HEART RATE REDUCTION WITH IVABRADINE IN THE EARLY PHASE
OF ATHEROSCLEROSIS IS PROTECTIVE IN THE ENDOTHELIUM
OF ApoE-DEFICIENT MICE
INTRODUCTION
Epidemiological studies have shown that elevated heart rate (HR) is a marker of coronary artery disease (CAD) progression and correlates with cardiovascular mortality (1-4). In accordance, experimental data suggest that reduction in resting HR slows endothelial cell (EC) replication, a marker of endothelial dysfunction, and reduces the progression of atherosclerosis in the cynomolgus monkeys fed a high fat diet (5).
Ivabradine decreases HR by selective inhibition of the sinus node funny current (If) with no effect on blood pressure or contractility (6-9), thus is the ideal tool to study the effects of HR reduction on the vessels. Pre-clinical studies have demonstrated a protective effect of ivabradine on the vasculature (6). In mouse models of mild (10) or severe dyslipidemia (6, 8, 11-13), ivabradine prevented endothelial dysfunction and reduced the aortic plaque area (6, 11, 12) and counteracted dyslipidemia-induced reduction of both endothelium-dependent vasorelaxation (8) and aortic distensibility and circumferential cyclic strain (14). In wild type (WT) mice the above cited parameters were unaffected, either in the presence or absence of ivabradine, even though treatment with this drug induced a HR reduction comparable to the reduction observed in ApoE–/– mice fed a high-fat diet (8). These studies suggest that the protective effect of ivabradine on endothelial function and vascular wall distensibility is measurable in the presence of an endothelium-damaging agent, such as dyslipidemia, that triggers the onset of atherosclerosis. Ivabradine treatment has been associated with a reduction of vascular oxidative stress (decrease of NADPH oxidase activity (6, 13)), prevention of eNOS uncoupling (13), as well as with a lower expression of pro-inflammatory chemokine MCP-1 (6) and of angiotensin II receptor 1 (AT1-R) (13). Since all the existing studies have tested the beneficial effects of ivabradine treatment in the context of advanced stages of atherosclerosis, it is not known if ivabradine, under dyslipidemic conditions, has an effect on endothelial function before plaque formation.
The mechanisms underlying ivabradine protection are not completely understood. It is thought that an improvement of shear stress, consequent to the HR reduction, is implicated. It is well known that the ECs exposed to different shear stress patterns undergo complex transcriptional modifications (15) which modulate their proliferation, survival and activation. In arterial regions with not-disturbed flow, the ECs express various atheroprotective genes and suppress several pro-atherogenic ones, leading to endothelial stability. In regions with disturbed flow, the atheroprotective genes are suppressed, whereas the pro-atherogenic genes are upregulated, thereby promoting atherosclerosis (16, 17). In vivo (6, 10, 13) and ex vivo studies (10) seem to support this hypothesis and rule out a direct action of ivabradine on the vessels.
The aim of our study was to investigate the possibility that treatment with ivabradine, in the early phase of atherosclerotic process, before plaque formation, could result in transcriptional changes leading to maintenance of normal endothelial function. The study was conducted in 6 weeks old apoliprotein E-deficient (ApoE–/–) mice fed a chow diet (instead of a western diet) and subjected to early ivabradine treatment.
MATERIALS AND METHODS
Animal treatment
Animal studies were carried out according to the guidelines of the European (2010/63/EU) and the Italian (D.L. 26/2014) laws and after approval by the local ethical review panel of University Animal House and by the Italian Ministry of University and Research. Five-weeks old C57BL6/J and ApoE–/– mice (C57/Bl6 genetic background), purchased from Charles River Laboratories (Wilmington, MA, US), were exposed to artificial day/night cycle (13 hours light and 11 hours darkness with light on at 7 a.m.) and were caged, in group of 3, at room temperature (21 – 23°C) with 55 – 60% of humidity. Ivabradine (30 mg/kg/day, ivabradine hydrochloride, S 16257-2, Servier, France) was administered in drinking water available ad libitum. Water consumption was registered on each water renewal by visual inspection of ivabradine or placebo solution level in the bottles. HR, measured before the beginning of treatment and one week before sacrifice, was monitored by counting the number of waveforms registered per minute by Doppler echocardiography (Vivid ECG, GE Healthcare Worldwide) and using a pediatric probe (Vivid cardiovascular ultrasound 12S R-S, GE Healthcare Worldwide). Animals were not anesthetized during HR monitoring to avoid interferences with HR values. To reduce stress, which could affect HR, at least 2 days before HR measurement, mice chest was shaved at third and fourth intercostal space, next to the sternum. Mice were handled carefully, gently held by the neck and placed with their abdomen up. Nevertheless, this method cannot completely prevent stress, which could explain the high HR measured at baseline. The calculated HR was the mean of 20 consecutive measurements. HR results were expressed as mean ± SEM. Differences between groups were analyzed by one-way ANOVA followed by Dunnett’s test and P < 0.05 was considered significant. For microarray analysis, 6-weeks old ApoE–/– mice (n = 24) were randomly assigned to four groups receiving ivabradine or vehicle for 2 or 4 weeks. For blood sampling and Western blotting analysis, 6-weeks old ApoE–/– mice (n = 12) were randomly assigned to two treatment groups receiving ivabradine or vehicle for 4 weeks. For the analysis of endothelium damages, Hes5 expression levels and plaque deposition in the aortic root, 6-weeks old ApoE–/– mice (n = 16) were randomly assigned to two treatment groups receiving ivabradine or vehicle for 19 weeks.
For blood samples collection mice were sedated with an intraperitoneal injection of Zoletil (60 mg/kg; Parnell Laboratories, Alexandria, NSW, Australia) and Dexdomitor (10 mg/kg; Zoetis, Florham Park, NJ, US). For aortic arch isolation, mice, after being sedated, were euthanized by an overdose of Zoletil (120 mg/kg; Parnell Laboratories, Alexandria, NSW, Australia).
Endothelial enriched-RNA extraction
In order to increase the specificity of microarrays, RNA was isolated from endothelium of mice aortic arch only instead of whole aorta, adapting the method described by Krenek P et al., (18). After euthanasia, mice’s hearts were perfused through the left ventricle with ice-cold saline. When the effluent was completely clear, aorta was quickly and carefully isolated and placed into a Petri dish containing RNAlater solution (RNA stabilization solution, Ambion). Under stereomicroscope (SMZ745T 6x-50x, Nikon, Chiyoda, Japan) the adventitial layer and fat portion of the aorta was cleaned off by straining the aorta oppositely with angled forceps. Aortic arch was isolated and a microloader tip (Eppendorf - Germany) was adapted to an insulin syringe and filled with 300 µl of Qiazollysis solution (Qiagen, USA). The microloader tip was carefully inserted in the aortic arch until it reached the left common artery, entering from the previously removed portion of thoracic aorta. Aortic arch was flushed with Qiazol lysis solution for 30 seconds and the eluate was collected into a RNase-free microfuge tube. As a control, RNA was isolated from another whole arch after mechanical disruption of the tissue using a Dounce homogenizer. In both cases, RNA was extracted using a commercially available kit (miRNeasyMini kit - Qiagen, CA, USA). RNA concentration and purity were determined by Agilent 2100 Bioanalyzer (Agilent Technologies).
Validation of endothelial cells-enriched RNA extraction
Aortic arch of six 10 weeks old C57BL6/J mice were flushed with different volumes of a phenol containing solution to achieve efficient lysis of ECs only: more than 350 µl of Qiazol lysis buffer caused a disgregation of the whole arch, conversely smaller volume of lysis buffer resulted in insufficient RNA yield (data not shown). Noteworthy, to obtain an efficient RNA extraction and to avoid aorta dissolution, it was needed to constantly perfuse the aorta for not more than thirty seconds. As expected, this method gave a low yield of RNA (150 – 600 ng from one arch, n = 3–10 weeks old C57BL6/J mice) nevertheless every target gene was successfully amplified by qRT-PCR. RNA was also isolated from the whole arch (n = 3) after mechanical disruption of the tissue using a Dounce homogenizer and the same lysis buffer used for flushing the aorta. This method gave good yield and good quality RNA (1.5 µg total RNA from one arch). Successful endothelial enriched-RNA extraction was confirmed by measuring mRNA levels of endothelial and smooth muscle specific genes (eNOS and SM22 respectively) (Fig. 1).
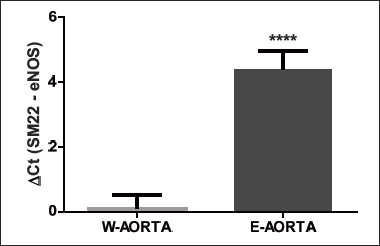 |
Fig. 1. Enrichment of endothelial specific markers in RNA extracted by using the ‘flushing technique’. Enrichment in eNOS and SM22 mRNA levels is shown by the difference in the relative Ct numbers between the RNA extracted from whole aortic arch (W-AORTA, n = 3) and RNA extracted by the ‘flushing’ technique (E-AORTA, n = 3). Differences between groups were analyzed by unpaired t-test and P < 0.05 was considered significant (****P < 0.0001). |
Microarray procedures
Total RNA from 12 samples (each containing 70 ng of RNA from 2 mice for treatment group) was hybridized on Agilent Whole Mouse Gene Expression Microarray (#G4122F, 60K transcripts, Agilent Technologies, Palo Alto, CA, USA). One-color gene expression was performed according to the manufacturer’s procedure. Briefly, labeled cRNA was synthesized from total RNA using the Low RNA Input Linear Amplification Kit (Agilent Technologies, Palo Alto, CA, USA) in the presence of cyanine 3-CTP (Perkin-Elmer Life Sciences, Boston, MA, USA). Hybridizations were performed at 65°C for 17 hours in a rotating oven. Images at 5 µm resolution were generated by Agilent scanner and the Feature Extraction 10.5 software (Agilent Technologies, Palo Alto, CA, USA) was used to obtain the microarray raw data. Data transformation was applied to set all the negative raw values at 1.0 followed by a quantile normalization. A filter on low gene expression was used to keep only the probes expressed (Detected) in at least one sample (n = 36, 013).
Microarray data analysis and bioinformatics
Unsupervised principal component analysis (PCA) was performed to assess sample similarity and to determine the contribution of age and treatment to global gene expression changes (10K expressed probes, (Qlucore software, Qlucore, Lund, Sweden)). Differentially expressed genes (DEG) were selected as having a ≥ 1.5-fold expression difference between treated and untreated groups with a P-value £ 0.01 at moderated t-test (False Discovery Rate, FDR, 7%). Hierarchical clustering was performed with GeneSpring Clustering tool (Agilent Technologies, Palo Alto, CA, USA) using the list of DEG and the Pearson centered correlation as a measure of similarity between samples. Correlations between continuous variables were tested by Pearson analysis. The Database for Annotation, Visualization and Integrated Discovery (DAVID) (19) was used to perform Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) and Biocarta pathways enrichment analyses (threshold P-value < 0.1 and enrichment gene count > 2).
Reverse transcription and real time PCR
Total RNA from 6 samples (each containing 70 ng of RNA from 2 mice for treatment group) was reverse transcribed in a volume of 25 µl using 250 units of SuperScript III reverse transcriptase and 50 ng of random hexamers. 2 µl of the cDNA mixture were used for real-time qPCR. Real-time qPCR reactions (final volume of 25 µl) were performed on an Applied Biosystems 7500 Fast Real-Time PCR System using PerfeCta SYBR Green SuperMix with ROX kit (Quanta Biosciences, Gaithersburg MD, USA) according to the manufacturer’s protocol. Changes in gene expression were calculated by the 2–ΔΔCt formula using Rpl13a as reference gene. The following primers were used:
RPL13: forward 5’-AGCCCAGGGTGCTTTGCGG-3’,
reverse 5’-GCGCCATGGCTGCCTCCTATAC-3’;
SM22: forward: 5’-GGGCGGCAGAGGGGTGACAT-3’,
reverse: 5’-TGAGGCAGAGAAGGCTTGGTCGT-3’;
NPPC: forward 5’-ACACCACCGAAGGTCCCG-3’,
reverse: 5’-TCGGTCTCCCTTGAGATTGG-3’;
OLR1: forward 5’-TGCAAACTTTTCAGGTCCTTGT-3’,
reverse: 5’-AACTGGCCACCCAAAGATTG-3’;
eNOS: forward: 5’-TTCCCCGCCTAGTCCTCGCC-3’,
reverse: 5’-CCGGGGGTCCTGGCTGAGAG-3’.
Results were expressed as mean ± SEM. Differences between groups were analyzed by unpaired t-test, P-value ≤ 0.05 was considered significant.
Western blotting
Immediately after euthanasia, the mouse aorta was perfused with saline solution then excised, cleaned of fat and fibrous material in a Petri dish containing ice cold saline solution. Segments of aortic arch were pooled (n = 6 per treatment group) and immersed in 0.5 ml of RIPA buffer (0.05% sodium deoxycholate was freshly added) containing 10 µg/ml of aprotinin, 10 µg/ml of leupeptin, 10 µg/ml of pepstatin A, 1 mM PMSF, and 1 mM sodium orthovanadate. Samples were homogenized by Dounce homogenizer on ice for 30 min. Protein concentration was quantified by by Pierce BCA Protein Assay Kit (Thermo Scientific, Wilmington, DE). Immunoblotting was performed as described in (20, 21), using the following primary antibodies: rabbit anti-OLR-1 (Abcam, Cambridge, UK, 1:1000, Cat. ab60178), rat anti-VE-cadherin (BD Pharmigen, San Diego, CA, USA, 1:500, Cat. 555289) and rabbit anti-a-smooth muscle actin (a-SMA) (Novus Biologicals, Littleton, CO, USA, 1:500, Cat. NB600).
En-face analysis of vascular endothelial-cadherin and Hes5 staining in the endothelium of the lesser curvature of the aortic arch
Immediately after mouse euthanasia, the aorta was perfused with saline solution and fixed for 10 minutes with 4% paraformaldehyde. Aorta was then excised, cleaned of fat and fibrous material in a Petri dish containing ice cold saline solution. Segments of aortic arch corresponding to lesser curvature (22) were immersed in 1 ml of blocking buffer (PBS 1X, 0.1% Triton X-100, 2% BSA) and left rocking at room temperature for 1.5 hours. Primary antibody incubation (rat anti-VE-cadherin, BD Pharmigen, San Diego, CA, USA, 1:100, Cat. 555289; rabbit anti-Hes5, Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:100, Cat. sc-25395) was performed overnight at 4°C. After washing the aortic segments three times in washing buffer (PBS 1X, 0.1% Triton X-100), secondary antibody, previously diluted in blocking buffer, was added and incubated for 1.5 hours at 4°C. The secondary antibody used were Alexa Fluor 546 Goat Anti-Rat IgG (Life Technologies, CA, USA, 1:500, Cat. A11081) and Alexa Fluor 633 Goat Anti-Rabbit IgG (Life Technologies, CA, USA, 1:500, Cat. A21070). After washing three times, aortic specimens were placed on a glass slide with the intima side up and, before covering samples with a coverslip, a drop of mounting media was applied directly to fluorescently labeled tissue samples (ProLong Antifade Reagents, Life Technologies, CA, USA). Five images of the endothelial monolayer were obtained using a Zeiss LSM 510 confocal microscope (Zeiss, Jena, Germany, 40 × magnification). Lack of VE-cadherin staining was the criteria used to identify the areas with damaged endothelium. Hes5 protein levels and the damaged endothelium area were calculated by using Image J software (Image J analysis software - http://imagej.nih.gov/ij/). Results were expressed as mean ± SEM. Differences between groups were analyzed by unpaired t-test and P < 0.05 was considered significant.
Plaque size analysis in section of aortic root
Hearts of 25 weeks old ApoE–/– mice, following 19 weeks treatment with ivabradine (30 mg/kg/day, started at 6 weeks of age) or vehicle, were snap-frozen and samples were cut on a Leica (Wetzlar, Germany) cryostat into 10 µm sections, starting at the apex until was reached the aortic valve area. At least four consecutive sections were fixed in Baker’s fixative, stained in Oil Red O and hematoxylin/eosin solution and mounted on glass slides. Images were acquired by Aperio microscope (Leica, Wetzlar, Germany). Percentage of cross-sectional aortic valve area occupied by plaque was quantified by using Image J software (Image J analysis software - http://imagej.nih.gov/ij/). Results were expressed as mean ± SEM. Differences between groups were analyzed by unpaired t-test and P < 0.05 was considered significant.
Serum analysis
Blood samples were collected from mice tail. Serum was prepared and stored at –80°C. A commercially available ELISA kit was used to measure levels of HDL and LDL/VLDL cholesterol (Cat. Ab 65390, Applied Biosystems, Waltham, MA, USA). Results were expressed as mean ± SEM. Differences between groups were analyzed by unpaired t-test and P < 0.05 was considered significant.
RESULTS
Heart rate, body weight and lipid levels
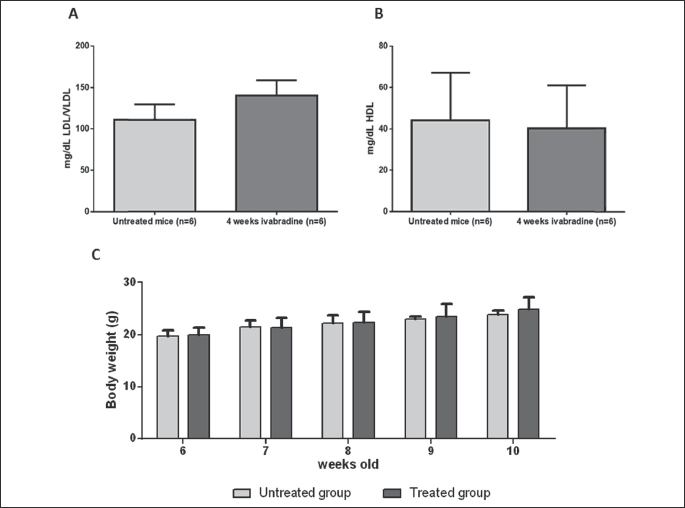
We chose to start the treatment in 6 week-old ApoE–/– mice as this age corresponds to the early stage atherosclerosis development, i.e. before monocyte adhesion (23). For microarray analysis, treatment was administered for 2 and 4 weeks and HR reduction was confirmed for each mouse in both treatments groups (data not shown). HR decreased from 751.5 ± 4.8 bpm to 604 ± 12.7 bpm (19.6%) after 2 weeks treatment (P < 0.0001) and to 550 ± 15 bpm (27%) after 4 weeks (P < 0.0001) (Fig. 2). The dose of 30 mg/kg/day of ivabradine was chosen as in preliminary dose finding experiments, it ensured a reduction of HR in the desired range of 13 – 23% (6, 14). Similar HR reduction was obtained in 6 week-old ApoE–/– mice, treated with ivabradine for 4 weeks, used for protein levels assessment, and 19 weeks, used for endothelium function assessment (data not shown). Ivabradine had not effect on body weight after 2 and 4 weeks treatment (Fig. 3A) and on serum levels of LDL/VLDL (110.6 ± 18 mg/dl and 140 ± 19 mg/dl, control versus 4 weeks treatment group) and HDL (44 ± 23 mg/dl and 40 ± 20 mg/dl, control versus 4 weeks treatment group) (Fig. 3B). Lastly, during the 19 weeks of ivabradine treatment there was no statistically significant differences in body weight between untreated and treated animals (data not shown).
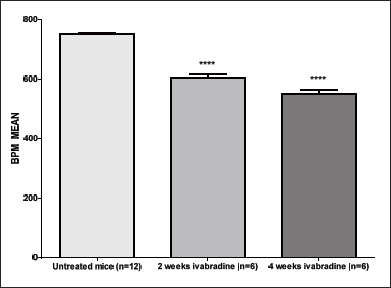 |
Fig. 2. Heart rate reduction induced by ivabradine treatment. Heart rate (BPM) in ApoE–/– mice before and after two or four weeks of ivabradine treatment. Results are expressed as mean ± SEM. Differences between groups were analyzed by one-way ANOVA followed by Dunnett’s test and P < 0.05 was considered significant (****P-value < 0.0001). |
Identification of differentially expressed genes in endothelium
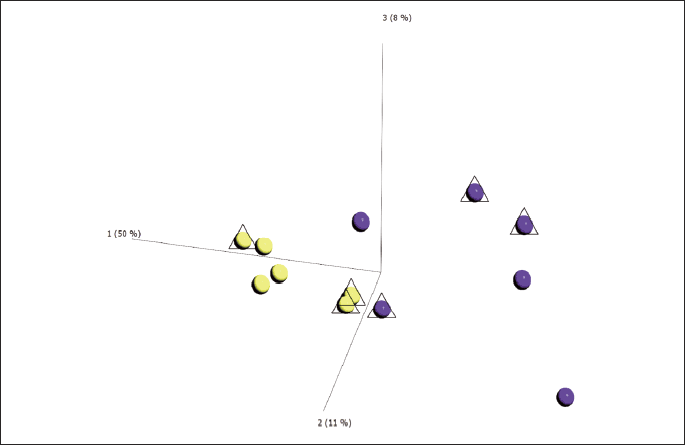
The gene expression profile of treated or placebo mice was analyzed after 2 or 4 weeks treatment in 8 and 10 week-old ApoE–/– mice, respectively. Unsupervised PCA showed that most of the variation in gene expression was due to ivabradine treatment and it was independent of treatment duration and age of the mice (Fig. 4). Based on the PCA results, a list of differentially expressed genes (DEG) was obtained by comparing the gene expression profile between the pool of treated (2- or 4- weeks treatment, 2T and 4T, respectively) and untreated mice (2NT and 4NT). The treatment altered the expression of 930 transcripts, 630 were up-regulated and 300 down-regulated (P-value cut-off: 0.01, fold change ≥ 1.5). Among the modulated sequences we identified 73 uncharacterized expressed sequence tags (ESTs), 61 Riken cDNA sequences and 314 long intergenic non-coding RNAs (lincRNA). Fig. 5 shows that the heatmap of hierarchical clustering of gene expression in samples from untreated and ivabradine-treated mice. To exclude that the differences in gene expression between groups were due to a different content of vascular smooth muscle cell (VSMCs) mRNA, we performed a clustering analyses for selected VSMCs-specific genes (Acta2, Tagln and Cnn1). These analyses showed that treated and untreated groups were not separated when the specific VSMCs transcripts were considered (Fig. 6A). In addition, the three VSMCs-specific gene expression values co-correlated (Tagln, Cnn1, R2 = 0.93, P < 0.0001 and Cnn1, Acta2, R2 = 0.902, P < 0.0001) (Fig. 6B and 6C), while there was no significant correlation between the expression levels of ivabradine-modulated transcripts and VSMCs-specific transcripts (i.e. Rasgrf2, Cnn1, R2 = 0.0119) (Fig. 6D).
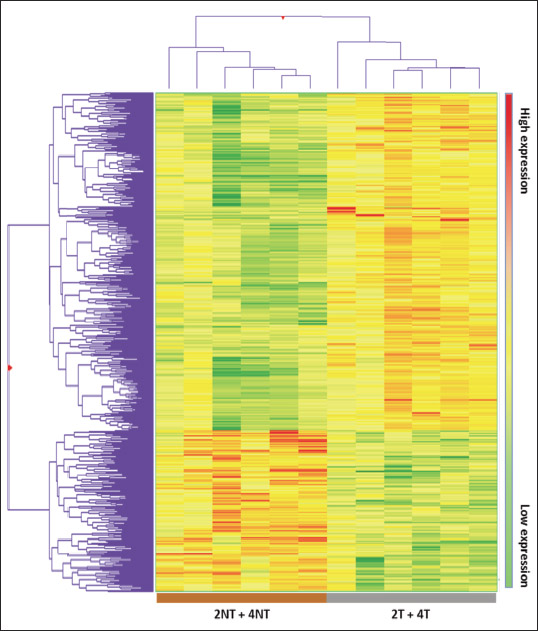 |
Fig. 5. Heatmap of hierarchical clustering of gene expression in samples from untreated and ivabradine-treated mice. Heatmap of untreated and ivabradine-treated mice (each group contains mice treated for 2 and 4 weeks) obtained using 930 differentially expressed genes. The represented genes (rows) and the experimental groups (columns) were sorted out by hierarchical clustering. High- and low-expression is referred to the treated group. |

Therefore, the analysis strongly suggests that the differences in gene expression profile between treated and untreated mice cannot be explained by the different contribution of VSMCs transcripts.
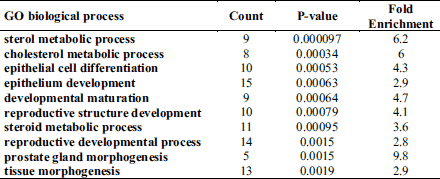
Gene ontology functional and pathway enrichment analysis of differentially expressed genes
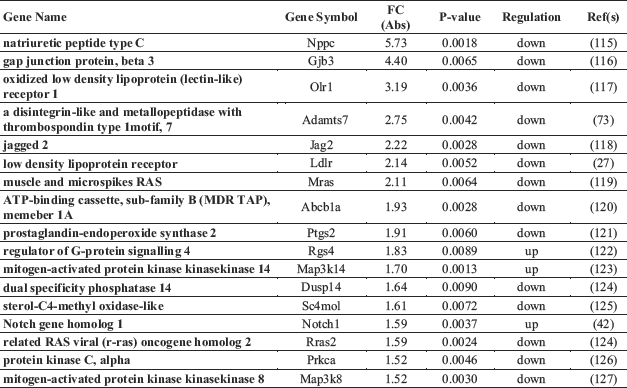
A total of 140 gene ontology (GO) biological processes were enriched by ivabradine treatment. The most significantly biological process affected were those related to sterol metabolism (P = 0.000097), cholesterol metabolic process (P = 0.00034), epithelial cells differentiation (P = 0.00053) and epithelium development (P = 0.00063) (Table 1). The KEGGS and BIOCARTA pathways affected by treatment are shown in Table 2. The MAPK signalling pathway and the steroid biosynthesis (P = 0.0013 and P = 0.0032, respectively), as well as the Notch signalling pathway (P = 0.009) were among the most significantly regulated. Tables 3 and 4 show individual genes belonging to the above cited pathways and the fold-changes in their expression following ivabradine treatment.

Ivabradine modulates expression of genes regulating epithelial cells functions and inflammation
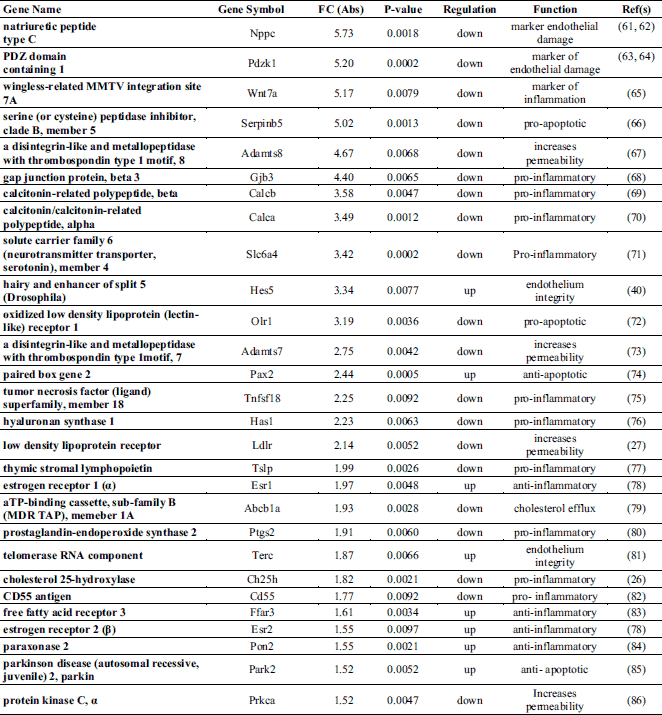
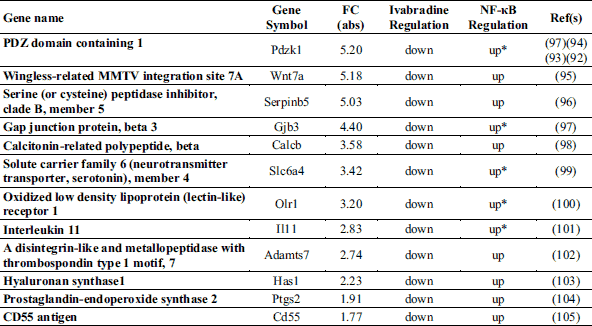
To understand the possible functional significance of the genes affected by the treatment in the atherosclerotic process, we performed a literature search which showed that an elevated number of genes linked to ECs dysfunctions and/or inflammation are present among the DEG (Table 5). More specifically, ivabradine downregulated the expression of pro-inflammatory (Calca, Calcb, Tnfsf, Has1, Tslp, Ptgs2, Wnt7A) and pro-apoptotic (Serpinb5, Olr1) genes, whereas anti-inflammatory (Ers1, Ers2, Ffar3) and anti-apototic genes (Park2, Pax2, Dad1) were upregulated. Furthermore, genes associated to increased endothelium permeability (Prkca, Ldlr1, Adamts7, Adamts8) were downregulated, whereas genes involved in endothelium repair (Hes5, Terc) were induced by treatment. Nppc and Pdzk1, both coding for proteins secreted by the endothelium to limit damages induced by inflammatory conditions, were among the genes more affected by ivabradine treatment. Quantitative RT-PCR confirmed the down-regulation of Olr1 and Nppc following 4 weeks treatment with ivabradine (Fig. 7A and 7B). Additionally, Western blot analysis showed that ivabradine treatment also downregulated OLR-1 protein (Fig. 7C).

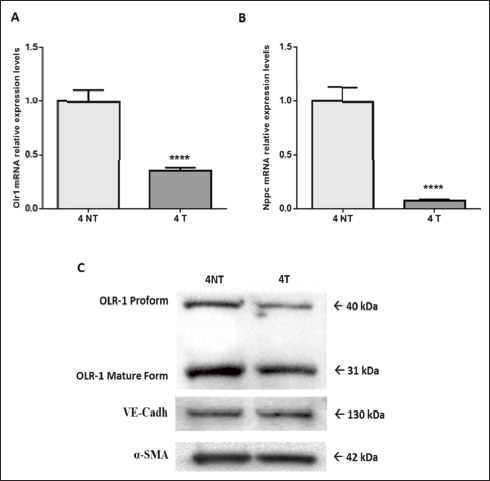 |
Fig. 7. Ivabradine-mediated down-regulation of Olr1 and Nppc in 10 week-old ApoE–/– mice. qRT-PCR analysis of Olr1 (A) and Nppc (B) expression in 10 week-old ApoE–/– mice, following 4 weeks of treatment with ivabradine (30 mg/kg/day in drinking water, n = 6) or no treatment (n = 6). Relative changes in mRNA expression levels were calculated according to the 2–ΔΔCt method using RPL13 as reference gene. Results are expressed as mean ± SEM (n = 3, each sample is a pool of two ApoE–/– mice aortic arch ECs enriched-RNA). Differences between groups were analyzed by unpaired t-test and P < 0.05 was considered significant (****P < 0.0001). T, treated; NT, untreated. (C) Western blot analysis of OLR-1 in 10 week-old ApoE–/– mice, following 4 weeks of treatment with ivabradine (30 mg/kg/day in drinking water, n = 6, 4T) or no treatment (n = 6, 4NT). VE-cadherin and a-SMA were used for equal protein loading. |
The analysis of the genes involved in cholesterol metabolic process which resulted to be modulated by ivabradine showed downregulation of the low density lipoprotein receptor gene (Ldlr) and of several other genes involved in cholesterol metabolism (Hmgcs1, Nsdhl, Cyp51, Ch25h, Sc4mol) and upregulation of Hnf1A and Cyp46a1, which are involved in the regulation of HDL cholesterol levels and cholesterol degradation (Table 6). The genes reported in Table 6, are also involved in atherosclerosis onset and progression. For instance, Ldlr, downregulated by ivabradine treatment, induces uptake of LDL in endothelial cells leading to alteration of lipid dynamic in cell membrane and increased monocytes adherence (24). Ch25h, downregulated by the treatment, plays a role in inflammation, in addition to the more established role in the regulation of cholesterol homeostasis (25). This gene encodes for an enzyme which catalyzes the formation of 25-hydroxycholesterol from cholesterol, which stimulates macrophage foam cell formation in Apoe–/– mice (26). Sc4mol, Cyp51, Hmgcs1 and Ldlr are target of SREBP1 (sterol regulatory element-binding proteins), a family of transcription factors transiently induced by laminar flow but continuously unpregulated under disturbed flow. Their downregulation could be an indication of ivabradine-mediated modulation of shear stress and reduced disturbed flow (27-29). Finally, Lepr (upregulated by ivabradine treatment) seems to confer protection against atherosclerosis, since mice defective in all leptin receptor signaling pathways develop severe obesity, hypercholesterolemia, and increased atherosclerosis (30).

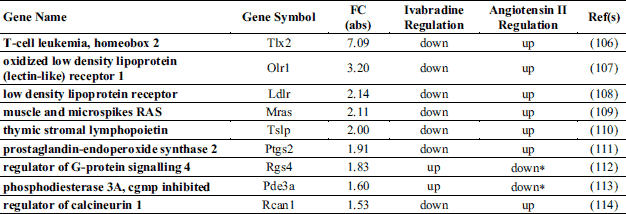
Pubmed search also showed that many genes involved in inflammation and endothelial dysfunction, which resulted to be downregulated by ivabradine, are targets of the nuclear factor-kappaB (NF-κB) (Table 7) and angiotensin II (AngII) signalling pathways (Table 8). Surprisingly, ivabradine modified the expression of these genes but not the expression of genes coding for NF-κB transcription factors (Rela, Relb, Nfkb1, Nfkb2) and for angiotensin II receptor 1 (Agtr1) or angiotensin I converting enzyme (Ace). Interestingly, Table 9 shows that several genes known to be shear stress-regulated were affected by treatment (31).
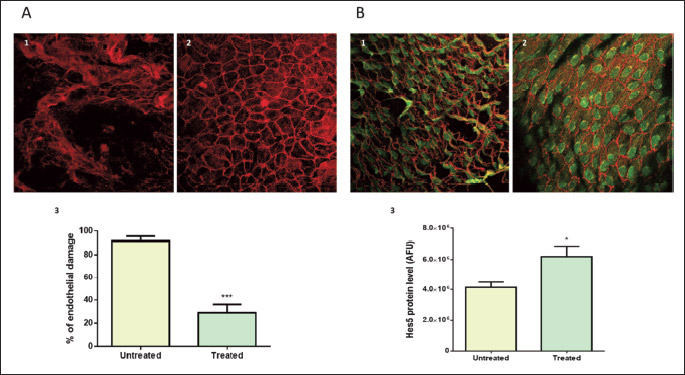
Ivabradine induces Notch signalling, reduces atherosclerotic lesion formation in the aortic root and limits endothelial damage in ApoE–/– mice aortic arch
Confocal microscopy-aided en face analysis of the VE-cadherin staining in the lesser curvature of aortic arch showed a difference in the percentage of area characterized by damaged endothelium in favour of treated mice (91.74 ± 4.084 % versus 28.98 ± 7.279 %, untreated and treated, respectively, P < 0.0001) (Fig. 8A). We also performed en face analysis of HES5 expression levels in the areas of the endothelium of untreated mice still partially intact (about 10% of the analysed area). The analysis showed that treatment induced HES5, a marker of Notch signalling activation, in the endothelium of the lesser curvature (4.1 × 106 ± 0.3 × 106 AFU versus 6.2 × 106 ± 0.6 × 106 AFU, untreated and treated, respectively, P < 0.05) (Fig. 8B). Oil Red O staining of sections of aortic root showed that the atherosclerotic plaque area was reduced by 44% in the aortic root of ivabradine-treated ApoE–/– mice compared with untreated ApoE–/– mice (18.81 ± 0.6% versus 10.48 ± 0.97 %, untreated and treated, respectively, P < 0.0001) (Fig. 9).
 |
Fig. 9. Plaque area reduction in aortic root by ivabradine treatment in ApoE–/– mice aortic arch. Hematoxylin and Oil Red staining in 25 weeks old ApoE–/– mice aortic valves following treatment with (1) vehicle or (2) ivabradine (30 mg/kg/day, started at 6 weeks of age). Both groups were fed standard diet. (3) Difference in % aortic root plaque area between untreated (n =4) and treated (n = 4) mice. Results are expressed as mean ± SEM. Differences between groups were analyzed by unpaired t-test and P < 0.05 was considered significant (*****P < 0.0001). |
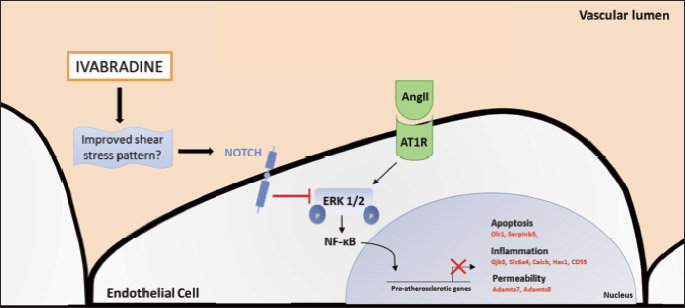
DISCUSSION
The main finding of this study is that early HR reduction with ivabradine induces an atheroprotective gene expression profile in the aortic arch endothelium of ApoE–/– mice fed a low fat diet.
The atheroprotective milieu induced by ivabradine consists in an enhancement of anti-apoptotic, anti-inflammatory and pro-endothelium repair genes with a concomitant decrease of expression of genes that increase endothelium permeability and favor inflammation and apoptosis. These effects of early treatment are linked with maintenance of endothelial integrity and reduction in plaque area in the aortic root, detected at a later stage (after 19 weeks). These results are in agreement with those of other authors which have shown a reduction of lesions in the aorta of older and dyslipidemic mice treated with ivabradine (6).
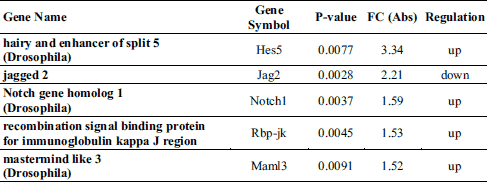
Our data also show, for the first time, that early treatment with ivabradine upregulated Hes5, Notch1, Maml1, Rbp-jk, suggestive of the activation of the Notch signalling pathway. This hypothesis is reinforced by the downregulation induced by ivabradine of Jagged2, coding for a Notch ligand. The Notch ligand Jagged1 (a member of the Jagged/Serrate family to which belongs Jagged2) is a weaker activator of endothelial Notch signalling in comparison to Delta-like ligand 4 (32). The reduction of Jagged2 by ivabradine could lead to increased Delta-like ligand 4 mediated-signalling and, therefore, to activation of the Notch pathway. Changes in expression levels of Nanog and Tlx2, Notch target genes in breast cancer (33) and neuronal cells (34) respectively, are also suggestive of an activation of Notch pathway by ivabradine treatment. Also consistent with the activation of Notch signalling, Hes5 protein was found upregulated in the endothelium of the lower aortic arch of ApoE–/– treated with ivabradine for 19 weeks.
During the last 10 years, the role of Notch in the endothelium has been actively investigated. Except for two studies showing a proatherosclerotic role of Notch (35, 36), there is growing evidence for a protective role of this pathway in the endothelium (37-41). To this end, it is relevant that laminar shear stress-modulated transcription of genes belonging to the Notch pathway has been found in ECs cultures (42, 43) and in the aorta of the mouse (40, 41, 44), where Notch signalling is uniquely positioned to attenuate the calcific response to turbulent shear stress within the aortic valves by preventing the expression of mediators of inflammation and osteogenic markers leading to aortic calcification (41). The role of Notch as mechanosensor in arteries has been recently confirmed by a study showing that Notch1 activation by shear stress directly regulates vascular barrier function and it is indispensable for the maintenance of junctional integrity, and suppression of proliferation, induced by laminar shear stress (45, 46). Oppositely to laminar shear stress, dyslipidemia reduces Notch1 in the endothelium leading to increased hypercholesterolemia-induced atherosclerosis in the descending aorta (46).
In dyslipidemic mice, expression of Hes5 mediates the proliferation of cells required to repair the endothelium damaged by dyslipidemia (40). Additionally, Notch1 activation in ECs, interferes with NF-κB activity by blocking the transcription of miR155 (39, 47) and by inhibiting ERK1/2 activation (48). Taken all together these data suggest that activation of the Notch pathway, possibly through inhibition of ERK/NF-κB signalling, could be responsible for the beneficial effects of ivabradine on the endothelium in aortic arch of ApoE-deficient mice. The late increase of Hes5 protein level concomitant to the endothelial protection that we found is supportive of the hypothesis.
The hypothesis of a Notch-mediated inhibition of the NF-κB pathway is interesting. This pathway is ‘primed’ for over-activation in atheroprone regions and this ‘priming’ is induced by disturbed/low shear stress which causes an increase in the expression of NF-κB components (49). Our analyses show that early ivabradine treatment did not alter the expression of NF-κB components, but clearly reduced the expression of several pro-inflammatory genes known to be activated by NF-κB, as demonstrated by decreased Olr1 gene and protein expression levels. It is possible that HR reduction with ivabradine reduces NF-κB activity acting by other mechanisms than downregulation of transcription of its components. Similarly, our data suggest that ivabradine counteracts AngII-signalling. This is not new, as Custodis et al., showed that in ApoE–/– mice fed a high cholesterol diet, treatment with ivabradine for 6 weeks, starting at 12 weeks of age, reduces the expression of the AT-1 receptor in the vascular wall (14). We did not observe similar changes in Agtr1 expression; this discrepancy, likely due to the difference in diet and treatment duration, suggests, once again, that in early stages, HR reduction with ivabradine modulates other genes involved in the activation of the AngII-pathway. Ang II signalling is activated by dyslipidemic conditions and it requires ERK1/2-mediated activation of NF-κB to induce its downstream genes (50, 51). In agreement with other studies (6), and similarly to other anti-atherosclerotic agents, like modified pullulan (52), ivabradine did not alter the serum lipid levels, neither we observed changes in the expression of Ace, but, we did observe a downregulation of genes belonging to the MAPK pathway such as Prkca, Rras2, Map3k8 and Mras which, if confirmed by studies on protein levels, may suggest that ivabradine may antagonize both AngII and NF-κB signalling by interfering with the phosphorylation cascade which leads to ERK1/2 activation (50, 51). Thus, it is possible that early administration of ivabradine results in an intriguing interaction between Notch activation and antagonization of AngII and NF-κB pro-atherosclerotic signaling (Fig. 10).
There is not conclusive evidence on how ivabradine protects the vasculature. Unlike cardiac-specific b-blockers (8, 10, 53), like nebivolol which increases vascular nitric oxide production (54), there is no evidence of a direct effect of ivabradine on endothelium (6, 10). Several studies show that ivabradine doesn’t affect diastolic function, cardiac output, or blood pressure both in ApoE–/– and WT mice (7-9). It is worth mentioning that in other animal models, i.e. L-NAME-induced hypertensive rats, ivabradine, besides lowering HR, is also able to reduce systolic blood pressure (55). Custodis et al. reported that HR reduction with ivabradine improves aortic distensibility and circumferential cyclic strain in ApoE mice, which might, in turn, modify shear stress pattern (14). More recently, it has been shown that treatment of low-density lipoprotein receptor (LDLr)-deficient mice with ivabradine alters local mechanical conditions and improves shear stress conditions in the aorta, without changes in blood pressure, leading to the reduction of the expression of inflammatory marker (7). In this study, we did not investigate the mechanism underlying the effects of ivabradine on the gene expression in the endothelium. In support with studies suggesting an effect mediated by shear stress alteration, we report that the expression of many shear stress-regulated genes, including Notch1, was modified by treatment.
The recent clinical trial SIGNIFY (Study Assessing the Morbidity-Mortality Benefits of the If Inhibitor Ivabradine in Patients with Coronary Artery Disease) in patients with CAD and preserved left ventricular function, specifically tested the hypothesis that HR reduction with ivabradine reduces the progression of coronary atherosclerosis and prevents myocardial infarction and cardiovascular death. Contrary to the previous data in patients with left ventricular dysfunction and heart failure showing a clear beneficial outcome from ivabradine treatment (56), and contrary to our experimental findings, in SIGNIFY study ivabradine failed to improve patients’ outcome (57). Obviously, it is difficult, if at all possible, to extrapolate experimental into clinical data but it could be that in CAD patients enrolled in SIGNIFY study, treatment with ivabradine was applied too late, when atherosclerotic plaques were already developed as this was an entry criteria to the trial. Interestingly, recent data on patients with microvascular angina in whom atherosclerosis of the major coronary artery was excluded, ivabradine improved endothelial function and coronary reserve (58-60).
In conclusion, we report that ivabradine treatment, initiated before plaque formation, induces an atheroprotective gene expression profile and it reduces endothelial damage in the aortic arch of ApoE–/– mice. Potential mechanisms involve activation of Notch pathway and inhibition of ERK/NF-κB.
Acknowledgements: This work was supported by grant from Servier (France) to P.R.
Conflict of interests: Roberto Ferrari reported that he received honorarium from Servier for steering committee membership consulting and speaking, and support for travel to study meetings from Servier. In addition, he received personal fees from Boehringer-Ingelheim, Novartis, Merck Serono, Micom, Cipla, and Menarini. Finally, he is a stockholder in Medical Trials Analysis.
REFERENCES
- Custodis F, Schirmer SH, Baumhakel M, Heusch G, Bohm M, Laufs U. Vascular pathophysiology in response to increased heart rate. J Am Coll Cardiol 2010; 56: 1973-1983.
- Fox KM, Ferrari R. Heart rate: a forgotten link in coronary artery disease? Nat Rev Cardiol 2011; 8: 369-379.
- Kaplan JR, Manuck SB, Clarkson TB. The influence of heart rate on coronary artery atherosclerosis. J Cardiovasc Pharmacol 1987; 10: S100-S103.
- Perski A, Olsson G, Landou C, de Faire U, Theorell T, Hamsten A. Minimum heart rate and coronary atherosclerosis: independent relations to global severity and rate of progression of angiographic lesions in men with myocardial infarction at a young age. Am Heart J 1992; 123: 609-616.
- Beere PA, Glagov S, Zarins CK. Retarding effect of lowered heart rate on coronary atherosclerosis. Science 1984; 226: 180-182.
- Custodis F, Baumhakel M, Schlimmer N, et al. Heart rate reduction by ivabradine reduces oxidative stress, improves endothelial function, and prevents atherosclerosis in apolipoprotein E-deficient mice. Circulation 2008; 117: 2377-2387.
- Luong L, Duckles H, Schenkel T, et al. Heart rate reduction with ivabradine promotes shear stress-dependent anti-inflammatory mechanisms in arteries. Thromb Haemost 2016; 116: 181-190.
- Schirmer SH, Degen A, Baumhakel M, et al. Heart-rate reduction by If-channel inhibition with ivabradine restores collateral artery growth in hypercholesterolemic atherosclerosis. Eur Heart J 2012; 33: 1223-1231.
- Lauzier B, Vaillant F, Gelinas R, et al. Ivabradine reduces heart rate while preserving metabolic fluxes and energy status of healthy normoxic working hearts. Am J Physiol Heart Circ Physiol 2011; 300: H845-H852.
- Drouin A, Gendron ME, Thorin E, Gillis MA, Mahlberg-Gaudin F, Tardif JC. Chronic heart rate reduction by ivabradine prevents endothelial dysfunction in dyslipidaemic mice. Br J Pharmacol 2008; 154: 749-757.
- Bolduc V, Drouin A, Gillis MA, et al. Heart rate-associated mechanical stress impairs carotid but not cerebral artery compliance in dyslipidemic atherosclerotic mice. Am J Physiol Heart Circ Physiol 2011; 301: H2081-H2092.
- Baumhakel M, Custodis F, Schlimmer N, Laufs U, Bohm M. Heart rate reduction with ivabradine improves erectile dysfunction in parallel to decrease in atherosclerotic plaque load in ApoE-knockout mice. Atherosclerosis 2010; 212: 55-62.
- Kroller-Schon S, Schulz E, Wenzel P, et al. Differential effects of heart rate reduction with ivabradine in two models of endothelial dysfunction and oxidative stress. Basic Res Cardiol 2011; 106: 1147-1158.
- Custodis F, Fries P, Muller A, et al. Heart rate reduction by ivabradine improves aortic compliance in apolipoprotein E-deficient mice. J Vasc Res 2012; 49: 432-440.
- Resnick N, Gimbrone MA. Hemodynamic forces are complex regulators of endothelial gene expression. FASEB J 1995; 9: 874-882.
- Foteinos G, Hu Y, Xiao Q, Metzler B, Xu Q. Rapid endothelial turnover in atherosclerosis-prone areas coincides with stem cell repair in apolipoprotein E-deficient mice. Circulation 2008; 117: 1856-1863.
- Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J 2005; 19: 1178-1180.
- Krenek P, Hamaide MC, Morel N, Wibo M. A simple method for rapid separation of endothelial and smooth muscle mRNA reveals Na/K+-ATPase alpha-subunit distribution in rat arteries. J Vasc Res 2006; 43: 502-510.
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44-57.
- Caliceti C, Aquila G, Pannella M, et al. 17beta-estradiol enhances signalling mediated by VEGF-A-delta-like ligand 4-notch1 axis in human endothelial cells. PLoS One 2013; 8: e71440. doi: 10.1371/journal.pone.0071440
- Fortini F, Vieceli Dalla SF, Caliceti C, et al. Estrogen receptor beta-dependent Notch1 activation protects vascular endothelium against tumor necrosis factor alpha (TNFalpha)-induced apoptosis. J Biol Chem 2017; 292: 18178-18191.
- Iiyama K, Hajra L, Iiyama M, et al. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res 1999; 85: 199-207.
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 1994; 14: 133-140.
- Pritchard KA, Schwarz SM, Medow MS, Stemerman MB. Effect of low-density lipoprotein on endothelial cell membrane fluidity and mononuclear cell attachment. Am J Physiol 1991; 260: C43-C49.
- Diczfalusy U, Olofsson KE, Carlsson AM, et al. Marked upregulation of cholesterol 25-hydroxylase expression by lipopolysaccharide. J Lipid Res 2009; 50: 2258-2264.
- Gold ES, Ramsey SA, Sartain MJ, et al. ATF3 protects against atherosclerosis by suppressing 25-hydroxycholesterol-induced lipid body formation. J Exp Med 2012; 209: 807-817.
- Liu Y, Chen BP, Lu M, et al. Shear stress activation of SREBP1 in endothelial cells is mediated by integrins. Arterioscler Thromb Vasc Biol 2002; 22: 76-81.
- Gargalovic PS, Gharavi NM, Clark MJ, et al. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol 2006; 26: 2490-2496.
- Kallin A, Johannessen LE, Cani PD, et al. SREBP-1 regulates the expression of heme oxygenase 1 and the phosphatidylinositol-3 kinase regulatory subunit p55 gamma. J Lipid Res 2007; 48: 1628-1636.
- Luo W, Bodary PF, Shen Y, et al. Leptin receptor-induced STAT3-independent signaling pathways are protective against atherosclerosis in a murine model of obesity and hyperlipidemia. Atherosclerosis 2011; 214: 81-85.
- Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, Stone PH. Role of endothelial shear stress in the natural history of coronary atherosclerosis and vascular remodeling: molecular, cellular, and vascular behavior. J Am Coll Cardiol 2007; 49: 2379-2393.
- Benedito R, Roca C, Sorensen I, et al. The Notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 2009; 137: 1124-1135.
- Simmons MJ, Serra R, Hermance N, Kelliher MA. NOTCH1 inhibition in vivo results in mammary tumor regression and reduced mammary tumorsphere-forming activity in vitro. Breast Cancer Res 2012; 14: R126. doi: 10.1186/bcr3321
- Lu TM, Luo YJ, Yu JK. BMP and Delta/Notch signaling control the development of amphioxus epidermal sensory neurons: insights into the evolution of the peripheral sensory system. Development 2012; 139: 2020-2030.
- Nus M, Martinez-Poveda B, MacGrogan D. Endothelial Jag1-RBPJ signalling promotes inflammatory leucocyte recruitment and atherosclerosis. Cardiovasc Res 2016; Aug. 5: cvw193. Epub ahead of print.
- Liu ZJ, Tan Y, Beecham GW, et al. Notch activation induces endothelial cell senescence and pro-inflammatory response: implication of Notch signaling in atherosclerosis. Atherosclerosis 2012; 225: 296-303.
- Briot A, Civelek M, Seki A, et al. Endothelial NOTCH1 is suppressed by circulating lipids and antagonizes inflammation during atherosclerosis. J Exp Med 2015; 212: 2147-2163.
- Quillard T, Charreau B. Impact of notch signaling on inflammatory responses in cardiovascular disorders. Int J Mol Sci 2013; 14: 6863-6888.
- Wang L, Zhang H, Rodriguez S, et al. Notch-dependent repression of miR-155 in the bone marrow niche regulates hematopoiesis in an NF-kappaB-dependent manner. Cell Stem Cell 2014; 15: 51-65.
- Schober A, Nazari-Jahantigh M, Wei Y, et al. MicroRNA-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing Dlk1. Nat Med 2014; 20: 368-376.
- Theodoris CV, Li M, White MP, et al. Human disease modeling reveals integrated transcriptional and epigenetic mechanisms of NOTCH1 haploinsufficiency. Cell 2015; 160: 1072-1086.
- Masumura T, Yamamoto K, Shimizu N, Obi S, Ando J. Shear stress increases expression of the arterial endothelial marker ephrinB2 in murine ES cells via the VEGF-Notch signaling pathways. Arterioscler Thromb Vasc Biol 2009; 29: 2125-2131.
- Walshe TE, Connell P, Cryan L, et al. Microvascular retinal endothelial and pericyte cell apoptosis in vitro: role of Hedgehog and Notch signaling. Invest Ophthalmol Vis Sci 2011; 52: 4472-4483.
- Morelli MB, Aquila G, Bonora M, et al. Differential expression of Notch pathway components in atheroprotected versus atherosusceptible regions of endothelium of mouse aorta. [abstract]. Eur Heart J 2014; 35 (Suppl.): 704.
- Polacheck WJ, Kutys ML, Yang J, et al. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017; 552: 258-262.
- Mack JJ, Mosqueiro TS, Archer BJ, et al. NOTCH1 is a mechanosensor in adult arteries. Nat Commun 2017; 8: 1620. doi: 10.1038/s41467-017-01741-8
- Lee KS, Kim J, Kwak SN, et al. Functional role of NF-kappaB in expression of human endothelial nitric oxide synthase. Biochem Biophys Res Commun 2014; 448: 101-107.
- Liu ZJ, Xiao M, Balint K, et al. Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. FASEB J 2006; 20: 1009-1011.
- Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappaB signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci USA 2000; 97: 9052-9057.
- Alonso F, Krattinger N, Mazzolai L, et al. An angiotensin II- and NF-kappaB-dependent mechanism increases connexin 43 in murine arteries targeted by renin-dependent hypertension. Cardiovasc Res 2010; 87: 166-176.
- Indra MR, Karyono S, Ratnawati R, Malik SG. Quercetin suppresses inflammation by reducing ERK1/2 phosphorylation and NF kappa B activation in leptin-induced human umbilical vein endothelial cells (HUVECs). BMC Res Notes 2013; 6: 275. doi: 10.1186/1756-0500-6-275
- Stefan J, Kus K, Wisniewska A, et al. New cationically modified pullulan attenuates atherogenesis and influences lipid metabolism in apoE-knockout mice. J Physiol Pharmacol 2016; 67: 739-749.
- Garlichs CD, Zhang H, Mugge A, Daniel WG. Beta-blockers reduce the release and synthesis of endothelin-1 in human endothelial cells. Eur J Clin Invest 1999; 29: 12-16.
- Kus K, Wisniewska A, Toton-Zuranska J, Olszanecki R, Jawien J, Korbut R. Significant deterioration of anti-atherogenic efficacy of nebivolol in a double (apolipoprotein E and endothelial nitric oxide synthase) knockout mouse model of atherosclerosis in comparison to single (apolipoprotein E) knockout model. J Physiol Pharmacol 2014; 65: 877-881.
- Aziriova S, Repova K, Krajcirovicova K, et al. Effect of ivabradine, captopril and melatonin on the behaviour of rats in L-nitro-arginine methyl ester-induced hypertension. J Physiol Pharmacol 2016; 67: 895-902.
- Swedberg K, Komajda M, Bohm M, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet 2010; 376: 875-885.
- Fox K, Ford I, Steg PG, Tardif JC, Tendera M, Ferrari R. Ivabradine in stable coronary artery disease without clinical heart failure. N Engl J Med 2014; 371: 1091-1099.
- Skalidis EI, Hamilos MI, Chlouverakis G, Zacharis EA, Vardas PE. Ivabradine improves coronary flow reserve in patients with stable coronary artery disease. Atherosclerosis 2011; 215: 160-165.
- van der Hoeven NW, van Royen N. The effect of heart rate reduction by ivabradine on collateral function in patients with chronic stable coronary artery disease, another funny aspect of the funny channel? Heart 2014; 100: 98-99.
- Gloekler S, Traupe T, Stoller M, et al. The effect of heart rate reduction by ivabradine on collateral function in patients with chronic stable coronary artery disease. Heart 2014; 100: 160-166.
- Passino C, Del Ry S, Severino S, et al. C-type natriuretic peptide expression in patients with chronic heart failure: effects of aerobic training. Eur J Cardiovasc Prev Rehabil 2008; 15: 168-172.
- Chen G, Zhao J, Yin Y, et al. C-type natriuretic peptide attenuates LPS-induced endothelial activation: involvement of p38, Akt, and NF-kappaB pathways. Amino Acids 2014; 46: 2653-2663.
- Zhu W, Saddar S, Seetharam D, et al. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res 2008; 102: 480-487.
- Turner EC, Mulvaney EP, Reid HM, Kinsella BT. Interaction of the human prostacyclin receptor with the PDZ adapter protein PDZK1: role in endothelial cell migration and angiogenesis. Mol Biol Cell 2011; 22: 2664-2679.
- Al-Aly Z, Shao JS, Lai CF, et al. Aortic Msx2-Wnt calcification cascade is regulated by TNF-alpha-dependent signals in diabetic Ldlr–/– mice. Arterioscler Thromb Vasc Biol 2007; 27: 2589-2596.
- Li Z, Shi HY, Zhang M. Targeted expression of maspin in tumor vasculatures induces endothelial cell apoptosis. Oncogene 2005; 24: 2008-2019.
- Salter RC, Ashlin TG, Kwan AP, Ramji DP. ADAMTS proteases: key roles in atherosclerosis? J Mol Med (Berl) 2010; 88: 1203-1211.
- Yuan D, Sun G, Zhang R, et al. Connexin 43 expressed in endothelial cells modulates monocyteendothelial adhesion by regulating cell adhesion proteins. Mol Med Rep 2015; 12: 7146-7152.
- Huang J, Stohl LL, Zhou X, Ding W, Granstein RD. Calcitonin gene-related peptide inhibits chemokine production by human dermal microvascular endothelial cells. Brain Behav Immun 2011; 25: 787-799.
- Hernanz A, De Miguel E, Romera N, Perez-Ayala C, Gijon J, Arnalich F. Calcitonin gene-related peptide II, substance P and vasoactive intestinal peptide in plasma and synovial fluid from patients with inflammatory joint disease. Br J Rheumatol 1993; 32: 31-35.
- Savas S, Hyde A, Stuckless SN, Parfrey P, Younghusband HB, Green R. Serotonin transporter gene (SLC6A4) variations are associated with poor survival in colorectal cancer patients. PLoS One 2012; 7: e38953. doi: 10.1371/journal.pone.0038953
- Morawietz H, Duerrschmidt N, Niemann B, Galle J, Sawamura T, Holtz J. Induction of the oxLDL receptor LOX-1 by endothelin-1 in human endothelial cells. Biochem Biophys Res Commun 2001; 284: 961-965.
- Bond AR, Hultgardh-Nilsson A, Knutsson A, Jackson CL, Rauch U. Cartilage oligomeric matrix protein (COMP) in murine brachiocephalic and carotid atherosclerotic lesions. Atherosclerosis 2014; 236: 366-372.
- Torban E, Eccles MR, Favor J, Goodyer PR. PAX2 suppresses apoptosis in renal collecting duct cells. Am J Pathol 2000; 157: 833-842.
- Kim WJ, Bae EM, Kang YJ, et al. Glucocorticoid-induced tumour necrosis factor receptor family related protein (GITR) mediates inflammatory activation of macrophages that can destabilize atherosclerotic plaques. Immunology 2006; 119: 421-429.
- Marzoll A, Nagy N, Wordehoff L, et al. Cyclooxygenase inhibitors repress vascular hyaluronan-synthesis in murine atherosclerosis and neointimal thickening. J Cell Mol Med 2009; 13: 3713-3719.
- Lin J, Chang W, Dong J, et al. Thymic stromal lymphopoietin over-expressed in human atherosclerosis: potential role in Th17 differentiation. Cell Physiol Biochem 2013; 31: 305-318.
- Jia M, Dahlman-Wright K, Gustafsson JA. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab 2015; 29: 557-568.
- Hassan HH, Denis M, Krimbou L, Marcil M, Genest J. Cellular cholesterol homeostasis in vascular endothelial cells. Can J Cardiol 2006; 22 (Suppl. B): 35B-40B.
- Burleigh ME, Babaev VR, Yancey PG, et al. Cyclooxygenase-2 promotes early atherosclerotic lesion formation in ApoE-deficient and C57BL/6 mice. J Mol Cell Cardiol 2005; 39: 443-452.
- Bhayadia R, Schmidt BM, Melk A, Homme M. Senescence-induced oxidative stress causes endothelial dysfunction. J Gerontol A Biol Sci Med Sci 2016; 71: 161-169.
- Lewis RD, Perry MJ, Guschina IA, Jackson CL, Morgan BP, Hughes TR. CD55 deficiency protects against atherosclerosis in ApoE-deficient mice via C3a modulation of lipid metabolism. Am J Pathol 2011; 179: 1601-1607.
- Cox MA, Jackson J, Stanton M, et al. Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E(2) and cytokines. World J Gastroenterol 2009; 15: 5549-5557.
- Garcia-Heredia A, Marsillach J, Rull A, et al. Paraoxonase-1 inhibits oxidized low-density lipoprotein-induced metabolic alterations and apoptosis in endothelial cells: a nondirected metabolomic study. Mediators Inflamm 2013; 2013: 156053. doi: 10.1155/2013/156053
- Charan RA, Johnson BN, Zaganelli S, Nardozzi JD, LaVoie MJ. Inhibition of apoptotic Bax translocation to the mitochondria is a central function of parkin. Cell Death Dis 2014; 5: e1313. doi: 10.1038/cddis.2014.278
- Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis CA, Tiruppathi C. Ca(2+) signalling and PKCalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J Physiol 2001; 533: 433-445.
- DeBose-Boyd RA. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res 20018; 18: 609-621.
- Lorbek G, Lewinska M, Rozman D. Cytochrome P450s in the synthesis of cholesterol and bile acids - from mouse models to human diseases. FEBS J 2012; 279: 1516-1533.
- Shih DQ, Bussen M, Sehayek E, et al. Hepatocyte nuclear factor-1alpha is an essential regulator of bile acid and plasma cholesterol metabolism. Nat Genet 2001; 27: 375-382.
- O’Rourke L, Yeaman SJ, Shepherd PR. Insulin and leptin acutely regulate cholesterol ester metabolism in macrophages by novel signaling pathways. Diabetes 2001; 50: 955-961.
- Liu XY, Dangel AW, Kelley RI, et al. The gene mutated in bare patches and striated mice encodes a novel 3beta-hydroxysteroid dehydrogenase. Nat Genet 1999; 22: 182-187.
- Boussicault L, Alves S, Lamaziere A, et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 2016; 139: 953-970.
- Lund EG, Kerr TA, Sakai J, Li WP, Russell DW. cDNA cloning of mouse and human cholesterol 25-hydroxylases, polytopic membrane proteins that synthesize a potent oxysterol regulator of lipid metabolism. J Biol Chem 1998; 273: 34316-34327.
- Luo M, Chodisetti G, Riederer B, Tian DA, Yeruva S, Seidler U. Interleukin-1b mediated transcriptional downregulation of NHERF family PDZ adaptor protein PDZK1 in CACO-2BBE cells. Acta Physiol 2013; 207 (Suppl. 694): P163.
- Qin S, Taglienti M, Cai L, Zhou J, Kreidberg JA. c-Met and NF-kappaB-dependent overexpression of Wnt7a and -7b and Pax2 promotes cystogenesis in polycystic kidney disease. J Am Soc Nephrol 2012; 23: 1309-1318.
- Guo F, Kang S, Zhou P, Guo L, Ma L, Hou J. Maspin expression is regulated by the non-canonical NF-kappaB subunit in androgen-insensitive prostate cancer cell lines. Mol Immunol 2011; 49: 8-17.
- Gabriel HD, Strobl B, Hellmann P, Buettner R, Winterhager E. Organization and regulation of the rat Cx31 gene. Implication for a crucial role of the intron region. Eur J Biochem 2001; 268: 1749-1759.
- Bowen EJ, Schmidt TW, Firm CS, Russo AF, Durham PL. Tumor necrosis factor-alpha stimulation of calcitonin gene-related peptide expression and secretion from rat trigeminal ganglion neurons. J Neurochem 2006; 96: 65-77.
- Flattem NL, Blakely RD. Modified structure of the human serotonin transporter promoter. Mol Psychiatry 2000; 5: 110-115.
- Nagase M, Abe J, Takahashi K, Ando J, Hirose S, Fujita T. Genomic organization and regulation of expression of the lectin-like oxidized low-density lipoprotein receptor (LOX-1) gene. J Biol Chem 1998; 273: 33702-33707.
- Bitko V, Velazquez A, Yang L, Yang YC, Barik S. Transcriptional induction of multiple cytokines by human respiratory syncytial virus requires activation of NF-kappa B and is inhibited by sodium salicylate and aspirin. Virology 1997; 232: 369-378.
- Lai Y, Bai X, Zhao Y, et al. ADAMTS-7 forms a positive feedback loop with TNF-alpha in the pathogenesis of osteoarthritis. Ann Rheum Dis 2014; 73: 1575-1584.
- Saavalainen K, Tammi MI, Bowen T, Schmitz ML, Carlberg C. Integration of the activation of the human hyaluronan synthase 2 gene promoter by common cofactors of the transcription factors retinoic acid receptor and nuclear factor kappaB. J Biol Chem 2007; 282: 11530-11539.
- Ackerman WE, Summerfield TL, Vandre DD, Robinson JM, Kniss DA. Nuclear factor-kappa B regulates inducible prostaglandin E synthase expression in human amnion mesenchymal cells. Biol Reprod 2008; 78: 68-76.
- Izban MG, Nowicki BJ, Nowicki S. 1,25-Dihydroxyvitamin D3 promotes a sustained LPS-induced NF-kappaB-dependent expression of CD55 in human monocytic THP-1 cells. PLoS One 2012; 7: e49318. doi: 10.1371/journal.pone.0049318
- Zhang M, Zhou SH, Zhao SP, Liu QM, Li XP, Shen XQ. Irbesartan attenuates Ang II-induced BMP-2 expression in human umbilical vein endothelial cells. Vasc Med 2008; 13: 239-245.
- Morawietz H, Rueckschloss U, Niemann B, et al. Angiotensin II induces LOX-1, the human endothelial receptor for oxidized low-density lipoprotein. Circulation 1999; 100: 899-902.
- Sendra J, Llorente-Cortes V, Costales P, Huesca-Gomez C, Badimon L. Angiotensin II upregulates LDL receptor-related protein (LRP1) expression in the vascular wall: a new pro-atherogenic mechanism of hypertension. Cardiovasc Res 2008; 78: 581-589.
- Romero DG, Plonczynski M, Vergara GR, Gomez-Sanchez EP, Gomez-Sanchez CE. Angiotensin II early regulated genes in H295R human adrenocortical cells. Physiol Genomics 2004; 19: 106-116.
- Zhao H, Li M, Wang L, et al. Angiotensin II induces TSLP via an AT1 receptor/NF-KappaB pathway, promoting Th17 differentiation. Cell Physiol Biochem 2012; 30: 1383-1397.
- Schlondorff D, DeCandido S, Satriano JA. Angiotensin II stimulates phospholipases C and A2 in cultured rat mesangial cells. Am J Physiol 1987; 253: C113-C120.
- Takata Y, Liu J, Yin F, et al. PPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin II-accelerated atherosclerosis. Proc Natl Acad Sci USA 2008; 105: 4277-4282.
- Iwaya S, Oikawa M, Chen Y, Takeishi Y. Phosphodiesterase 3A1 protects the heart against angiotensin II-induced cardiac remodeling through regulation of transforming growth factor-beta expression. Int Heart J 2014; 55: 165-168.
- Esteban V, Mendez-Barbero N, Jimenez-Borreguero LJ, et al. Regulator of calcineurin 1 mediates pathological vascular wall remodeling. J Exp Med 2011; 208: 2125-2139.
- Zhang Z, Xiao Z, Diamond SL. Shear stress induction of C-type natriuretic peptide (CNP) in endothelial cells is independent of NO autocrine signaling. Ann Biomed Eng 1999; 27: 419-426.
- Avvisato CL, Yang X, Shah S, et al. Mechanical force modulates global gene expression and beta-catenin signaling in colon cancer cells. J Cell Sci 2007; 120: 2672-2682.
- Ding Z, Liu S, Deng X, Fan Y, Wang X, Mehta JL. Hemodynamic shear stress modulates endothelial cell autophagy: role of LOX-1. Int J Cardiol 2015; 184: 86-95.
- Brooks AR, Lelkes PI, Rubanyi GM. Gene expression profiling of vascular endothelial cells exposed to fluid mechanical forces: relevance for focal susceptibility to atherosclerosis. Endothelium 2004; 11: 45-57.
- Dolan JM, Sim FJ, Meng H, Kolega J. Endothelial cells express a unique transcriptional profile under very high wall shear stress known to induce expansive arterial remodeling. Am J Physiol Cell Physiol 2012; 302: C1109-C1118.
- Vion AC, Ramkhelawon B, Loyer X, et al. Shear stress regulates endothelial microparticle release. Circ Res 2013; 112: 1323-1333.
- Passerini AG, Polacek DC, Shi C, et al. Coexisting proinflammatory and antioxidative endothelial transcription profiles in a disturbed flow region of the adult porcine aorta. Proc Natl Acad Sci USA 2004; 101: 2482-2487.
- Peters DG, Zhang XC, Benos PV, Heidrich-O’Hare E, Ferrell RE. Genomic analysis of immediate/early response to shear stress in human coronary artery endothelial cells. Physiol Genomics 2002; 12: 25-33.
- White SJ, Hayes EM, Lehoux S, Jeremy JY, Horrevoets AJ, Newby AC. Characterization of the differential response of endothelial cells exposed to normal and elevated laminar shear stress. J Cell Physiol 2011; 226: 2841-2848.
- Andersson M, Karlsson L, Svensson PA, et al. Differential global gene expression response patterns of human endothelium exposed to shear stress and intraluminal pressure. J Vasc Res 2005; 42: 441-452.
- Chiu JJ, Lee PL, Chang SF, et al. Shear stress regulates gene expression in vascular endothelial cells in response to tumor necrosis factor-alpha: a study of the transcription profile with complementary DNA microarray. J Biomed Sci 2005; 12: 481-502.
- Ni CW, Wang DL, Lien SC, Cheng JJ, Chao YJ, Hsieh HJ. Activation of PKC-epsilon and ERK1/2 participates in shear-induced endothelial MCP-1 expression that is repressed by nitric oxide. J Cell Physiol 2003; 195: 428-434.
- Glossop JR, Cartmell SH. Effect of fluid flow-induced shear stress on human mesenchymal stem cells: differential gene expression of IL1B and MAP3K8 in MAPK signaling. Gene Expr Patterns 2009; 9: 381-388.
- Lenz M, Muller FJ, Zenke M, Schuppert A. Principal components analysis and the reported low intrinsic dimensionality of gene expression microarray data. Sci Rep 2016; 6: 25696. doi: 10.1038/srep25696
A c c e p t e d : February 15, 2018