucosal injuries in stomach and small intestine. J Clin Biochem Nutr 2011; 48: 107-111.
- Zhang Y, Bradley AD, Wang D, Reinhardt RA. Statins, bone metabolism and treatment of bone catabolic diseases. Pharmacol Res 2014; 88: 53-61.
- Chen HH, Lin MC, Muo CH, Yeh SY, Sung FC, Kao CH. Combination therapy of metformin and statinm decrease hepatocellular carcinoma among diabetic patients in Asia. Medicine (Baltimore) 2015; 94: e1013. doi: 10.1097/MD.0000000000001013
- Makk L, Creech JL, Whelan JG, Johnson MN. Liver damage and angiosarcoma in vinyl chloride workers. A systematic detection program. JAMA 1974; 230: 64-68.
- Campbell BA. Infantile amnesia and the etiology of alcoholism. Ann NY Acad Sci 1972; 197: 170-171.
- Bansback N, Ara R, Ward S, Anis A, Choi HK. Statin therapy in rheumatoid arthritis: a cost-effectiveness and value-of-information analysis. Pharmacoeconomics 2009; 27: 25-37.
- Park JH, McMillan DC, Horgan PG, Roxburgh CS. The impact of anti-inflammatory agents on the outcome of patients with colorectal cancer. Cancer Treat Rev 2014; 40: 68-77.
- Bereswill S, Munoz M, Fischer A, et al. Anti-inflammatory effects of resveratrol, curcumin and simvastatin in acute small intestinal inflammation. PLoS One 2010; 5: e15099. doi: 10.1371/journal.pone.0015099
- Lee JY, Kim JS, Kim JM, Kim N, Jung HC, Song IS. Simvastatin inhibits NF-kappaB signaling in intestinal epithelial cells and ameliorates acute murine colitis. Int Immunopharmacol 2007; 7: 241-248.
- Hatsukami D, Hughes JR, Pickens R. Characterization of tobacco withdrawal: physiological and subjective effects. NIDA Res Monogr 1985; 53: 56-67.
- Moran JP, Walter J, Tannock GW, Tonkonogy SL, Sartor RB. Bifidobacterium animalis causes extensive duodenitis and mild colonic inflammation in monoassociated interleukin-10-deficient mice. Inflamm Bowel Dis 2009; 15: 1022-1031.
- Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell 2004; 6: 203-208.
- Ghosh S, May MJ, Kopp EB. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol 1998; 16: 225-260.
- Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol 2000; 279: L1005-L1028.
- Fukumoto K, Naito Y, Takagi T, et al. Role of tumor necrosis factor-alpha in the pathogenesis of indomethacin-induced small intestinal injury in mice. Int J Mol Med 2011; 27: 353-359.
- Nandi J, Saud B, Zinkievich JM, Yang ZJ, Levine RA. TNF-αlpha modulates iNOS expression in an experimental rat model of indomethacin-induced jejunoileitis. Mol Cell Biochem 2010; 336: 17-24.
- Musumba C, Jorgensen A, Sutton L, et al. The relative contribution of NSAIDs and Helicobacter pylori to the aetiology of endoscopically-diagnosed peptic ulcer disease: observations from a tertiary referral hospital in the UK between 2005 and 2010. Aliment Pharmacol Ther 2012; 36: 48-56.
- Hunt RH, Bazzoli F. Review article: should NSAID/low-dose aspirin takers be tested routinely for H. pylori infection and treated if positive? Implications for primary risk of ulcer and ulcer relapse after initial healing. Aliment Pharmacol Ther 2004; 19 (Suppl. 1): 9-16.
- Seibert K, Masferrer JL. Role of inducible cyclooxygenase (COX-2) in inflammation. Receptor 1994; 4: 17-23.
- Lichtenberger LM. The hydrophobic barrier properties of gastrointestinal mucus. Annu Rev Physiol 1995; 57: 565-583.
- McCafferty DM, Granger DN, Wallace JL. Indomethacin-induced gastric injury and leukocyte adherence in arthritic versus healthy rats. Gastroenterology 1995; 109: 1173-1180.
- Lee SW, Choi DW, Park SC, et al. Expression of heat shock proteins and cytokines in response to ethanol induced damage in the small intestine of ICR mice. Intest Res 2014; 12: 205-213.
- Tulassay Z, Herszenyi L. Gastric mucosal defense and cytoprotection. Best Pract Res Clin Gastroenterol 2010; 24: 99-108.
- Wallace JL, Chin BC. Inflammatory mediators in gastrointestinal defense and injury. Proc Soc Exp Biol Med 1997; 214: 192-203.
- Wallace JL. Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterology 1997; 112: 1000-1016.
- Tomita T, Sadakata H, Tamura M, Matsui H. Indomethacin-induced generation of reactive oxygen species leads to epithelial cell injury before the formation of intestinal lesions in mice. J Physiol Pharmacol 2014; 65: 435-440.
- Ock CY, Kim EH, Choi DJ, Lee HJ, Hahm KB, Chung MH. 8-Hydroxydeoxyguanosine: not mere biomarker for oxidative stress, but remedy for oxidative stress-implicated gastrointestinal diseases. World J Gastroenterol 2012; 18: 302-308.
- Satoh H, Kondo R, Shinoda T, Idaka S, Ishigami K, Shiotani S. Diets with no or low amounts of dietary fiber can reduce small intestinal ulcers induced by non-steroidal anti-inflammatory drugs in dogs. J Physiol Pharmacol 2016; 67: 563-573.
- Hagiwara M, Kataoka K, Arimochi H, Kuwahara T, Nakayama H, Ohnishi Y. Inhibitory effect of fluvastatin on ileal ulcer formation in rats induced by nonsteroidal antiinflammatory drug. World J Gastroenterol 2005; 11: 1040-1043.
- Szary N, Rector RS, Uptergrove GM, et al. High intrinsic aerobic capacity protects against ethanol-induced hepatic injury and metabolic dysfunction: study using high capacity runner rat model. Biomolecules 2015; 5: 3295-3308.
- Frisell A, Lagergren J, de Boniface J. National study of the impact of patient information and involvement in decision-making on immediate breast reconstruction rates. Br J Surg 2016; 103: 1640-1648.
- Cortes-Bergoderi M, Pineda AM, Santana O. The pleiotropic effects and therapeutic potential of the hydroxy-methyl-glutaryl-CoA reductase inhibitors in gastrointestinal tract disorders: a comprehensive review. J Gastrointest Liver Dis 2013; 22: 199-204.
- de Freitas IN, de Campos FG, Alves VA, et al. Proficiency of DNA repair genes and microsatellite instability in operated colorectal cancer patients with clinical suspicion of lynch syndrome. J Gastrointest Oncol 2015; 6: 628-637.
- Jahovic N, Gedik N, Ercan F, et al. Effects of statins on experimental colitis in normocholesterolemic rats. Scand J Gastroenterol 2006; 41: 954-962.
- Heeba GH, Hassan MK, Amin RS. Gastroprotective effect of simvastatin against indomethacin-induced gastric ulcer in rats: role of nitric oxide and prostaglandins. Eur J Pharmacol 2009; 607: 188-193.
- Tariq M, Khan HA, Elfaki I, et al. Gastric antisecretory and antiulcer effects of simvastatin in rats. J Gastroenterol Hepatol 2007; 22: 2316-2323.
- Baraka AM, Guemei A, Gawad HA. Role of modulation of vascular endothelial growth factor and tumor necrosis factor-alpha in gastric ulcer healing in diabetic rats. Biochem Pharmacol 2010; 79: 1634-1639.
- Mason RP, Corbalan JJ, Jacob RF, Dawoud H, Malinski T. Atorvastatin enhanced nitric oxide release and reduced blood pressure, nitroxidative stress and rantes levels in hypertensive rats with diabetes. J Physiol Pharmacol 2015; 66: 65-72.
- Lefer AM, Campbell B, Shin YK, Scalia R, Hayward R, Lefer DJ. Simvastatin preserves the ischemic-reperfused myocardium in normocholesterolemic rat hearts. Circulation 1999; 100: 178-184.
- Huang W, Shang WL, Li DH, Wu WW, Hou SX. Simvastatin protects osteoblast against H2O2-induced oxidative damage via inhibiting the upregulation of Nox4. Mol Cell Biochem 2012; 360: 71-77.
A c c e p t e d : February 24, 2017