
GUANYLYATE CYCLASES
Guanylate cyclases are a family of enzymes that catalyze the conversion of GTP to cGMP. The family comprises both membrane-bound and soluble isoforms that are expressed in nearly all cell types. Enzyme activity is stimulated by diverse extracellular agonists that include peptide hormones, neurotransmitters, cotransmitters as adenine nucleotides, bacterial toxins, free radicals and liberated under these conditions nitric oxide (NO). Moreover agonists transducing information by receptors coupled with G protein regulated phospholipase C enhance calcium concentration that stimulates GCs.
Mammalian pGC are large transmembrane molecules, consisted of seven different isoforms: from GC-A to GC-G. In human there are two known loci for genes encoding pGC isoforms. Retina specific GC-D is located on chromosome 17 and GC-F on chromosome X. All isoforms exhibit highly conserved domain structures, including an extracellular receptor domain at the N terminus, a single transmembrane domain, a cytoplasmic juxtamembrane domain, a regulatory domain that shares significant homology with protein kinases, a hinge region, and a C-terminal catalytic domain (1). Particulate guanylate cyclases schematic structure is presented on Fig.1a.
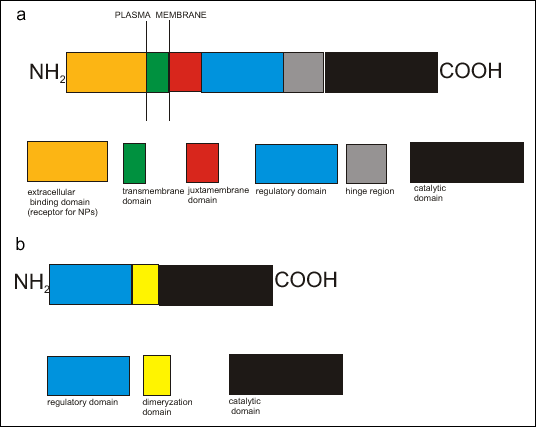 |
| Fig.
1. The schematic structure of guanylate cyclase (GC) isoforms a) particulate guanylate cyclase (pGC) b) soluble guanylate cyclase (sGC) |
Soluble guanylate cyclase (sGC) is expressed in the cytoplasm of a large number of mammalian cells and this isoform is mainly present in cells of central nervous system (CNS). This enzyme is a heterodimeric protein consisting of alpha- and ß-subunits. The expression of both subunits is required for catalytic activity. Analysis of sGC from different tissues demonstrated multiple isotypes with different subunit composition. In the brain the most abundant subunits are alpha1 and ß1. In human there are 3 known loci for genes encoding sGC subunits alpha2 is localized on chromosome 11, alpha3 on chromosome 4 and ß2 is localized on chromosome 13. Each subunit has an N-terminal regulatory domain and a C-terminal catalytic domain. A hem-binding domain is located at the N terminus of each subunit. The presence of the hem prosthetic group is required for activation of sGC by NO. In cell models expression of alpha1 or ß1 individually does not exhibit catalytic activity, coexpression of the subunits is necessary for the stimulation by NO and for the function of sGC. NO activates sGC directly by binding to hem molecule and forming a ferrous-nitrosyl-heme complex. The half- life of this complex depending on temperature and is 4 min at 37°C and 3h at 20°C. sGC require divalent cations Mn2+ or Mg2+as cofactors and allosteric modulators to express maximum catalytic activity (1). In the retina guanylate cyclase is regulated by GC-activating proteins (GC-AP1, GC-AP2) and by recoverin. All of them are Ca2+ -binding proteins specifically expressed in rods and cones of vertebrates retinas. Soluble guanylate cyclase schematic structure is demonstrated on Fig.1b.
Studies on cerebellar astrocytes showed a desensitizing profile of sGC activity. Analysis of NO-induced cGMP accumulation in the presence of a PDE inhibitor indicated that sGC underwent marked desensitization. However, the desensitization kinetics determined under these conditions described poorly the cGMP profile observed in the absence of the PDE inhibitor. An explanation was that cGMP determines the level of sGC desensitization. In support, tests in cerebellar astrocytes indicated an inverse relationship between cGMP level and recovery of sGC from its desensitized state. On the basis of this observation sGC desensitization is related to the cGMP concentration and that this effect is not mediated by (de)phosphorylation (5). On the other hand there are results obtained in experiments on chromatin cells, which indicate that the catalytic activity of sGC is closely coupled to the phosphorylation state of its beta subunit and that the tonic activity of PKG or its stimulation regulates sGC activity. (6).
CYCLIC GMP-DEPENDENT PROTEIN KINASE(S)
Protein phosphorylation mediated by cGMP is catalyzed by cGMP-dependent protein kinases (PKG). Two different genes for PKG have been identified in mammals. The gene located on human chromosome 10 encodes Ialpha and Iß isoforms of PKG I, which arise from alternative splicing of the N-terminal region. The other gene is located on human chromosome 4 and encodes PKG II.
PKG I is a cytosolic 76-kDa homodimer widely expressed in the brain and in the other mammalian tissues. In the brain the highest level of PKG was observed in the Purkinje cells of the cerebellum, and lower level was found in neurons in the dorsomedial nucleus of the hypothalamus (7). The difference in the N-terminal domain between two PKG subtypes confers different binding affinities for cGMP. PKG Ialpha has high and low affinity binding sites that display positive cooperative behavior. PKG Iß has two cGMP binding sites characterized by lower affinity and cooperativity comparing to PKG Ialpha. Fig.2a.presentes PKG Ialpha schematic structure and Fig.2b. PKG Iß structure.
 |
| Fig.
2. The schematic structure of cGMP dependent protein kinase (PKG)
isoforms a) PKG I alfa b) PKG I beta c) PKG II |
PKG II is an 86-kDa membrane-bound homodimer. It is abundant in brain and intestine, and is expressed in lung, kidney, bone but is absent in the cardiovascular system. In brain PKG II is highly expressed in the thalamus in the outer layers of the brain cortex, in the septum, amygdala, and olfactory bulb. High amount of PGK II mRNA is also found in specific brain stem loci, including the medial habenula, the subthalamic nucleus, the locus ceruleus, the pontine nucleus, the inferior olivary nuclei, and the nucleus of the solitary tract. Low levels of PGK II mRNA were detected in the striatum, cerebellum, and hippocampus (8). The amino acid sequence of PKG II differs from the sequence of PKG I principally at the N terminus. PKG II contains a myristoylation site that is required for membrane association, whereas PKG I contains an acetylation site. All known PKGs have regulatory site at the N-terminal and the catalytic domain is at the C-terminal side. The N-terminal domain contains five regulatory sites: 1) the subunit dimerization site, consisting of an alpha-helix with a conserved leucine/ isoleucine heptad repeat; 2) autoinhibitory sites, involved in the inhibition of the catalytic domain in the absence of cGMP; 3) autophosphorylation sites, which in the presence of cGMP may increase the basal catalytic activity and the affinity of PKGs for cAMP; 4) a site regulating the affinity and the cooperative behavior of the cGMP binding sites; 5) the intracellular localization site determines the interaction of the enzyme with specific subcellular structures. The regulatory domain contains two cyclic nucleotide binding sites that allow for full activation of the enzyme after specific binding of two molecules of cGMP. Finally, the catalytic domain, located at the C-terminus of PKGs, contains the binding sites for Mg2+ , ATP and the target protein (2). On Fig.2c. PKG II schematic structure is shown. Biological substrates for PKG I are: Inositol (1, 4, 5) triphosphate (IP3) receptor, phospholamban, G-protein(s), dopamine- and cAMP-regulated phosphoprotein (DARP-32) and phospholipase C (1, 9). PKG in brain is involved in regulation of neurotransmitter release and uptake, neuronal differentiation and gene expression, in learning and memory processes. On the other hand is suggested to be involved brain seizure activity and neurotoxity (2). De Vente et al. (10) demonstrated the widespread distribution of PKG II in the cerebral cortex, brainstem and cerebellum. Also, colocalization of PKG with its activator, cGMP, was examined in several brain regions after in vitro incubation of brain slices with sodium nitroprusside. PKG II was observed in all brain regions examined, although cerebellar cortex and pituitary gland contained comparatively less of PKG II. Immunocytochemistry revealed PKG II mainly in neurons, and occasionally in oligodendrocytes and astrocytes. PKG II appeared to be easily transported to nerve endings. Immunocytochemical labeling of PKG II often did not colocalize with PKG mRNA observed in in situ hybridization studies. In contrast to PKG I, which is mainly localized in cerebellar Purkinje cells, PKG II is a very ubiquitous brain protein kinase and thus a more likely candidate for cGMP mediated effects in brain requiring protein phosphorylation (10). Dinerman et al. (11) indicated that cGMP by PKG is involved in regulation of NOS activity. Phosphorylation of purified NOS by PKG similarly to PKC and PKA diminishes its catalytic activity.
cGMP REGULATED PHOSPHODIESTERASES AND THEIR INHIBITORS
At least 11 different gene families of PDEs in mammals were identified and characterized. Each member contains a conserved catalytic domain of about 270 amino acids at the C-terminus. These enzymes cleave the phosphodiester bond, hydrolyzing the 3', 5'-cyclic nucleotide to its corresponding 5' monophosphate. All PDEs contain heterogeneous regulatory domains at the N-terminus and function as dimmer. PDE families 1, 2, 3 and 10 hydrolyze both cGMP and cAMP; PDE families 4, 7 and 8 preferentially cleave cAMP and PDE families 5, 6 and 9 specifically hydrolyze cGMP. The activity of PDEs is crucial for regulation of the intracellular concentration of cyclic nucleotides (1). Table1.
| Table 1. Characteristics of phosphodiesterases and their inhibitors |
 |
All PDE1 isoforms are activated by calmodulin in the presence of Ca2+. They are encoded by three different genes (PDE1A, PDE1B and PDE1C), that undergo alternative splicing also. PDE1 is highly expressed in the brain, PDE1A is present in hippocampal CA1 and CA2 cells layer. In the striatum PDE1B plays a role in dopamine receptors signaling (12). PDE1 activity was found in the cytosol of cerebellar granule cells and astrocytes (13) and PDE1B is highly expressed in Purkinje cerebellar neurons (14). There is only one known gene encoding PDE2, the two isoforms are the result of alternative splicing. In the brain PDE2 is expressed in cerebral cortex (15), it is also expressed in olfactory neurons and cilia (16). Molecular weight of PDE2 is 100 - 105 kDa. PDE5 is also the product of one gene. In the human the gene for PDEs 5 is localized on chromosome 4 and give rise to three variants by alternative splicing, which differ in N-terminal regions. PDE5 expression in the brain is found in cerebellar Purkinje cells (14, 17) . PDE6 is encoded by three different genes, which are exclusively expressed in the retina, and has an important role in the phototransduction process. There is only one known gene for PDE10, but it has 5 variants due to the alternative splicing. In human it's localized on chromosome 6. The PDE 10 is phosphorylated by PKC and PKA (18). In brain PDE10A is highly expressed in caudate putamen and in a lower extent in hippocampus and cerebral cortex (19). PDE11 is encoded by one gene and 4 isoforms are due to the alternative splicing. PDE11 is not expressed in the brain on a high level. These enzymes are phosphorylated by PKG and PKA. PDE9 differs from the other PDEs with respect to the lack of GAF domain at N-terminal. It consists of the product of two genes, which are expressed mainly in brain, intestine and lung. PDE9 has the highest affinity for cGMP (20). In the brain the highest expression of PDE9A occurs in cerebellum and olfactory bulb and it's also present in basal forebrain, cortex, medulla, midbrain, hippocampus, thalamus and pons (21). cGMP regulated PDEs structure is presented on Fig.3. PDEs are engaged in many important cellular processes, by regulation of cyclic nucleotides concentration in cells. Their inhibitors are widely used in medicine for the treatment of asthma, dermatological and cardiovascular diseases. One of the most well known examples is PDE5 inhibitor sidenafil (Viagra), used as a drug for erectile dysfunction.
 |
| Fig. 3. The schematic structure of cGMP-regulated phosphodiesterases (PDEs) |
Wirtz-Brugger and Giovanni (22) described that propentofylline, inhibitor of PDEs inhibits apoptotic cells death by cGMP/PKG evoked supression of caspase-3 activity. Moreover propentofylline decreases also free radicals formation and lipids peroxydation (22). Clayton et all. (23) have found that selective inhibitor of PDE4 (RO 20-1724) has an anti-inflammatory effect in murine model of allergic asthma and this effect is attenuated by administration of PDE5 inhibitor (sidenafil) and PDE3 inhibitor (cilostazol). PDE4 inhibitor rolipram attenuates also formation of reactive oxygen metabolites protecting cells against injury (24) and improves long-term memory retention in the mice hippocampus (25). Another selective PDE4 inhibitors (EMD249615, EMD219906, EMD273316 and EMD95833) can promote the recruitment of bone marrow osteoprogenitor cells (26). In Tab.1 characteristic of PDEs and their inhibitors are described according to Wróblewska, Gorczyca (12) and Sung at al. (27) and others (18, 28-35).
cGMP IT'S LOCALIZATION AND ROLE IN NERVOUS SYSTEM PHYSIOLOGY
The immunohistochemical localization of GCs, cGMP and PKG in brain has been examined during the last three decades. However, only a few data were published till now. Ariano et al. (36) have studied GC immunoreactivity in rat neocortex, caudate-putamen, and cerebellum. Immunofluorescence was found within somata and proximal dendrites of neurons in these regions. The staining pattern for GC was coincident with localizations of cyclic GMP immunofluorescence within medium spiny neurons of the caudate-putamen and pyramidal cells of the neocortex. Cerebellar GC immunoreactivity was observed in Purkinje cells and their primary dendrites, similar to the pattern reported for cGMP and PKG localization. It was found that cGMP and PKG are synthesized at the highest level in the cerebellum (37, 38). Localization and the nature of cGMP synthesizing structures and cells in the developing brain was investigated by Tanaka et al. (39). They have observed that morphology and the distribution of the cGMP-positive cells were consistent with the criteria for oligodendrocytes. On the basis of their study it was concluded that the cGMP-positive cells in immature (1-28 days old) brain are mainly oligodendrocytes. A subpopulation of the oligodendrocytes was found to be cGMP-immunoreactive even when slices were incubated in the absence of NO donor. Furthermore, incubation of slices in the presence of an inhibitor of NOS, and in the absence of NO donor abolished cGMP immunostaining. In addition, some populations of neurons and astrocytes in restricted brain areas produced cGMP in response to the incubation with NO donor, whereas microglial cells did not respond to the treatment. Atrial natiuretic peptide (ANP), a stimulator of pGC, enhanced cGMP synthesis in astrocytes in some brain regions but not in oligodendrocytes. These findings indicated that those cells in the immature rat brain express sGC. However cGMP-positive oligodendrocytes were not found in the mature rat brain, suggesting that cGMP may mediate signals related to myelinogenesis in the rat brain (39). The developmental study on NO dependent cGMP synthesis in cholinergic system of the rat brain was done by Domek et al. (40). It was observed that cholinergic cells in the diagonal band of Broca and caudate putamen synthesize high levels of cGMP on the beginning of postnatal development and that this ability is lost during development and aging of the brain. NO-mediated cGMP synthesis is localized throughout the rat brain in close proximity to the NOS -containing structures (41). Using double immunostaining for cGMP and the vesicular acetylcholine transporter, it was presented that all of the cholinergic fibers in the cerebral cortex and the majority of the cholinergic fibers in the basal ganglia accumulate cGMP in response to a NO donor. Colocalization between cGMP and the vesicular acetylcholine transporter was observed in ventral forebrain, in hippocampus, reticular thalamic nucleus, and in nucleus ambiguous. There was no association of cGMP synthesis with the cholinergic system to a similar extent in the other brain areas (41). Liu et al. (42) presented the connection between muscarinic cholinergic receptors stimulation and cGMP formation. Acetylcholine was suggested to be implicated in nocturnal phase of circadian rhythms, but the results are controversial. The suprachiasmatic nucleus (SCN), site of the circadian clock receives cholinergic projections from basal forebrain and mesopontine tegmental nuclei, the structure contributing to sleep and wakefulness. They have demonstrated coupling of muscarinic cholinergic receptor (mAChRs) and cGMP in nocturnal regulation of suprachiasmatic circardian clock. They have used this paradigm to probe the muscarinic signal transduction mechanism and the site(s) gating nocturnal responsiveness. The cholinergic agonist carbachol altered the circadian rhythm of SCN activity in a pattern closely resembling that for analogs of cGMP. Specific inhibitors of GC and PKG blocked events induced by carbachol. Further, carbachol administration to the SCN at night increased cGMP production and PKG activity. The carbachol-induced increase in cGMP was blocked both by atropine, a mAChRs antagonist, and by GC inhibitor - LY83583. Authors conclude that (1) mAChR effect is mediated via GC/cGMP/PKG, (2) nocturnal gating of this pathway is controlled by the circadian clock, and (3) a gating site is positioned downstream from cGMP. This study was among the first to identify a functional role for mAChR-cGMP coupling in the CNS (42). The further studies indicated that pharmacological inhibition of kinases PKG, CAMK, but not cAMP-dependent kinase (PKA), block the circadian responses to light in in vivo studies. Ferreyra and Golombek (43) showed a diurnal and circadian rhythm of cGMP levels and PKG II activity in the hamster SCN. This rhythm depends on phosphodiesterases but not on GC activity. Inhibition of PKG or GC during in vivo experiments significantly attenuated light-induced phase shifts. The results suggest that cGMP and PKG are related to SCN responses to light and undergo diurnal and circadian changes (43). Stimulation of glutamatergic N-methyl-D-asparaginian acid (NMDA) receptor in brain is the most important and well known pathway for NO/cGMP/PKG signaling. NMDA receptor activation induces statistically significant Ca2+-dependent nitric oxide (NO)-activated cGMP synthesis (44). Release of NO is completely blocked by APV and MK-801, the competitive and noncompetitive NMDA receptor antagonists. NMDA-mediated cGMP elevation depends on NO, and is abolished by N-nitro-L-arginine (NNLA), the specific inhibitor of NO synthase (NOS). NNLA inhibits both constitutive isoforms of NOS, neuronal nNOS and endothelial isoform eNOS. The action of excitatory amino acid for NO/cGMP synthesis in rat hippocampus is potently antagonized by serotonin, through 5-HT1A receptor (45). The role of NO/cGMP in glutamatergic neurotransmission is presented on Fig.4. The stimulation of NMDA/NO/cGMP pathway play important role in long term potentiation (LTP) and long term depression (LTD), suggesting their involvement in learning and memory mechanism. However excessive stimulation of NMDA receptor and NO release leads to excitotoxity and to the cell death. Infusion of cGMP into the hippocampus directly reveals a role in early stages of memory consolidation. LTP is primarily a postsynaptic event but must be maintained presynaptically by increasing the release of glutamate. This requires that presynaptic cell receives a signal from the postsynaptic cell. NO is one possible candidate for this retrograde signal that increases neurotransmitter(s) release in LTP. Inhibitors of GC or PKG block LTP, whereas injection of cGMP into the presynaptic neuron produced an activity-dependent long-lasting potentiation involving increased neurotransmitter(s) release (2). However during the events involved in the induction of late phase of LTP, NO, cGMP and PKG cause release of Ca2+ from ryanodine-sensitive stores, causing phosphorylation of cAMP regulatory element binding protein (CREB) in parallel with PKA (46). But in contrast to these data Kleppisch et al. (47) showed that LTP is not altered in mice lacking genes for PKG I and PKG II. Moreover, sGC inhibitor (ODQ) also failed to reduce LTP in wt mice, whereas ADP-ribosylotransferase inhibitor nicotinamide, effectively suppressed LTP in wt and mutant mice. These findings argue for cGMP-independent NO signaling in hippocampus. Now it is well established that NO/cGMP/PKG signaling plays a role in LTD. In hippocampus Schaffer collateral CA1 synapses, NO/cGMP/PKG, is necessary to cause the release of calcium from ryanodine receptor gated stores, that is important for induction of LTD. It was found by Harde et al. (48) that the molecule which acts between cGMP and ryanodine receptor is cyclic ADP riboze cADPR. It was shown in hippocampal slices that cGMP increases cADPR concentration, leading to the activation of ryanodine receptor and calcium release from the presynaptic stores (48, 49).
 |
| Fig. 4. The scheme of NO/cGMP role in glutamatergic signaling pathway |
Pickaerts et al. (50) administered into the hippocampus of rats the analogue of cGMP or cAMP (8-Br-cGMP, 8-Br-cAMP) to investigate the role of cyclic nucleotides in the object recognition memory. 8-Br-cGMP in a dose-dependent manner improved object recognition memory, but not 8-Br-cAMP. These results indicate that exclusively hippocampal cGMP is involved in early stages of consolidation of object memory. It was also found that PDE5 inhibitors (sildenafil and vardenafil) improved memory performance in the object recognition task, that supported the observation described for cGMP analogue (51). Physiological aging significantly affects NO/cGMP pathway (9, 52). Alteration in NOS isoforms and IBMX sensitive PDEs in aged brain was observed by Chalimoniuk and Strosznajder (9), Jesko et al. (52).
cGMP AND Ca2+ HOMEOSTASIS
Two major pathways have been reported by which a cGMP increases intracellular Ca2+ concentration [Ca2+]i. cGMP can activate Ca2+ influx, by a process involving PKG regulation of ion channels. It can also activate Ca2+ release from ryanodine-sensitive intracellular stores by a pathway involving PKG and cyclic ADP-ribose. The cholinergic and ß-adrenergic receptors stimulated production of NO that activates a rise in cGMP concentration. Activation of phospholipase C (PLC) leads to Inositol (1,4,5) triphosphate (IP3) production resulting in Ca2+ release from endoplasmic reticulum and to NOS activation. The data of Ishikawa et al. (53) supported the view that NO/cGMP signal transduction has a crucial role in Ca2+ homeostasis. The stimulation of IP3 production by muscarinic agonists causes both intracellular Ca2+ release and activation of a voltage-independent cation current in neuroblastoma cell line N1E-115 cells. It was showed that the membrane current requires an increase in cGMP produced through the NOS /GC cascade and suggested that cells may express cyclic nucleotide-gated ion channels (CNGs). Using patch clamp method it was demonstrated that 8-Br-cGMP, activates Na+ permeable channels in cells. cGMP-dependent channel activity consists of prolonged bursts of openings and closings. The rate of the burst length is cGMP concentration dependent and is independent of voltage in the range -50 to +50 mV. The dose response curve relating cGMP concentration to channel opening by the Hill equation, assuming an apparent Km of 10 µM and a Hill coefficient of 2. In contrast, cAMP activates the channel at concentrations higher that 100 µM. CNG channels in N1E-115 cells share a number of properties with CNG channels in sensory receptors. Their presence in neuronal cells provides a mechanism by which activation of the NO/cGMP pathway by G-protein-coupled neurotransmitter receptors can directly modify Ca2+ influx and electrical excitability. In N1E-115 cells, Ca2+ entry by this pathway is necessary to refill the IP3-sensitive intracellular Ca2+ pool during repeated stimulation and CNG channels may play a similar role in different types of neurons (3).Calcium is involved in induction of transcription activation and in some cells its effect is enhanced synergistically by cGMP (54). Stimulation of Ca2+ release and other second messengers induces the expression of immediate early genes such as c-fos, c-jun, junB and junD, but till now little is known about the role of cGMP. Haby et al. (4) found that expression of c-fos and junB but not of c-jun or junD is increased upon activation of cGMP pathway. Moreover they show that NO promotes AP1 binding enhancement trough the stimulation of cGMP pathway. There was also shown, that NO/cGMP/PKG pathway is involved in regulation of CREB transcriptional activity and in anti-apoptotic Bcl-2 gene expression (55).
cGMP IN PHOTOTRANSDUCTION
cGMP play a key role in the visual system by regulating the recovery phase of visual excitation and adaptation to background light. Both rods and cones contain unique proteins that act cooperatively to control both key second messengers concentration [cGMP]i and [Ca2+]i. These, in turn regulate entire mechanism underlying phototransduction and determine physiological response to light. Retinal cells contain two isoforms of membrane-bound GC: GC-E (present in rods and cones) and GC-F (present only in rod cells). These enzymes form homodimers that are activated by interaction with specific Ca2+-binding proteins (GCAP's) in the cytoplasmic compartment. Cyclic nucleotide gated ion channel (CNG) is another essential component of the phototransduction mechanisms and the principal molecular target of cGMP in the plasma membrane of rods and cones. Two distinct heterotetrameric CNG channels are present in mammalian retina. CNG3, with high Ca2+ conductance in cones and CNG1 with low Ca2+ conductance in rods. Therefore, in cone photoreceptors, Ca2+ influx during the dark phase is significantly higher (about two times) that in rods. These inward Ca2+ currents are proportionally counteracted by outward Ca2+ currents carried by Na+/ Ca2+/K+-exchangers in the plasma membrane of photoreceptors and provide molecular basis for the difference in timing of photoresponses in cones and rods. The last component of cGMP-related photodransduction machinery is PDE6, the enzyme that degrades cGMP in photoreceptors. The enzyme is maximally activated by a cooperative mechanism that involves activated transducin, the
 |
| Fig. 5. The scheme of cGMP/PKG signaling pathways |
great number of physiological pathways, in which cGMP is involved, has been described above. In mature brain cGMP acts mainly in cortex, caudate- putamen, cerebellum and hippocampus. In immature brain cGMP is involved in guiding neurons to achieve their destination and it plays important role in myelinogenesis. In mature brain the colocalization of cGMP with glutamatergic and cholinergic system in hippocampus and brain cortex seems to have a functional implication in the view of its role in LTP and LTD. Nitric oxide and cGMP are the main messengers in glutamatergic system. It seems that cGMP is also a very important messenger for cholinergic system, where it could be involved in cross- talk between cholinergic and other neurotransmitters mediated pathways. However, the exact role of cGMP in many processes has not been yet clearly established. Also in the senescent brain, where memory processes are getting worst, cGMP turnover is changed. In accordance to most of data the NOS-containing neurons are resistant to degeneration in Alzheimer's, Parkinson's and Huntington's diseases, whereas the surrounding neurons are destroyed. However, there are also results indicating that NOS containing neurons are undergoing degeneration and cGMP concentration and cGMP regulated processes are affected.
The role of NO/cGMP in neurodegeneration and aging is the new area of investigation for the last two decades. Discovery of the new family members of cGMP phosphodiesterases and their specific inhibitors offers now new strategy for improvement of brain function and for the new therapy in the future.
- Lucas KA, Pitari GM, Kazerounian S, et al. Guanylyl cyclases and signaling by cyclic GMP. Pharmacol Rev 2000; 52(3): 375-414.
- Wang X, Robinson PJ. Cyclic GMP-dependent protein kinase and cellular signaling in the nervous system. J Neurochem, 1997; 68: 443-456.
- Thompson SH. Cyclic GMP-gated channels in a sympathetic neuron cell line. J Gen Physiol, 1997; 110: 155-164.
- Haby C, Lisovoski F, Aunis D, Zwiller J. Stimulation of the cyclic GMP pathway by NO induces expression of the immediate early genes c-fos and junB in PC12 cells. J Neurochem, 1994; 62: 496-501.
- Wykes V, Bellamy TC, Garthwaite J. Kinetics of nitric oxide-cyclic GMP signalling in CNS cells and its possible regulation by cyclic GMP. J Neurochem, 2002; 83: 37-47.
- Ferrero R, Rodriguez-Pascual F, Miras-Portugal MT, Torres M. Nitric oxide-sensitive guanylyl cyclase activity inhibition through cyclic GMP-dependent dephosphorylation. J Neurochem, 2000; 75: 2029-2039.
- El-Husseini AE, Williams J, Reiner PB, Pelech S, Vincent SR. Localization of the cGMP-dependent protein kinases in relation to nitric oxide synthase in the brain. J Chem Neuroanat, 1999; 17: 45-55.
- el-Husseini AE, Bladen C, Vincent SR. Molecular characterization of a type II cyclic GMP-dependent protein kinase expressed in the rat brain. J Neurochem, 1995; 64: 2814-2817.
- Chalimoniuk M, Strosznajder JB. Aging modulates nitric oxide synthesis and cGMP levels in hippocampus and cerebellum. Effects of amyloid beta peptide. Mol Chem Neuropathol, 1998; 35: 77-95.
- de Vente J, Asan E, Gambaryan S et al. Localization of cGMP-dependent protein kinase type II in rat brain. Neuroscience, 2001; 108: 27-49.
- Dinerman JL., Steiner JP, Dawson TM, Dawson V, Snyder SH. Cyclic nucleotide dependent phosphorylation of neuronal nitric oxide synthase inhibits catalytic activity. Neuropharmacology, 1994; 33: 1245-1251.
- Wroblewska H, Gorczyca WA. (Phosphodiesterases of cyclic GMP). Postepy Hig Med Dosw, 2001; 55: 611-627.
- Agullo L, Garcia A. Ca2+/calmodulin-dependent cyclic GMP phosphodiesterase activity in granule neurons and astrocytes from rat cerebellum. Eur J Pharmacol, 1997; 323: 119-125.
- Shimizu-Albergine M, Rybalkin SD, Rybalkina IG, Feil R, Wolfsgruber W, Hofmann F, Beavo JA. Individual cerebellar Purkinje cells express different cGMP phosphodiesterases (PDEs): in vivo phosphorylation of cGMP-specific PDE (PDE5) as an indicator of cGMP-dependent protein kinase (PKG) activation. J Neurosci, 2003; 23: 6452-6459.
- Noyama K, Maekawa S. Localization of cyclic nucleotide phosphodiesterase 2 in the brain-derived Triton-insoluble low-density fraction (raft). Neurosci Res, 2003; 45: 141-148.
- Juilfs DM, Fulle HJ, Zhao AZ, Houslay MD, Garbers DL, Beavo JA. A subset of olfactory neurons that selectively express cGMP-stimulated phosphodiesterase (PDE2) and guanylyl cyclase-D define a unique olfactory signal transduction pathway. Proc Natl Acad Sci U S A, 1997; 94: 3388-3395.
- Giordano D, De Stefano ME, Citro G, Modica A, Giorgi M. Expression of cGMP-binding cGMP-specific phosphodiesterase (PDE5) in mouse tissues and cell lines using an antibody against the enzyme amino-terminal domain. Biochim Biophys Acta, 2001; 1539: 16-27.
- Soderling SH., Bayuga SJ, Beavo JA. Isolation and characterization of a dual-substrate phosphodiesterase gene family: PDE10A. Proc Natl Acad Sci U S A, 1999; 96: 7071-7076.
- Seeger TF, Bartlett B, Coskran TM et al. Immunohistochemical localization of PDE10A in the rat brain. Brain Res, 2003; 985: 113-26.
- Fisher DA, Smith JF, Pillar JS, SH St Denis, Cheng JB. Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J Biol Chem, 1998; 273: 15559-15564.
- Andreeva SG, Dikkes P, Epstein PM, Rosenberg PA. Expression of cGMP-specific phosphodiesterase 9A mRNA in the rat brain. J Neurosci, 2001; 21: 9068-9076.
- Wirtz-Brugger F, Giovanni A. Guanosine 3',5'-cyclic monophosphate mediated inhibition of cell death induced by nerve growth factor withdrawal and beta-amyloid: protective effects of propentofylline. Neuroscience, 2000; 99: 737-750.
- Clayton RA, Dick CA, Mackenzie A et al. The effect of selective phosphodiesterase inhibitors, alone and in combination, on a murine model of allergic asthma. Respir Res, 2004. 5: 4.
- Matousovic K, Grande JP, Chini CC, Chini EN, and Dousa TP, Inhibitors of cyclic nucleotide phosphodiesterase isozymes type-III and type-IV suppress mitogenesis of rat mesangial cells. J Clin Invest, 1995; 96: 401-410.
- Barad M, Bourtchouladze R, Winder DG, Golan H, Kandel E. Rolipram, a type IV-specific phosphodiesterase inhibitor, facilitates the establishment of long-lasting long-term potentiation and improves memory. Proc Natl Acad Sci USA, 1998. 95(25): p. 15020-15025.
- Scutt A, Beier N, Fittschen C, EMD273316 & EMD95833, type 4 phosphodiesterase inhibitors, stimulate fibroblastic-colony formation by bone marrow cells via direct inhibition of PDE4 and the induction of endogenous prostaglandin synthesis. BMC Pharmacol, 2004; 4: 10.
- Sung BJ, Yeon Hwang K, Ho Jeon Y et al. Structure of the catalytic domain of human phosphodiesterase 5 with bound drug molecules. Nature, 2003; 425: 98-102.
- Hagiwara M, Endo T, Hidaka H. Effects of vinpocetine on cyclic nucleotide metabolism in vascular smooth muscle. Biochem Pharmacol, 1984; 33: 453-457.
- Wells JN, Miller JR, Methylxanthine inhibitors of phosphodiesterases. Methods Enzymol, 1988; 159: 489-496.
- Tang KM., Jang EK, Haslam RJ, Photoaffinity labelling of cyclic GMP-inhibited phosphodiesterase (PDE III) in human and rat platelets and rat tissues: effects of phosphodiesterase inhibitors. Eur J Pharmacol, 1994; 268: 105-114.
- Galvan M, Schudt C, Actions of the phosphodiesterase inhibitor zardaverine on guinea-pig ventricular muscle. Naunyn Schmiedebergs Arch Pharmacol, 1990; 342: 221-227.
- Soderling SH, Bayuga SJ, Beavo JA. Cloning and characterization of a cAMP-specific cyclic nucleotide phosphodiesterase. Proc Natl Acad Sci USA, 1998; 95: 8991-8996.
- Takase Y, Saeki T, Watanabe N, Adachi H, Souda S, Saito I. Cyclic GMP phosphodiesterase inhibitors. 2. Requirement of 6-substitution of quinazoline derivatives for potent and selective inhibitory activity. J Med Chem, 1994; 37: 2106-2111.
- Hidaka H, Endo T. Selective inhibitors of three forms of cyclic nucleotide phosphodiesterase--basic and potential clinical applications. Adv Cyclic Nucleotide Protein Phosphorylation Res, 1984; 16: 245-259.
- Fujishige K, Kotera J, Michibata H. Cloning and characterization of a novel human phosphodiesterase that hydrolyzes both cAMP and cGMP (PDE10A). J Biol Chem, 1999; 274: 18438-18445.
- Ariano MA, Lewicki JA, Brandwein HJ, Murad F, Immunohistochemical localization of guanylate cyclase within neurons of rat brain. Proc Natl Acad Sci USA, 1982; 79: 1316-1320.
- Schlichter DJ, Detre JA, Aswad DW, Chehrazi B, Greengard P. Localization of cyclic GMP-dependent protein kinase and substrate in mammalian cerebellum. Proc Natl Acad Sci USA, 1980; 77: 5537-41.
- Lohmann SM, Walter U, Miller PE, Greengard P, De Camilli P. Immunohistochemical localization of cyclic GMP-dependent protein kinase in mammalian brain. Proc Natl Acad Sci USA, 1981; 78: 653-657.
- Tanaka J, Markerink-van Ittersum M, Steinbusch HW, De Vente J. Nitric oxide-mediated cGMP synthesis in oligodendrocytes in the developing rat brain. Glia, 1997; 19: 286-297.
- Domek K, Waarenburg van de M, Markerink van Ittersum M, Steinbusch HWM, Vente de J. Nitric oxide-induced cGMP synthesis in cholinergic neurons in the developing rat brain. International Symposium on nitric oxide-cyclic GMP signal transduction in brain, abstract book, 2003. abstract nr 12.
- de Vente J.M.-v.I., Axer M, H. Steinbusch HW, Nitric -oxide-induced cGMP synthesis in cholinergic neurons in the rat brain. Exp Brain Res, 2001; 136 480-491.
- Liu C, Ding JM, Faiman LE, Gillette MU. Coupling of muscarinic cholinergic receptors and cGMP in nocturnal regulation of the suprachiasmatic circadian clock. J Neurosci, 1997; 17: 659-666.
- Ferreyra GA, Golombek DA. Rhythmicity of the cGMP-related signal transduction pathway in the mammalian circadian system. Am J Physiol Regul Integr Comp Physiol, 2001; 280: R1348-1355.
- Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature, 1988; 336: 385-388.
- Strosznajder J, Chalimoniuk M, Samochocki M. Activation of serotonergic 5-HT1A receptor reduces Ca(2+)- and glutamatergic receptor-evoked arachidonic acid and No/cGMP release in adult hippocampus. Neurochem Int, 1996; 28: 439-444.
- Lu YF, Hawkins RD. Ryanodine receptors contribute to cGMP-induced late-phase LTP and CREB phosphorylation in the hippocampus. J Neurophysiol, 2002; 88: 1270-1278.
- Kleppisch T, Pfeifer A, Klatt P et al. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci, 1999; 19: 48-55.
- Reyes-Harde M, Empson R, Potter BV, Galione A, Stanton PK, Evidence of a role for cyclic ADP-ribose in long-term synaptic depression in hippocampus. Proc Natl Acad Sci USA, 1999; 96: 4061-4066.
- Reyes-Harde M, Potter BV, Galione A, Stanton PK. Induction of hippocampal LTD requires nitric-oxide-stimulated PKG activity and Ca2+ release from cyclic ADP-ribose-sensitive stores. J Neurophysiol, 1999; 82: 1569-1576.
- Prickaerts J, de Vente J, Honig W, Steinbusch HW, Blokland A. cGMP, but not cAMP, in rat hippocampus is involved in early stages of object memory consolidation. Eur J Pharmacol, 2002; 436: 83-87.
- Prickaerts J, van Staveren WC, Sik A, Markerink-van Ittersum M, Niewohner U, van der Staay FJ, Blokland A, de Vente J. Effects of two selective phosphodiesterase type 5 inhibitors, sildenafil and vardenafil, on object recognition memory and hippocampal cyclic GMP levels in the rat. Neuroscience, 2002; 113: 351-361.
- Jesko H, Chalimoniuk M, Strosznajder JB, Activation of constitutive nitric oxide synthase(s) and absence of inducible isoform in aged rat brain. Neurochem Int, 2003; 42: 315-222.
- Ishikawa Y, Iida H. Ishida H. The muscarinic acetylcholine receptor-stimulated increase in aquaporin-5 levels in the apical plasma membrane in rat parotid acinar cells is coupled with activation of nitric oxide/cGMP signal transduction. Mol Pharmacol, 2002; 61: 1423-34.
- Chen Y, Zhuang S, Cassenaer S et al. Synergism between calcium and cyclic GMP in cyclic AMP response element-dependent transcriptional regulation requires cooperation between CREB and C/EBP-beta. Mol Cell Biol, 2003. 23: 4066-4082.
- Ciani E, Guidi S, Bartesaghi R, Contestabile A. Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J Neurochem, 2002. 82: 1282-1289.