IN VIVO MODELS FOR CANCER STEM CELL RESEARCH: A PRACTICAL GUIDE FOR FREQUENTLY USED ANIMAL MODELS AND AVAILABLE BIOMARKERS
2Institute of Pharmaceutical Technology, Johann Wolfgang Goethe University, Frankfurt/Main, Germany
INTRODUCTION
Stem cell biology is one of the most challenging and exciting fields of modern science, and at the same time one of the most controversial. Attitudes toward stem cell research have changed significantly in recent decades as a result of the complexity of the subject. This complexity is reflected in the diversity of hypotheses and methods of analysis using various cell lines, laboratory animals, and human tissue samples. Stem cell research has accelerated even more vigorously since the first population of tumor-initiating cells, also known as cancer stem cells (CSCs), was identified in human leukemia (1). According to the CSCs hypothesis, a small self-renewing population of cells within hematologic cancers or solid tumors plays a critical role in cancer initiation, progression, and metastasis (2). Despite the significant progress to date, the nature of many biological processes which account for CSC properties still remains unclear. There are several pertinent topics currently under active investigation including the molecular pathways regulating CSCs functions, the role of the CSC’s niche and its “plasticity” characteristics, specific CSCs biomarkers in all cancer types, ideal marker(s) and ideal assay(s) for CSC markers, variable frequency of CSCs between tumors, instability of the CSC’s phenotype, the existence of multiple CSC pools within individual tumors, the nature of metastatic CSCs, the resistance of CSCs to cytotoxic chemotherapy and low-dose radiation treatment, and the prognostic significance of CSCs. More details about the biological properties of CSCs can be found in the comprehensive review published by Visvader et al. (3). Scientific attention is also currently focused on the discovery and development of novel drugs that can selectively target and destroy CSCs. In the face of these challenges, many biopharmaceutical companies around the globe are investing in new anti-CSC drug programs. Numerous anti-CSC compounds have already entered human clinical trials, and some have successfully completed different phases of testing (4-6). Since it is beyond the scope of this review to cover the detailed evaluation of each anti-CSC drug candidate we will highlight only the most important properties of anti-CSC drugs and discuss how these properties relate to the drug development process.
THERAPEUTIC APPROACHES TO TARGET CANCER STEM CELLS
The different types of anti-cancer stem cells therapeutics and their characteristics
The majority of anti-CSC drugs can be divided into three major groups: the first group of therapeutic biologics includes monoclonal antibodies, human vaccines, cellular immunotherapeutics, therapeutic peptides, and microRNA-based therapeutics (7-12). The second group of therapeutics that target CSC is small molecules (13-15), and the third is chemical compounds that not specifically designed for use against CSCs, but which have recently been found to have anti-CSC properties (16-22).
The complex pharmacokinetic behaviour, various modes of actions, and toxicity of therapeutic biologics are mainly determined by their various physicochemical properties. For example, therapeutic monoclonal antibodies (mAbs), the most common type of biologics, do not readily cross cell membranes due to their large size, usually resulting in low tumor tissue penetration. However, mAbs interact with a wide range of extracellular signaling molecules and cell-surface proteins triggering various events involved in cancer cell death. The presence of two identical antigen-binding sites in mAbs offers an exceedingly high selectivity and binding affinity for the intended target tumor cell antigens, including CSCs antigens. In addition, one of mAbs’ important modes of action is their interaction with the immune system. An example is the immune-mediated cell killing mechanisms, which include induction of phagocytosis, complement activation, antibody-dependent cell-mediated cytotoxicity, target gene-modified T cells, and activation of T cells through inhibition of T cell inhibitory receptors and antibody-mediated cross presentation of antigen to DC-cells (23). Some biologics such as RNA-based therapeutics can have off-target effects (24, 25) and typically require special delivery systems for efficient transport to the target cells (26). Another type of biologic is therapeutic vaccines. The specific immune responses caused by vaccines are often weak due to the lack of co-stimulatory signals and specificity to antigens (27). In general, biologics are sensitive to temperature changes, susceptible to microbial contamination, and easily degrade in the gastrointestinal (GI)-tract before reaching the systemic circulation. Furthermore, such therapeutics are often difficult and expensive to manufacture (28, 29).

In contrast to biologics, the common characteristics of small molecules include a low molecular weight, rapid entry into systemic circulation through blood capillaries, usually a short half-life, poor selectivity, and typically a lack of high specificity. However, current data are ambiguous and biased toward certain target classes, most prominently human G-protein-coupled receptors and kinases, specifically tyrosine kinases (30). Unlike many biologics, small molecules readily diffuse across cell membranes and reach intracellular targets. However, they are often limited to protein targets due to small binding pockets with which they can bind and alter the rates of specific intracellular signaling pathways (31). A major safety concern with small molecules is toxicity to different organs and systems where they potentially accumulate. Additionally, the therapeutic applicability of small molecules is often limited by their low aqueous solubility. Because of this, they require stable aqueous formulations for the intravenous administration. In case of oral administrations, special delivery systems are needed to maintain, enhance, or facilitate small molecule absorption from the GI-tract (30, 31).
As mentioned above, the third group may include a variety of chemical compounds alone and in combinations with drugs from the first or second group or well-known drugs at novel dosage forms and drug delivery systems such as polymeric micelles, liposomes, nanoparticle drug carriers etc. Because of the diversity of drugs, their possible combinations, dosage forms and delivery systems in the third group, it is difficult to make generalizations about them. Obviously, the third group may include some common characteristics of the first and second group. However, compounds of the third group may differ in their biodistribution, toxicity and efficacy. Examples of different types of drugs, which represent all three groups, can be found in Table 1 and Table 2.
Drug delivery targeting cancer stem cells
It is well-known that the accumulation of macromolecular drugs within a tumors is predominantly a result of ”leaky” microvasculature and impaired lymphatic support, known as the enhanced permeability and retention (EPR) effect (32, 33). This abnormality allows various types of anti-cancer drugs, alone or in association with polymeric delivery systems such as nanoparticles, to exhibit selectivity of action (34). In this context, we have previously reported that micelles prepared from conjugates of polyethylene glycol (PEG) and diacyllipids, such as phosphatidylethanolamine (PE) can be used for an efficient solubilization of poorly soluble substances and provides aqueous stability to the micelles (35-37). Small size of PEG-PE-micelles facilitates their “passive” targeting into tumors via the EPR effect. In contrast, active targeting of drugs can be achieved using various specific ligands attached to nanoparticle surfaces (33, 35-38). Therefore, certain drug delivery platforms for targeting CSCs, such as polymeric nanoparticle formulation of curcumin (18), triblock polymeric micelles with doxorubicin (39), lipid nanocapsules functionalized with a monoclonal antibody (40), all-trans retinoic acid stealth liposomes (16), poly(benzylglutamate)-hyaluronan polymerosomes with doxorubicin (41), paclitaxel-hyaluronan bioconjugate (42) make it one of the promising approaches in anti-CSC therapy. More examples of strategies for passive or active CSC targeting can be found elsewhere (43).
In summary, modern drug delivery technologies which have been designed to dose drugs continually for prolonged time periods and in a controlled fashion have a high potential for enhancing accumulation of drug in tumors. However, even after these drugs reach therapeutic levels in tumors, it may not be enough for them to act specifically on one particular targeted type of cell - for example, to selectively seek and destroy CSCs. While current efforts are focused on passive or active targeting of tumors, where many drugs have extracellular and/or intracellular compartments as their site of action, much less is known about efficient and selective therapies that target CSCs preferentially.
GENERAL PRINCIPLES OF ANTI-CANCER STEM CELL DRUG DEVELOPMENT
Traditionally, the preclinical drug development process meets several critical experimental requirements. These include: the availability of prospective drug candidates in large enough quantities for comprehensive in vitro analysis, and the ability to identify potential drug targets (e.g. these targets can be outside the cell, inside the cell, or on the cell surface) that are specifically involved in a particular metabolic or signaling pathway. Another requirement is the accessibility of suitable clinically relevant animal models for in vivo studies. The animal models must have a histotype for the evaluation of modes of action, quantitative pharmacology studies (e.g. pharma- and toxicokinetics), toxicity, and efficacy studies, and accessibility of suitable biomarkers that can serve as marker(s) of efficacy. One of the most distinctive characteristics of in vivo efficacy studies for the identification of potent anti-CSC drugs is that they usually consist of two steps. The first step in this approach is to administer drugs into the tumor-bearing animals for the inhibition of malignant ascites production or inhibition of solid tumor growth and/or metastasis. Researchers then use other types of in vivo assays to confirm the stem cell behaviors of self-renewal and their long-term repopulating ability. The diagram in Fig. 1 shows a typical in vivo study for the identification of a highly selective anti-CSC drug.
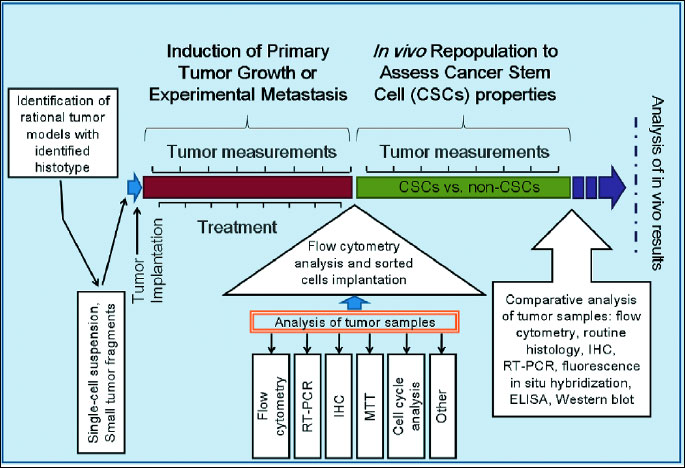
CSC: cancer stem cells; RT-PCR: reverse transcriptase - polymerase chain reaction; IHC: immunohistochemistry; MTT: (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay.
In preclinical studies, the anti-tumor effectiveness of the applied drugs can be evaluated by direct caliper measurement of tumor size, calculation of visible surface metastasis, or Kaplan-Meier survival analysis of animals harboring various hematological and solid tumors. Alternatively, tumor growth and metastasis can be visualized in live animals using X-ray, ultrasound, CT, SPECT, PET, MRI, and other in vivo imaging modalities to detect green fluorescent protein- and luciferase-expressing tumor models. However, in some cases an in vivo anti-cancer therapy may be considered efficacious based on the significant reduction of primary tumor size and metastasis, but there is still a high risk that after treatment the cancer could relapse due to the survival of sufficient number of CSCs. In addition, many traditional cytostatic or cytotoxic and even novel molecularly-targeted drugs may be highly toxic to healthy cells, including normal stem cells. Therefore, new anti-CSCs drugs are still urgently needed.
Tumor microenvironment and cancer stem cell markers in preclinical cancer stem cell research
It has been shown that solid tumors are a complex mix of phenotypically and functionally heterogeneous neoplastic cells including cancer cells, CSCs, and various normal stromal cells and bioactive molecules (e.g. cytokines and chemokines) that surround tumor cells, also referred to as the tumor microenvironment (44). Interestingly, normal stem cells and CSC share common features that include activation of the same molecular pathways that regulate their stemness and their ability to self-renew. For example, many cell surface markers such as EpCAM, CD24, CD44, and CD133 that currently are associated with CSCs were also found in normal stem cells in the tissue-of-origin (45-48). Due to the similarities between CSCs and normal stem cells, the majority of current therapies that target CSCs are not selective exclusively to CSC, they may also eliminate normal stem cells. On the other hand, one of the differences between normal stem cells and CSC is their degree of dependence on the stem cell niche, a specialized microenvironment in which stem cells reside (49). Thus, the microenvironment seems to be of crucial importance for primary tumor growth as well as metastasis formation (50).
The interactions between cellular components of the stroma and the factors that they express during cancer initiation and progression are critically important. For example, the chemokine stroma-derived factor (SDF-1/CXCL12) is of particular interest because the binding of this ligand to chemokine receptor CXCR4 in CSCs is implicated in tumor proliferation, survival, and metastasis (51). Indeed, CXCL12 contributes to tumor immunosuppression through recruiting of specific immune cell populations, enhances tumor neovascularization, and mediates tumor cell migration, adhesion, and invasion. In fact, various components of the tumor microenvironment including cancer-associated fibroblasts, endothelial cells, pericytes, cancer cells, and immune inflammatory cells have a direct influence on CSC properties (2, 44, 52). On the other hand, using the same chemokine receptors to get into the bone marrow, CSCs may use the normal hematopoietic stem cell microenvironment or “niche” which houses hematopoietic stem cells in the bone marrow to their advantage (53). It is therefore obvious that the understanding of the specific diversity of cell morphology and molecular profiling reveals the similarities and differences between CSCs and other cell types in the tumor stroma, and understanding their mechanisms of interaction will provide new potential targets for specific anti-CSC therapeutic intervention.
One way to enhance the efficacy and reduce the unwanted effects of anti-cancer drugs is to selectively target CSCs. In order to do this it is necessary to distinguish CSCs from other stromal components of the tumor. Distinct and specific markers, including molecules that are on the cell surface, cell signaling molecules (5, 6, 54-60) and differential expression of miRNAs in CSCs (61) can be used for this purpose. Although the prognostic value of CSC-associated markers is still under active investigation, many researchers believe that specific gene expression by CSCs may be able to predict clinical outcomes. More details about the current state of CSC biomarkers, CSC detection in patients, and discussion about future perspectives for the development of prognostic and predictive CSC biomarkers for routine clinical use can be found elsewhere (3, 54-56, 62-65).
It is important to emphasize that CSCs can also be distinguished from the bulk-tumor population by other characteristics such as high expression of cytoprotective enzymes, for instance, aldehyde dehydrogenase 1 (ALDH1) (66), ATP-binding cassette (ABC) transport proteins (4) or thymosin β4 (Tβ4), a small acidic actin binding peptide (67).
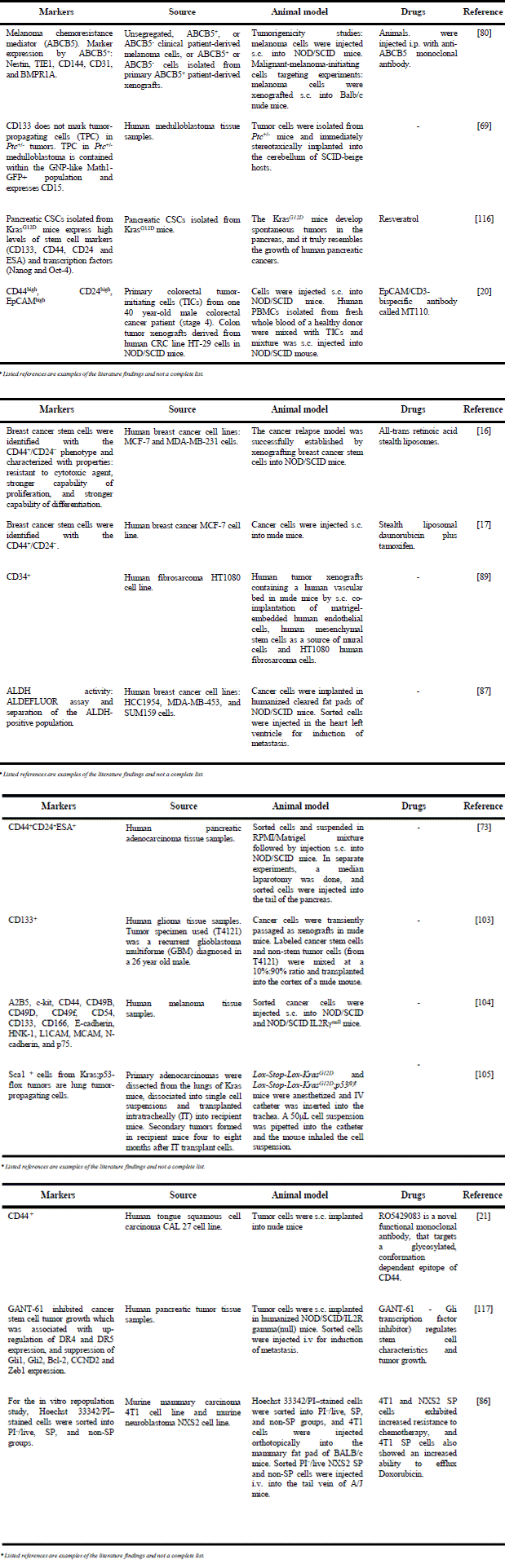
As mentioned earlier, the sorted subsets of CSCs can be used in both, in vitro clonogenic and in vivo tumorigenic assays to characterize CSCs. For example, in one in vivo tumorigenic assay where CSCs were sorted by CD133 expression it was found that 1 in 262 CD133+ cells had colon tumor formation capacity in immunodeficient mice, while only 1 in 57,000 unsorted tumor cells could form a tumor (68). Other researchers have identified CSCs in the most commonly used animal model of medulloblastoma (MB), the Patched mutant Ptc+/– mice. CSCs were propagated not by CD133+ cells but by cells expressing markers Math1+ and CD15+ (CD15+ -known as SSEA-1 or LeX). As a result, MB tumors from Ptc+/– mice were propagated by orthotopic transplantation of 300,000 unsorted and 300,000 CD15+ Ptc+/– MB sorted cells, and not by 300,000 CD15- cells (69). In these experiments the most common method used to identify CSC population is fluorescence-activated cell sorting (FACS). An overview of phenotypes of CSC markers in various cancer types (animal models) is provided in Table 2.
In recent decades, numerous therapeutic strategies for eradicating CSCs have been proposed. Some of the most promising approaches target cell surface marker(s) (e.g. CD33, CD44, CD90, CD133, CD166 and EpCAM etc.) (3, 55) and cell signal cascade(s) (e.g. Notch, Hedgehog, Wnt, NF-κB etc.) (4, 55). While other strategies are based on the targeting of specific microenvironments in solid tumors that form a permissive “niche” for the CSC (e.g. CXCL12/CXCR4, VEGF/VEGFR, weakly acid pH etc.) (3, 51, 55), metabolic activity of CSCs (e.g. ALDH1 etc.) (55, 66) or drug efflux transporters that are the “guardians” of the stem cell population (e.g. ATP-binding cassette (ABC) drug transporters)) (55, 70). Also, DNA repair mechanisms (e.g. CSCs use a network of activated DNA repair mechanisms to remove different types of DNA damage) (52, 71), and detoxifying enzymes that are responsible for the chemo-and radiation therapy resistance of CSCs can be considered (55, 56).
General characteristic of laboratory rodents and tumor material for the induction of tumor growth in rats and mice
Traditionally, there are three sources of tumor material used for the induction of tumor growth in laboratory rodents. To model and reproduce the variability of response of specific tumor types, researchers use stable, immortalized cancer cell lines, of which there are currently more than 1000 commercially available (70). The biggest disadvantage of using immortalized cancer cell lines is that they do not represent all of the biological and genomic properties of primary tumors (71, 72). In some studies, researchers may also use cancer cells isolated from primary tumors grown in vivo or primary patient tissue samples, which can be inoculated as single-cell suspensions or surgically resected human tumor fragment(s). Heterotransplantation of human tumor material into immunodeficient rodents is also known as human tumor xenografts. Notably, rodent xenograft models are critical for certain types of in vivo studies because of the difficulty in routinely obtaining primary human tumors from particular organs, especially the pancreas (73). Rapid engraftment of human tumor material from patient primary solid-tumor tissue at low passage is known as a patient-derived tumor xenograft (PDX). Currently, the inbred/outbred nude athymic mice that are T-cell deficient and suffer from a lack of cell-mediated immunity (74) and B-cell-deficient and T-cell-deficient severe combined immunodeficient (SCID) mice (74, 75) are widely used as xenograft hosts. In addition to the first two strains, human tumor xenografts can be successfully propagated in non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice that are B-cell-deficient and T-cell-deficient plus they are defective in DC-cell activity, and macrophages and natural killer (NK) cell activity. Also, these mice do not have a complement hemolytic activity due to the lack of the complement protein C5 (76). In contrast, the BUB/BnJ strain of mice is reported to have high serum complement hemolytic activity (77). It has been shown that NOD/SCID mice are an excellent model system to study the engraftment and mobilization of human peripheral blood stem cells (78) and appear to be more receptive for growing experimental leukemias and solid tumors (79, 80). Notably, the nude mice have intact innate immunity and they do not have a defect in T-cell precursors. Therefore, the chances of a successful transplantation of primary tumor cell into nude mice in some cases can be as low as <5% (81). In certain SCID mice have been reports of “leakiness”, meaning they have the ability to generate a small number of functional B and T cell clones (82). However, some adult SCID mice, specifically in the BALB/cBy and C57BL/6J genetic backgrounds have a high capacity for developing these clones and on the low end is the C3H/HeJ background (74). This should be taken into account when choosing immunodeficient animals for experiments. Some types of human tumor cells grow quite well in the basic nude and SCID mice, while others grow unacceptably slow with poor tumor “take”. For better engraftment of peripheral-blood mononuclear cells (PBMC) as well as human cancer cell lines, cancer cells isolated from primary tumors grown in vivo or patient-derived tissue, NSG mice (NOD-scid IL2Rgammanull) that are lacking mature T cells, B cells, NK cells, complement, and are defective in DC-cells and macrophages, and are deficient in cytokine signaling, can be successfully used for long or short-term in vivo experiments (83). Athymic nude rat that are T-cell deficient and show depleted cell populations in thymus-dependent areas of peripheral lymphoid organs are useful in the study of graft rejection in immunocompromised hosts since they can accept major histocompatibility complex mismatched organ allografts or xenografts for several months. Also, it has been shown that some human tumor cell lines grow well in such rats (84). It is believed that various immunodeficient strains have lymphoid tissue alterations, such as partial or total depletion of the thymus, spleen, and lymph nodes and that may hide potential toxic effects of applied anti-CSC drugs. This factor should be taken into account in designing in vivo experiments. Any new therapeutics, including anti-CSC drugs need to be tested in both normal and immunocompromised preclinical animal models. Therefore, the strains of mice and rats that researchers finally choose for in vivo studies will depend on the immunologic status and other characteristics, for instance, the type of therapeutics, duration of experiment, and tumor microenvironment that best fits with the aim of the study.
Examples of transplantable animal tumors in cancer stem cell research
For many years, laboratory animals have played a critical and necessary role in the anti-cancer drug development process. Despite that, however, no animal models can be referred to as “ideal model” that can fully recapitulate the genetic, epigenetic, and immune system complexity found in human cancer patients. In preclinical CSC research, there are now a variety of transplantable animal tumors which can be placed heterotopically (e.g. subcutaneous (s.c.), intraperitoneal (i.p.), and intramuscular (i.m.) administration of tumor cells into animals)) or orthotopically (i.e. into the organ of origin) in syngeneic (i.e. same species, genetically identical), allogeneic (i.e. same species, genetically different) or xenogeneic (i.e. different species and genetics)) host systems. Table 2 provides overviews of different host species, methods of inoculation of tumor cells, types of cancer for investigation and types of tumors that express proteins that can be important for CSC detection. More details about the types of transplantable tumor models that applicable for CSC research can be found in the review published by Talmadge (85). Moreover, for in vivo tumorigenic assays with sorted CSC populations, the model that belongs to the heterotopic group is particularly useful. By the intravenous (i.v.) or intracardiac (i.c.) administration of tumor cells, experimental metastasis in lungs and/or liver (by i.v.) or in bone, muscle, lung, and soft tissue (by i.c.) can be induced and studied or analyzed (15, 86, 87).
By leveraging lessons learned through testing and implementation of various models, the primary goal of this review is to provide practical solutions regarding the development of rational models for CSC-research, and highlight the advantages and disadvantages of each approach. For example, the s.c. inoculation of single-cell suspension(s) or tumor fragment(s) is still one of the more common techniques for the induction of tumor growth in heterotopic models for efficacy studies of novel anti-CSC candidates. The benefits of s.c. tumors are the simplicity of tumor inoculation, reproducibility and repeatability of experiments, and accessibility for direct tumor size measurements. The large s.c. space in rodents allows for continuous growth of the tumor, which can measure from approximately 50 mm3 up to 1500 mm3. This is beneficial for CSC research, especially when a large amount of tumor mass is needed for the isolation and molecular characterization of CSCs.
One drawback of the s.c. models is that it may not provide accurate information on the specific microenvironment (or “niche”) to support growth of CSC and may not represent the “true side” of human tumors. One possibility to overcome this problem is to s.c. administer the tumor cells suspended in the basement membrane matrix proteins CultrexBME or Matrigel (88). Tumor cells can also be co-injected with human fibroblasts, matrigel-embedded human endothelial cells, human mesenchymal stem cells, or other types of stromal cells that in some way mimic in vivo extracellular microenvironments. This will allow CSC to achieve sustainable tumor development, and enable the tumor to grow more rapidly (89). Table 2 shows several examples of s.c. models that are applicable in CSC-research. In addition, s.c. implantation of cells isolated from freshly dissociated human tumor tissues or small piece(s) of surgically resected human tumors into immunocompromised animals is often an initial step in the establishment of patient-derived xenografts (PDXs). The experimental benefit of s.c. PDXs is that they may maintain many aspects of the human microenvironment for several weeks, which offers a more accurate simulation of cancer cell behavior in patients. Moreover, orthotopic PDXs with organ-specific microenvironments for CSC might serve as an excellent source for the identification and validation of CSC-related markers with highly predictive potential for therapy. Unfortunately, it has been shown that after 2–3 passages the human microenvironment in such tumors can be substituted with elements of murine tumor stroma (90, 91). In the context of human tumor xenografts containing co-implanted matrigel-embedded human endothelial cells, endothelial cells have also been rapidly substituted by their murine counterparts, and by day 16 post-implantation human CD34-positive cells were almost undetectable in intratumoral vessels (89).
As noted above, s.c. models are a valuable tool, but they are limited by the lack of blood supply and lymphatic drainage that are so vastly different than those of the orthotopic site (92). Thus, s.c models cannot truly reproduce the patterns of development of distant metastasis often seen in cancer patients, for example, in sinonasal cancers (93). Importantly, the orthotopic model has all of the elements of the microenvironment that help maintain the CSC phenotype in a subset of cells, essential not only for primary tumor growth, but also in mediating tissue invasion and metastasis (94). It is possible that different organs that consist of epithelial tissue, such as the liver, kidneys, lungs, pancreas, and colon, harbor different microenvironments with distinct endothelial or stromal cell types and extracellular matrix components that promote or inhibit tumor growth (95). The authors suggested that the tumor initiation and maintenance at the primary site of disease or metastatic dissemination would depend on the interaction of CSCs with a particular niche. Recently, an orthotopic metastatic murine model for head and neck squamous cell carcinoma (HNSCC) using GFP-labeled cells and FACS for generation of a pure metastatic cell line has been developed (96). Moreover, this model also allowed the authors to identify a number of genes that may have an important role in metastasis in HNSCC, possibly through the induction of the epithelial-to-mesenchymal transition and formation of CSCs. Other authors suggested that CD44-positive CSC-like cells have enhanced ability for metastasis. Some researchers previously described the development of an orthotopic mouse model, in which an orthotopic implantation of CD44-positive subpopulation of tumor cells into the mammary gland (97) or into the cecal wall (98) of immunocompromised animals yielded local tumors, as well as distant metastasis in lungs or liver. Research also shows that in addition to genetic and epigenetic complexity and diversity, tumor cell plasticity may contribute to phenotypic and functional heterogeneity (2, 44, 52, 54, 64, 65, 99). Interestingly, CSC model of heterogeneity may apply more readily to early-stage rather than to advanced tumors due to the dominant clones that probably drive tumor progression (3). Also, advanced tumors often have some evidence of necrosis and bleeding that may have a significant impact on the quality of tumor samples for analysis.
The superiority of orthotopic xenograft models is due to the site-specific microenvironment in the host organ that closely mimics clinical pathology. However, orthotopic tumors in animal experiments grow much faster than in cancer patients and hence can be, at least hypothetically, more sensitive to targeted anti-CSC drugs. Therefore, preclinical in vivo data may not always translate well to the clinic. More information describing the translation of novel anticancer drugs into the clinic and highlighting the challenges, limitations and where there is a need for improvements can be found in (100-102).
As mentioned earlier, a CSC population may be confirmed by preferential tumor formation from cells directly isolated from primary tumors in drug-treated and untreated (control) animals. This requires the sacrifice of the animal to assess the treatment’s effect on both CSCs and other types of cancer cell populations. Also, site-specific tumor growth in orthotopic models limits the real time evaluation of the efficacy of novel anti-CSC drug candidates in living rodents. Alternatively, to evaluate the potential of CSCs in direct comparison to non-CSCs in the same microenvironment, researchers have recently developed techniques to use different fluorescent proteins for labeling of tumor cells to monitor their behavior over time using intravital microscopy (103).
The challenge of designing in vivo orthotopic model experiments is to develop sustainable growth of transplanted tumors. The initial engraftment rate with implantation of tumor material in immunocompromised mice has been shown to be as low as <5% (81), 20% (66) or 25% (73). In fact, many CSC results are derived from xenotransplantation experiments in which human cancer cells are grown in immunodeficient animals with different genetic backgrounds. Therefore, to increase the percentage of tumor “take,” the rational selection of the appropriate strains of rodents is necessary for success. For example, a group of authors showed that using the PDX melanoma model significantly increased the detectable frequency of CSCs through some modifications in xenotransplantation assays. In their study, the CSCs were more likely to be detected in more highly immunocompromised mice such as NOD/SCID/IL2Rgammanull mice (104). Other in vivo studies have shown that the genotype of tumor samples must be taken into account in the targeting of CSCs. For example, using orthotopic (intratracheal transplant) models of the most common form of human lung cancer, researchers found that lung adenocarcinomas of differing genotypes have CSCs with distinct markers that therefore require different strategies for isolation of CSCs from each model (105).
Other things to consider when using orthotopic models for CSC research include site-specific variation in response to therapy and accurate analysis of tumor growth and metastasis. Also, these models are labor-intensive and time-consuming, and some procedures require specialized training. Some animal models, especially PDXs, can cost tens of thousands of dollars per study, and therefore are not widely used in routine in vivo laboratory practices. Furthermore, the vast majority of internal orthotopic models provide only a small amount of tumor mass for analysis, which may be critical for the isolation of these rare populations of CSCs. The only exceptions to this are certain breast cancer models transplanted into a mammary gland fat pad, whose tumors are relatively superficial and accessible for routine tumor-size measurements, and, more importantly, are available for the collection of large amounts of tumor tissue. More details about models’ strengths, weaknesses, applicability, and translation to human cancer in the context of anti-cancer drug discovery can be found here (100, 106, 107).
It has long been recognized that the host immune status and the tumor microenvironment play a crucial role during tumor development in both animals and humans. Since immunocompromised rodents fail to functionally replicate the immune system characteristics that are present in humans, it can be assumed that such conditions may permit growth of tumors in rodents that would never occur in human patients. In addition, for some biologics such as humanized mAbs, the host immune system is needed to fully realize their anti-tumor potential, especially when the intact host immune system is required for the antibody-dependent cellular cytotoxicity (ADCC). To further explore the role of ADCC in anti-HER2/neu antibody therapy, researchers used Balb/c mice inoculated s.c. with neu-expressing TUBO breast carcinoma cells which are cells derived from BALB-neuT transgenic mice) (108). These researchers have clearly demonstrated that an activation of innate immunity and T-cells that was initiated by antibody treatment is necessary for significant tumor suppression. Furthermore, the possibility of targeting CSCs in DC cell-based vaccination, using an orthotopic (intracranial) 9L tumor in adult F344 Fisher rats has been explored (109). DC-based vaccination using CSC antigens elicited antigen-specific T-cell responses against CSCs. Because of the immune response required for the host (rodents) to realize the maximum therapeutic potential of particular types of drugs in cancer treatment, numerous experimental approaches have been developed and studied in preclinical practice. For example, the transfer of a functional human immune system, for instance, human PBMC (via i.v. or intrasplenic injections) to mice with severe combined immunodeficiency is a useful way to make “humanized mice” (110). Notably, humanized mice can be engineered by the engraftment of components of the human immune system not only from blood (110) or human bone marrow (111), but also by engraftment of primary human fetal lymph nodes (112). Alternatively, human fetal liver and thymus fragments can be implanted in some cases, under the renal capsule in adult SCID mice (113-115). Moreover, i.p administration of human haematopoietic stem cells into newborn or adult immunodeficient recipients usually resulted in very low engraftment (114).
One group of researchers have reported that humanized anti-CD44 antibody RO5429083 can effectively target the CSC population in CAL-27 human xenografts in humanized mice while boosting the number of NK cells, and thus may synergize with drugs that work via ADCC such as Cetuximab (116). In another study, humanized NOD/SCID/IL2Rgammanull mice were used to examine the effects of GANT-61 (Gli transcription factor inhibitor) on CSC’s tumor growth (117). Humanized mice serve not only for tumor grafting and testing of various anti-CSC drugs, but also allow researchers to study and manipulate the immune response in advanced cancer therapy (118). To explore the efficiency of anti-CSC drug treatments on tumor growth, researchers also used animal models in which tumor material or primary human tissue were transplanted orthotopically into the humanized cleared mammary fat pad of NOD/SCID mice, without cultivation in vitro (66). The humanized orthotopic mammary fat pad mouse model was first developed by Kuperwasser et al. (119) in which both the stromal and epithelial components of the reconstructed mammary gland are of human origin. Injection of immortalized human breast fibroblasts resulted in creation of a ‘humanized’ stroma that consist about 15% human fibroblasts after 8 weeks. This type of model is closer to the human tumor and more suitable for stem cell studies compared with s.c. injection. In addition, Stewart et al., (120) found that mammary fat pad xenografts recapitulated inter-and intratumor heterogeneity of primary serous ovarian cancer (CD133+), as assessed by histology, surface immunophenotype, and expression of p53, WT1, and CK7. Alternatively, cancer-initiating cells can be injected orthotopically either under the renal capsule, ovarian bursa, or peritoneum of NOD/SCID or NSG mice (68, 120). Interestingly, some authors suggested that the mammary fat pad might provide a microenvironment more similar to that of human serous ovarian cancer than “more relevant” sites such as peritoneal cavity (120).
In contrast, researchers have proposed that the CSC concept can be applied to syngenic tumor models that would provide alternative approaches for the in-depth characterization of CSCs in an immunocompetent tumor microenvironment, and for the understanding of complex behaviors in the interaction between anti-CSC drugs and the host’s immune system (86). In addition, syngenic animal models have the advantage of being widely available for preclinical studies and allowing researchers to avoid legal and ethical issues related to the sourcing of human tissue samples.
Targeted gene disruption in rodents is a powerful tool for generating an entirely new type of models for the development of novel anti-cancer drugs. This technology opens up the possibility of in vivo CSC research utilizing genetically engineered mouse models (GEMM) that include transgenic models, i.e. introduction of gene sequences into the mouse genome, or gene knock-out/knock-in models in which endogenous mouse genes are deleted and/or modified (69, 121, 122). These GEMM models would most likely be able to more accurately mimic the cancer-related disparities in humans (123). Today, GEMMs are becoming an important tool in the preclinical development of anti-CSC drugs. Recent research using GEMMs include the examination of the molecular mechanisms by which resveratrol inhibits stem cell characteristics of pancreatic CSCs derived from human primary tumors in KrasG12D transgenic mice (116), identification of cells initiating human melanomas using NOD/SCID/IL2Rgammanull chain knock-out mice (80), or evaluation of prominin-1/CD133 as a biomarker for stem cells and early progenitors in mouse small intestine, using transgenic Prom1/CD133 knock-in mice (124).
CONCLUSION
Preclinical animal research is an excellent instrument for the better understanding of biology, genetics, immunology, pharmacology, and other Life Science disciplines in novel investigative situations. In the context of in vivo cancer stem cell research, the robust understanding of rodent physiology, possible genetic variability in rodents strains and the effect this may have on the observed phenotype differences, types and methods of induction of experimental tumors as well as behavioral and molecular biomarkers in translational animal models are critically important for the development of rational in vivo models for anti-CSC drug testing. In addition to this, a deeper understanding of pharmacological properties of various types of anti-cancer drugs, and modern technologies which can increase the selectivity of drugs for their intended targets may ultimately lead to a greater understanding of the CSC biology and thus, ultimately help improving the development of predictive biomarkers and targeted therapeutic strategies for the eradication of CSCs.
Abbreviations: CSC: cancer stem cell; DC: dendritic cells; NK: natural killer cells; PBMC: peripheral blood mononuclear cells; mAbs: monoclonal antibodies; ADCC: antibody-dependent cellular cytotoxicity; GI-tract: gastrointestinal tract; EPR-effect: enhanced permeability and retention effect; ABC: ATP-binding cassette; MB: medulloblastoma; PDX: patient-derived xenograft; GEMM: genetically engineered mouse models; SCID mice: severe combined immunodeficient mice.
Conflict of interests: None declared.
REFERENCES
- Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994; 367: 645-648.
- Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell 2012; 21: 283-296.
- Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell 2012; 10: 717-728.
- Diaz A, Leon K. Therapeutic approaches to target cancer stem cells. Cancers (Basel) 2011; 3: 3331-3352.
- Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov 2009; 8: 806-823.
- Naujokat C. Targeting Human Cancer Stem Cells with Monoclonal Antibodies. J Clin Cell Immunol 2012; S5: 007.
- Perlstein B, Finniss SA, Miller C, et al. TRAIL conjugated to nanoparticles exhibits increased anti-tumor activities in glioma cells and glioma stem cells in vitro and in vivo. Neuro Oncol 2013; 15: 29-40.
- Zhou L, Yang ZX, Song WJ, et al. MicroRNA-21 regulates the migration and invasion of a stem-like population in hepatocellular carcinoma. Int J Oncol 2013; 43: 661-669.
- Liu C, Kelnar K, Liu B, et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med 2011; 17: 211-215.
- Singh BN, Fu J, Srivastava RK, Shankar S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS One 2011; 6: e27306.
- Luistro L, He W, Smith M, et al. Preclinical profile of a potent gamma-secretase inhibitor targeting notch signaling with in vivo efficacy and pharmacodynamic properties. Cancer Res 2009; 69: 7672-7680.
- Hart S, Novotny-Diermayr V, Goh KC, et al. VS-5584, a novel and highly selective PI3K/mTOR kinase inhibitor for the treatment of cancer. Mol Cancer Ther 2013; 12: 151-161.
- Lee H, Kim JB, Park SY, Kim SS, Kim H. Combination effect of paclitaxel and hyaluronic acid on cancer stem-like side population cells. J Biomed Nanotechnol 2013; 9: 299-302.
- Fang DD, Cao J, Jani JP, et al. Combined gemcitabine and CHK1 inhibitor treatment induces apoptosis resistance in cancer stem cell-like cells enriched with tumor spheroids from a non-small cell lung cancer cell line. Front Med 2013; 7: 462-476.
- Gupta PB, Onder TT, Jiang G, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009; 138: 645-659.
- Li RJ, Ying X, Zhang Y, et al. All-trans retinoic acid stealth liposomes prevent the relapse of breast cancer arising from the cancer stem cells. J Control Release 2011; 149: 281-291.
- Guo J, Zhou J, Ying X, et al. Effects of stealth liposomal daunorubicin plus tamoxifen on the breast cancer and cancer stem cells. J Pharm Pharm Sci 2010; 13: 136-151.
- Lim KJ, Bisht S, Bar EE, Maitra A, Eberhart CG. A polymeric nanoparticle formulation of curcumin inhibits growth, clonogenicity and stem-like fraction in malignant brain tumors. Cancer Biol Ther 2011; 11: 464-473.
- Burke AR, Singh RN, Carroll DL, et al. The resistance of breast cancer stem cells to conventional hyperthermia and their sensitivity to nanoparticle-mediated photothermal therapy. Biomaterials 2012; 33: 2961-2970.
- Herrmann I, Baeuerle PA, Friedrich M, et al. Highly efficient elimination of colorectal tumor-initiating cells by an EpCAM/CD3-bispecific antibody engaging human T cells. PLoS One 2010; 5: e13474.
- Perez, A, Neskey DM, Wen J, et al. Targeting CD44 in head and neck squamous cell carcinoma (HNSCC) with a new humanized antibody RO5429083. Cancer Res 2012; 72 (Suppl. 1): abstract 2521. doi: 10.1158/1538-7445.AM2012-2521
- Korbut E, Ptak-Belowska A, Brzozowski T. Mechanisms promoting physiological cells progression into tumorigenesis. J Physiol Pharmacol 2012; 63: 565-570.
- Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun 2012; 12: 14.
- Burnett JC, Rossi JJ. RNA-based therapeutics: current progress and future prospects. Chem Biol 2012; 19: 60-71.
- Jackson AL. Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov 2010; 9: 57-67.
- Seth S, Johns R, Templin MV. Delivery and biodistribution of siRNA for cancer therapy: challenges and future prospects. Ther Deliv 2012; 3: 245-261.
- Zaks TZ, Rosenberg SA. Immunization with a peptide epitope (p369-377) from HER-2/neu leads to peptide-specific cytotoxic T lymphocytes that fail to recognize HER-2/neu+ tumors. Cancer Res 1998; 58: 4902-4908.
- Sahr RN. The Biologics Price Competition and Innovation Act: Innovation Must Come Before Price Competition 2 (Boston College Intellectual Prop. & Tech. Forum 2009). http://bciptf.org/?s=The+biologics+price+competition+and+innovation+act%3A+Innovation+must+come+before+price+competition
- Materials at http://foundation.aarp.org.
- Kruger FA, Overington JP. Global analysis of small molecule binding to related protein targets. PLoS Comput Biol 2012; 8: e1002333.
- Hoelder S, Clarke PA, Workman P. Discovery of small molecule cancer drugs: successes, challenges and opportunities. Mol Oncol 2012; 6: 155-176.
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986; 46: 6387-6392.
- Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev 2011; 63: 131-135.
- Steiniger SC, Kreuter J, Khalansky AS, et al. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int J Cancer 2004; 109: 759-767.
- Skidan I, Miao B, Thekkedath RV, Dholakia P, Degterev A, Torchilin V. in vitro cytotoxicity of novel pro-apoptotic agent DM-PIT-1 in PEG-PE-based micelles alone and in combination with TRAIL. Drug Deliv 2009; 16: 45-51.
- Skidan I, Dholakia P, Torchilin V. Photodynamic therapy of experimental B-16 melanoma in mice with tumor-targeted 5,10,15,20-tetraphenylporphin-loaded PEG-PE micelles. J Drug Target 2008; 16: 486-493.
- Miao B, Skidan I, Yang J, et al. Small molecule inhibition of phosphatidylinositol-3,4,5-triphosphate (PIP3) binding to pleckstrin homology domains. Proc Natl Acad Sci USA 2010; 107: 20126-20131.
- Zamboni WC, Torchilin V, Patri AK, et al. Best practices in cancer nanotechnology: perspective from NCI nanotechnology alliance. Clin Cancer Res 2012; 18: 3229-3241.
- Kim TH, Mount CW, Gombotz WR, Pun SH. The delivery of doxorubicin to 3-D multicellular spheroids and tumors in a murine xenograft model using tumor-penetrating triblock polymeric micelles. Biomaterials 2010; 31: 7386-7397.
- Bourseau-Guilmain E, Bejaud J, Griveau A, et al. Development and characterization of immuno-nanocarriers targeting the cancer stem cell marker AC133. Int J Pharm 2012; 423: 93-101.
- Upadhyay KK, Bhatt AN, Mishra AK, et al. The intracellular drug delivery and anti tumor activity of doxorubicin loaded poly(gamma-benzyl L-glutamate)-b-hyaluronan polymersomes. Biomaterials 2010; 31: 2882-2892.
- Banzato A, Bobisse S, Rondina M, et al. A paclitaxel-hyaluronan bioconjugate targeting ovarian cancer affords a potent in vivo therapeutic activity. Clin Cancer Res 2008; 14: 3598-3606.
- Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine (Lond) 2012; 7: 597-615.
- Sounni NE, Noel A. Targeting the tumor microenvironment for cancer therapy. Clin Chem 2013; 59: 85-93.
- Mizrak D, Brittan M, Alison M. CD133: molecule of the moment. J Pathol 2008; 214: 3-9.
- Gires O, Klein CA, Baeuerle PA. On the abundance of EpCAM on cancer stem cells. Nature Rev Cancer 2009; 9: 143.
- Williams K, Motiani K, Giridhar PV, Kasper S. CD44 integrates signaling in normal stem cell, cancer stem cell and (pre)metastatic niches. Exp Biol Med (Maywood) 2013; 238: 324-338. Epub 2013/04/20.
- Sano A, Kato H, Sakurai S, et al. CD24 expression is a novel prognostic factor in esophageal squamous cell carcinoma. Ann Surg Oncol 2009; 16: 506-514.
- Li L, Neaves WB. Normal stem cells and cancer stem cells: the niche matters. Cancer Res 2006; 66: 4553-4557.
- Borovski T, De Sousa EM, Vermeulen L, Medema JP. Cancer stem cell niche: the place to be. Cancer Res 2011; 71: 634-639.
- Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol 2007; 292: C987-C995.
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144: 646-674.
- Shiozawa Y, Taichman RS. Cancer stem cells and the bone marrow microenvironment. Bonekey Rep 2012; 2012: 48.
- Keysar SB, Jimeno A. More than markers: biological significance of cancer stem cell-defining molecules. Mol Cancer Ther 2010; 9: 2450-2457.
- Chen K, Huang YH, Chen JL. Understanding and targeting cancer stem cells: therapeutic implications and challenges. Acta Pharmacol Sin 2013; 34: 732-740.
- Madka V, Rao CV. Cancer stem cell markers as potential targets for epithelial cancers. Indian J Exp Biol 2011; 49: 826-835.
- Han ME, Oh SO. Gastric stem cells and gastric cancer stem cells. Anat Cell Biol 2013; 46: 8-18.
- Riether C, Schurch C, Ochsenbein AF. From “magic bullets” to specific cancer immunotherapy. Swiss Med Wkly 2013; 143: w13734. doi: 10.4414/smw.2013.13734.
- Hill RP, Perris R. “Destemming” cancer stem cells. J Nat Cancer Inst 2007; 99: 1435-1440.
- Tu LC, Foltz G, Lin E, Hood L, Tian Q. Targeting stem cells-clinical implications for cancer therapy. Curr Stem Cell Res Ther 2009; 4: 147-153.
- Liu C, Tang DG. MicroRNA regulation of cancer stem cells. Cancer Res 2011; 71: 5950-5954.
- Klonisch T, Wiechec E, Hombach-Klonisch S, et al. Cancer stem cell markers in common cancers - therapeutic implications. Trends Mol Med 2008; 14:450-460.
- Rasheed ZA, Kowalski J, Smith BD, Matsui W. Concise review: Emerging concepts in clinical targeting of cancer stem cells. Stem Cells 2011; 29: 883-887.
- Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res 2007; 17: 3-14.
- Ponnusamy MP, Batra SK. Ovarian cancer: emerging concept on cancer stem cells. J Ovarian Res 2008; 1: 4.
- Ginestier C, Hur MH, Charafe-Jauffret E, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007; 1: 555-567.
- Steiniger SC, Coppinger JA, Kruger JA, Yates J, Janda KD. Quantitative mass spectrometry identifies drug targets in cancer stem cell-containing side population. Stem Cells 2008; 26: 3037-3046.
- O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007; 445: 106-110.
- Read TA, Fogarty MP, Markant SL, et al. Identification of CD15 as a marker for tumor-propagating cells in a mouse model of medulloblastoma. Cancer Cell 2009; 15: 135-147.
- Barretina J, Caponigro G, Stransky N, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483: 603-607.
- Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell 2006; 10: 515-527.
- Charafe-Jauffret E, Ginestier C, Monville F, et al. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006; 25: 2273-2284.
- Li C, Heidt DG, Dalerba P, et al. Identification of pancreatic cancer stem cells. Cancer Res 2007; 67: 1030-1037.
- Bosma MJ, Carroll AM. The SCID mouse mutant: definition, characterization, and potential uses. Annu Rev Immunol 1991; 9: 323-350.
- Ito M, Hiramatsu H, Kobayashi K, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood 2002; 100: 3175-3182.
- Shultz LD, Schweitzer PA, Christianson SW, et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol 1995; 154: 180-191.
- Ong GL, Baker AE, Mattes MJ. Analysis of high complement levels in Mus hortulanus and BUB mice. J Immunol Methods 1992; 154: 37-45.
- van der Loo JC, Hanenberg H, Cooper RJ, Luo FY, Lazaridis EN, Williams DA. Nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mouse as a model system to study the engraftment and mobilization of human peripheral blood stem cells. Blood 1998; 92: 2556-2570.
- Lock RB, Liem N, Farnsworth ML, et al. The nonobese diabetic/severe combined immunodeficient (NOD/SCID) mouse model of childhood acute lymphoblastic leukemia reveals intrinsic differences in biologic characteristics at diagnosis and relapse. Blood 2002; 99: 4100-4108.
- Schatton T, Murphy GF, Frank NY, et al. Identification of cells initiating human melanomas. Nature 2008; 451: 345-349.
- Pretlow TG, Wolman SR, Micale MA, et al. Xenografts of primary human prostatic carcinoma. J Nat Cancer Inst 1993; 85: 394-398.
- Bosma MJ. B and T cell leakiness in the scid mouse mutant. Immunodefic Rev 1992; 3: 261-276.
- Shultz LD, Lyons BL, Burzenski LM, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 2005; 174: 6477-6489.
- Rolstad B. The athymic nude rat: an animal experimental model to reveal novel aspects of innate immune responses? Immunol Rev 2001; 184: 136-144.
- Talmadge JE, Singh RK, Fidler IJ, Raz A. Murine models to evaluate novel and conventional therapeutic strategies for cancer. Am J Pathol 2007; 170: 793-804.
- Kruger JA, Kaplan CD, Luo Y, et al. Characterization of stem cell-like cancer cells in immune-competent mice. Blood 2006; 108: 3906-3912.
- Charafe-Jauffret E, Ginestier C, Iovino F, et al. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res 2009; 69: 1302-1313.
- Fridman R, Benton G, Aranoutova I, Kleinman HK, Bonfil RD. Increased initiation and growth of tumor cell lines, cancer stem cells and biopsy material in mice using basement membrane matrix protein (Cultrex or Matrigel) co-injection. Nat Protoc 2012; 7: 1138-144.
- Alonso-Camino V, Santos-Valle P, Ispizua MC, Sanz L, Alvarez-Vallina L. Engineered human tumor xenografts with functional human vascular networks. Microvasc Res 2011; 81: 18-25.
- Decaudin D. Primary human tumor xenografted models (‘tumorgrafts’) for good management of patients with cancer. Anticancer Drugs 2011; 22: 827-841.
- Moro M, Bertolini G, Tortoreto M, Pastorino U, Sozzi G, Roz L. Patient-derived xenografts of non small cell lung cancer: resurgence of an old model for investigation of modern concepts of tailored therapy and cancer stem cells. J Biomed Biotechnol 2012; 2012: 568567.
- Kim S, Park YW, Schiff BA, et al. An orthotopic model of anaplastic thyroid carcinoma in athymic nude mice. Clin Cancer Res 2005; 11: 1713-1721.
- Gelbard A, Kupferman ME, Jasser SA, et al. An orthotopic murine model of sinonasal malignancy. Clin Cancer Res 2008; 14: 7348-7357.
- Le NH, Franken P, Fodde R. Tumour-stroma interactions in colorectal cancer: converging on beta-catenin activation and cancer stemness. Br J Cancer 2008; 98: 1886-1893.
- Penchev VR, Rasheed ZA, Maitra A, Matsui W. Heterogeneity and targeting of pancreatic cancer stem cells. Clin Cancer Res 2012; 18: 4277-4284.
- Masood R, Hochstim C, Cervenka B, et al. A novel orthotopic mouse model of head and neck cancer and lymph node metastasis. Oncogenesis 2013; 2: e68.
- Yae T, Tsuchihashi K, Ishimoto T, et al. Alternative splicing of CD44 mRNA by ESRP1 enhances lung colonization of metastatic cancer cell. Nat Commun 2012; 3: 883. doi: 10.1038/ncomms1892
- Du L, Rao G, Wang H, et al. CD44-positive cancer stem cells expressing cellular prion protein contribute to metastatic capacity in colorectal cancer. Cancer Res 2013; 73: 2682-2694.
- La Porta CA. Thoughts about cancer stem cells in solid tumors. World J Stem Cells 2012; 4: 17-20.
- Ruggeri BA, Camp F. Miknyoczki S. Animal models of disease: pre-clinical animal models of cancer and their applications and utility in drug Discovery. Biochem Pharmacol 2013; 87: 150-161.
- van der Worp HB, Howells DW, Sena ES, et al. Can animal models of disease reliably inform human studies? PLoS Med 2010; 7: e1000245. doi: 10.1371/journal.pmed.1000245.
- Francipane MG, Chandler J, Lagasse E. Cancer stem cells: a moving target. Curr Pathobiol Rep 2013; 1: 111-118.
- Lathia JD, Gallagher J, Myers JT, et al. Direct in vivo evidence for tumor propagation by glioblastoma cancer stem cells. PloS One 2011; 6: e24807. doi: 10.1371/journal.pone.0024807.
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature 2008; 456: 593-598.
- Curtis SJ, Sinkevicius KW, Li D, et al. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell 2010; 7: 127-133.
- Cheng L, Ramesh AV, Flesken-Nikitin A, Choi J, Nikitin AY. Mouse models for cancer stem cell research. Toxicol Pathol 2010; 38: 62-71.
- Valent P, Bonnet D, De Maria R, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer 2012; 12: 767-775.
- Park S, Jiang Z, Mortenson ED, et al. The therapeutic effect of anti-HER2/neu antibody depends on both innate and adaptive immunity. Cancer Cell 2010; 18: 160-170.
- Xu Q, Liu G, Yuan X, et al. Antigen-specific T-cell response from dendritic cell vaccination using cancer stem-like cell-associated antigens. Stem Cells 2009; 27: 1734-1740.
- Mosier DE, Gulizia RJ, Baird SM, Wilson DB. Transfer of a functional human immune system to mice with severe combined immunodeficiency. Nature 1988; 335: 256-259.
- Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science 1992; 255: 1137-1141.
- Shih CC, Hu J, Arber D, LeBon T, Forman SJ. Transplantation and growth characteristics of human fetal lymph node in immunodeficient mice. Exp Hematol 2000; 28: 1046-1053.
- McCune JM, Namikawa R, Kaneshima H, Shultz LD, Lieberman M, Weissman IL. The SCID-hu mouse: murine model for the analysis of human hematolymphoid differentiation and function. Science 1988; 241: 1632-1639.
- Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nature Rev Immunol 2012; 12: 786-798.
- Brehm MA, Shultz LD, Greiner DL. Humanized mouse models to study human diseases. Curr Opin Endocrinol Diabetes Obes 2010; 17: 120-125.
- Shankar S, Nall D, Tang SN, et al. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and KrasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PloS One 2011; 6: e16530.
- Fu J, Rodova M, Roy SK, et al. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett 2013; 330: 22-32.
- Wege AK, Ernst W, Eckl J, et al. Humanized tumor mice—a new model to study and manipulate the immune response in advanced cancer therapy. Int J Cancer 2011; 129: 2194-2206.
- Kuperwasser C, Chavarria T, Wu M, et al. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc Nat Acad Sci USA 2004; 101: 4966-4971.
- Stewart JM, Shaw PA, Gedye C, Bernardini MQ, Neel BG, Ailles LE. Phenotypic heterogeneity and instability of human ovarian tumor-initiating cells. Proc Nat Acad Sci USA 2011; 108: 6468-6473.
- Doyle A, McGarry MP, Lee NA, Lee JJ. The construction of transgenic and gene knockout/knockin mouse models of human disease. Transgenic Res 2012; 21: 327-349.
- Wolfer DP, Crusio WE, Lipp HP. Knockout mice: simple solutions to the problems of genetic background and flanking genes. Trends Neurosci 2002; 25: 336-340.
- Politi K, Pao W. How genetically engineered mouse tumor models provide insights into human cancers. J Clin Oncol 2011; 29: 2273-2281.
- Snippert HJ, van Es JH, van den Born M, et al. Prominin-1/CD133 marks stem cells and early progenitors in mouse small intestine. Gastroenterology 2009; 136: 2187-2194.
- Gurney A, Axelrod F, Bond CJ, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Nat Acad Sci USA 2012; 109: 11717-11722.
- Dallas NA, Xia L, Fan F, et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res 2009; 69: 1951-1957.
A c c e p t e d : January 24, 2014