EFFECT OF VITAMIN E ON CEREBRAL CORTICAL OXIDATIVE STRESS AND BRAIN-DERIVED NEUROTROPHIC FACTOR GENE EXPRESSION INDUCED BY HYPOXIA AND EXERCISE IN RATS
INTRODUCTION
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is a key protein in the regulation of the maintenance, growth and survival of neurons (1). BDNF is important for cell proliferation, cell differentiation, neuronal protection, and the regulation of synaptic function in the central nervous system (CNS) via stimulating key intracellular signaling cascades (2). In addition, BDNF plays an important role in various aspects of neural plasticity, such as neurogenesis, long-term potentiation (LTP), learning and memory, and mood changes (3). The mutation or deletion of the BDNF gene in mice results in learning deficits and LTP impairment, whereas re-expression of BDNF restores LTP (4). In patients with Alzheimer’s disease or major depression, the brain or serum levels of BDNF are decreased (5, 6). Accumulated evidence has demonstrated that stress or corticosterone administration reduces BDNF mRNA expression in the brain (7). In addition, BDNF synthesis is centrally mediated by an activity-dependent process (8), and exercise enhances BDNF transcription in the brain (9). Therefore, in order to clarify the changes in BDNF after different stresses, we studied the expression of BDNF mRNA in the cerebral cortex of Sprague-Dawley rats following chronic exercise in normal and hypoxic conditions by real-time polymerase chain reaction (RT-PCR).
Under the hypoxic condition, the decreased partial pressure of O2 causes a reduction in available cellular O2, which is the ultimate electron acceptor in the electron transport chain (10). Due to low levels of O2, the electron accumulation is higher, and more electrons attack the ground state of available oxygen (O2) to form superoxide anion (O2–), in turn, H2O2 and hydroxyl radicals (OH–) are formed by chain reaction (11). In this way the reactive oxygen species (ROS) level is increased in hypoxia, which causes oxidative stress (12). In addition, under hypoxic stress, cellular defence systems such as antioxidant enzymes (glutathione peroxidase, glutathione reductase, superoxide dismutase) are disturbed and their activity decreases (13). The hypoxia-induced generation of ROS can cause protein oxidation, DNA and RNA oxidation, lipid peroxidation and neuronal dysfunction or death (14). Also, oxidative stress may be associated with various neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis (15). Therefore, it is crucial to identify how to secure the neuronal system from oxidative stress. Moreover, the brain is sensitive to oxidative stress due to (i) an abundant presence of polyunsaturated fatty acid, (ii) deficient antioxidant defence, (iii) a high rate of O2 utilisation due to the high metabolic rate and (iv) a high content of transition metals such as copper and iron in several regions, which could lead to the formation of hydroxyl radicals via the Fenton reaction (16). In addition, the cortex is particularly sensitive to hypoxia (17); therefore, the present study investigated the process of hypoxia-induced oxidative insult in the rat cortex and its possible effect on cortical BDNF mRNA expression in rats experiencing chronic exercise with or without vitamin E.
Accumulating evidence suggests that physical exercise is capable of beneficially affecting certain brain functions (18). It has been shown that voluntary running enhances brain plasticity and leads to an increase in the number of new hippocampal cells (19). However, we still lack a comprehensive picture regarding the relationships between exercise and oxidative stress in the brain. There are few studies on the effects of exercise on oxidative damage or the antioxidant brain state, and the available findings are conflicting (20). Radak et al. (21) showed that physical exercise, especially a single bout of exercise, could generate increased levels of ROS and result in oxidative damage to macromolecules. To date, the evidence for oxidative stress and tissue damage due to exercise remains incomplete because of the complexity of the chosen exercise models. Therefore, we tested the hypothesis that exercise can cause oxidative damage to the rat brain cortex in normal or hypoxic rats and its possible effect on cortical BDNF mRNA expression.
Vitamin E is a potent antioxidant that protects organisms against reactive oxygen species (ROS)- and reactive nitrogen species (RNS)-mediated damage. Epidemiological and clinical studies have suggested that vitamin E supplementation provides beneficial effects for neurodegenerative diseases such as Alzheimer’s disease (22) and Parkinson’s disease (23). Previous studies have indicated the susceptibility of cultured cortical neurons to oxidative stress, such as glutamate (24) and NO (25), which results in neuronal cell death. It has been reported that treatment with a-tocotrienol 5 min before or after glutamate stimulation protects cortical cells from glutamate-induced neurotoxicity (26). In another system, cotreatment with vitamin E analogs blocked NO or O2• donor-induced cell death in rat striatal cultures (27). These reports have suggested that vitamin E analogs acutely protect neuronal cells from oxidative stress.
Since there are few reports in the literature investigating the effects of sustained arterial hypoxia and exercise on cerebral cortical BDNF expression, so, the aim of the present work is to study the effects of chronic exercise, with or without vitamin E, on sustained hypoxia-induced oxidative stress and BDNF expression in rat cerebral cortex.
MATERIALS AND METHODS
Animals
Sixty four male Sprague-Dawley rats, 4 weeks old with a mean weight of 76.5 ± 3.7 g, were purchased from the Vaccine and Immunization Authority (Helwan, Cairo, Egypt) and housed (Animal House, Medical Physiology Department, Faculty of Medicine, Mansoura University, Egypt) under controlled conditions (23 ± 1°C and a 12 h light: 12 h dark cycle). The animals were allowed free access to food and tap water. The experiments were performed according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996). All experimental procedures in this study were approved by the Medical Research Ethics Committee of Mansoura University, Egypt (Protocol number: R/98).
Experimental design
At the age of four weeks, the rats were randomly divided into two groups of thirty two rats each. Group I rats were housed in the usual cages and provided with normal air (21% O2); these animals served as a normoxic (N) control. Group II rats were placed in cages supplied with 12% O2 (28). These rats served as the chronically hypoxic (CH) group. Animals from both groups were further subdivided into four subgroups of 8 rats each: sedentary, sedentary with vitamin E (100 mg/kg/day, i.p. injection) (29), chronic exercise and chronic exercise with vitamin E (100 mg/kg/day, i.p. injection) (29). Exercise program was started at the age of 4 weeks. Rats in CH group performed exercise at low PO2.
Exercise program
The animals in the chronic exercise subgroups were forced to perform exhaustive swimming in 26°C water in a 100 × 40 × 60 cm glass tank (35 cm depth of water) for 1 h/day at 8:30 am, 5 times/week, over a period of 8 weeks from the ages of 4 to 12 weeks old. The swimming duration increased by 30 min each week up to a maximum 4.5 h in the last week. At the end of each exercise session, animals were dried and kept in a warm environment. Sedentary controls were restricted to cage activity. However, on the days of exercise practice, the sedentary animals were removed from their cages and kept for 1 hour in the container (previously cleaned and dried) where the swimming sessions had taken place in order to handle stress (30).
Sampling protocol
Blood samples
At the end of the experimental period, arterial blood samples were taken from the rat tail artery cannula to analyse arterial blood PO2 using a laboratory blood gas analyser (Instrumentation Laboratory, MA, USA).
Brain samples
Each rat was anesthetised by ether, and the whole brain was immediately removed, washed with chilled saline, and weighed. The cortex was dissected on ice, frozen in liquid nitrogen, and stored at –80°C until biochemical analysis of the lipid peroxidation products (malondialdehyde, MDA), and antioxidants (reduced glutathione, catalase, superoxide dismutase).
Assessment of lipid peroxide, enzymatic and non-enzymatic antioxidants, and vitamin E in cerebral cortex
The cerebral cortex was removed and rinsed with phosphate-buffered saline (pH 7.4) to remove any red blood cells or clots. Next, the tissue was homogenised in 5–10 mL of cold 20 mmol/L HEPES buffer (pH 7.2) containing 1 mmol/L EGTA, 210 mmol/L mannitol, and 70 mmol/L sucrose per gram of tissue. The homogenates were centrifuged at 15,000 r/min (18 000 g) for 15 min at 4°C, and the supernatant was removed and stored at –80°C until further analysis of the glutathione (GSH), catalase (CAT), superoxide dismutase (SOD), MDA and vitamin E levels.
In the cerebral cortex tissue homogenate, the levels of GSH (Cat. No. 703002; Cayman Chemical, Ann Arbor, Mich., USA), SOD (Cat. No. 706002, Cayman Chemical, Ann Arbor, Mich., USA), CAT (Cat. No. NWK-CAT01, Northwest Life Science Specialties, Vancouver, Wash., USA) and vitamin E (Cat. No. C90922Ra, USCN Life Science Inc., USA) were measured according to the manufacturer’s instructions. In addition, MDA was analysed in the brain homogenate by measuring the production of thiobarbituric acid-reactive substances (TBARS) with a TBARS assay kit (Cat. No. 10009055, Cayman Chemical, Ann Arbor, Mich., USA).
Total RNA extraction from the brain tissue of rats
Total RNA extraction cerebral cortex with liquid nitrogen was carried out immediately using the TriFast TM reagent (PeqLab. Biotechnologie GmbH, Carl-Thiersch St. 2B 91052 Erlongen, Germany, Cat. No. 30-2010) according to the manufacturer’s instructions. The remaining DNA was removed by digestion with DNase I (Sigma). The concentration of isolated RNA was determined using a spectrophotometer that measures the optical density (OD) at 260 nm (Jenway, Genova Model, UK). A 10 µl aliquot of each sample was added to 990 µl of DEPC-treated water and by measuring the absorbance at 260 nm, the RNA yield (µg/ml) was quantified A260 X 40 X 100 (dilution factor). RNA purity was determined using formaldehyde agarose gel electrophoresis and ethidium bromide staining, which should produce 2 sharply purified bands representing 28S and 18S ribosomal RNA.
RT-PCR for the extracted RNA
RT-PCR was performed using Ready-to-Go RT-PCR beads (Amersham Biosciences England. Cat. No. 27-9266-01) for the first strand cDNA synthesis and PCR reaction according to the method of Berchtold.
Ready-to-Go RT-PCR beads utilise Moloney Murine Leukemia virus (M-MuLV) reverse transcriptase and Taq polymerase to generate the PCR product from an RNA template. Each bead is optimised to allow the first strand cDNA synthesis and PCR reaction to proceed sequentially as a single tube, single step reaction. The reaction proceeded as follows:
1. Synthesis of cDNA
The following materials were added to each tube along with the beads: 2 µl of first strand primer provided by the kit, 3 µl containing 30 pmol of PCR gene-specific primer (sense), 3 µl containing 30 pmol of PCR gene-specific primer (anti-sense), 25 µl containing 1 µg of total template RNA and 17 µl of DEPC-treated water to obtain a total volume of 50 µl. One tube was prepared as a negative control reaction to test for DNA contamination.
The dehydrated bead (without template and primers) was incubated at 95°C for 10 min to inactivate the M-MuLV reverse transcriptase. A 50-ul drop of mineral oil was added to overlay the reaction. The reactions were transferred to a thermal cycler and incubated at 40°C for 30 min for cDNA synthesis, followed by incubation at 95°C for 5 min to inactivate the reverse transcriptase and completely denature the template.
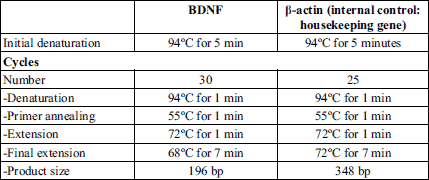
Gene specific primers were purchased from Biolegio (BV, PO Box 91, 5600 AB Nijmegen, Netherlands) as shown in Table 1.

2. Amplification of cDNA by PCR:
The thermal cycling reaction was performed using a thermal cycler (TECHEN TC-312, Model FTC3102D, Barloworld Scientific Ltd. Stone, Staffordshire, st 150 SA, UK) with the program shown in Table 2.
3. Detection of amplified RT-PCR products
For semi-quantitative RT-PCR, the amplification products were subjected to agarose gel electrophoresis using 2% agarose stained with ethidium bromide, visualised with UV light (Transilluminator, Model TUV-20, OWI. Scientific, Inc. 800 242-5560) and photographed under fixed conditions (constant distance, light and scale).
The resulting photos were analysed with Scion Image® release Alpha 4.0.3.2. software for Windows®, which performs band detection and peak conversion. The area under each peak was calculated in square pixels and used for quantification. The gene expression levels were determined by calculating the ratio between the square pixel values of the target gene in relation to the internal housekeeping control gene (β-actin).
The negative control tubes showed no PCR products, indicating that all of the reagents were free from target sequence contamination.
Quantitative real-time PCR
Reverse transcription (RT) of the extracted RNA was performed according to the manufacturer’s instructions using Maxima First Strand cDNA Synthesis Kit for RT-qPCR provided by ThermoScientific , U.S.A, cat No. #K1641.A control reaction containing 2 µl of purified mRNA is run without reverse transcription to confirm the DNA contamination, if any, in the purified mRNA samples. The synthesized cDNA is stored at –20°C before use.
1- Gene-specific PCR primers: Oligonucleotide primers designed to amplify rat BDNF were specific for the coding region of exon 5.BDNF-specific primers were 5’-CAGGGGCATAGACAAAAG-3’(forward); and 5’-CTTCCCCTTTTAATGGTC-3’ (reverse); (BDNF PCR product: 167 bp; (31), while for β-actin, used as internal control, 5’-CCTGTATGCCTCTGGTCGTA-3’ (forward) and 5’-CCATCTCTTGCTCGAAGTCT-3’ (reverse); β-actin PCR product: 260 bp; (32). The gene specific primers were purchased from Oligo™ Macrogen.
2- PCR reactions for optimization of primer annealing temperature were performed using DreamTaq™ Green PCR Master Mix (2X) provided by Thermo Scientific, USA, cat No. 1081.
The primer concentration is optimized for use in real time PCR to determine the minimum primer concentration giving the lowest CT (threshold cycle) while minimizing nonspecific amplification. Briefly, each amplification in duplicates of 25 µL reaction containing 12.5 µL 2X SYBR Green Master Mix, 2 µL cDNA and a variable concentration of forward and reverse primers is run with thermal cycling parameters as initial step of hold at 95°C for 10 min followed by 40 cycles of denaturation (hold at 95°C for 30 s) and annealing (hold at 60°C for 1 min). PCR products were subjected to agarose gel electrophoresis on 2% (w/v) agarose gels.
3- Quantitative real-time PCR analysis: the real time PCR assays were performed on real time PCR set (Applied Biosystem 7500, USA) with 96-well plates, using SybrGreen reagent (SYBR® Green PCR Master Mix (Applied Biosystem, USA, cat.No. 4344463)).
Amplification were performed, in duplicates, in a total of 25 µL reaction containing forward and reverse primers (10 pmol of each final concentration), 12.5 µL Power Sybr® Green PCR Master Mix reaction buffer (Applied Biosystems) and 2 µL cDNA. The cycling parameters are 95°C for 30 s, 60°C for 1 min, 40 cycles after one initial step of 95°C for 10 min, which is set to activate AmpliTaq Gold polymerase. The PCR products are monitored by measuring the increase in fluorescence caused by binding of SYBR Green Dye to the double stranded DNA and CT values (cycle threshold) are calculated. We performed melting curve analysis and agarose gel electrophoresis 2% to confirm the specificity of the PCR products obtained using each primer pair (Figs. 3). Samples containing no template were used as negative controls in each experiment.
The relative quantification of the BDNF gene in different tissue samples is doneusing a comparative method. β-actin is used as endogenous control to standardize the amount of DNA added to the reaction. Each analysis requires 4 reactions (2 for analysis of the target BDNF and 2 for analysis of the internal standard, β actin). The ΔCT per each sample is calculated and linearized using 2–ΔCT. Finally ΔΔCT between experimental and control samples is calculated and linearized using 2-ΔΔCT for overall change. Thus, the amount of target BDNF gene in the studied groups normalized to an endogenous reference (β actin) and relative to control group is given by an arithmetic formula.
Statistical analysis
The data were expressed as the mean ± standard deviation (S.D.). The data were processed and analysed using SPSS version 10.0 (SPSS, Inc., Chicago, Ill., USA). One-way ANOVA was done followed by Tukey’s post hoc test. Pearson correlation statistical analysis was utilised for the detection of a probable significance between two different parameters. The results were considered significant for P ≤0.05.
RESULTS
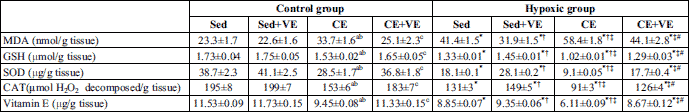
Exposure of rats to 12 % oxygen results in significant reduction in arterial PO2 as compared with the control group (61.2 ± 2.3 mmHg versus 91.8 ± 2.9 mmHg respectively). The changes in the activity of CAT and SOD and the levels of GSH, vitamin E and MDA in the cortex of control and hypoxic rats having experienced chronic exercise, are shown in Table 3. CAT and SOD activity and GSH, vitamin E and MDA levels were affected by hypoxia as well as chronic exercise. Hypoxia significantly increased MDA, decreased SOD and CAT activity and lowered the GSH and vitamin E levels (P <0.05) in the cortex of sedentary rats compared with the control group. Also, chronic exercise, in control or hypoxic rats, significantly increased MDA, decreased SOD and CAT activity and lowered the GSH and vitamin E levels in the cortex of control rats (P <0.05) compared to the sedentary control group. Administration of vitamin E significantly decreased the MDA level, increased SOD and CAT activity and boosted the GSH and vitamin E levels in the cortex of hypoxic sedentary group and control or hypoxic rats that experienced chronic exercise (P <0.05), but no changes were observed in control sedentary rats (P >0.05). A significant positive correlation was found between MDA and BDNF gene expression (r=0.7145) (P <0.00001) (Fig. 1).
a significant (P<0.05) as compared to control sedentary group; b significant (P <0.05) as compared to vitamin E (VE) group; c significant (P <0.05) as compared to control + chronic exercise (CE) group; * significant (P <0.05) as compared to the corresponding control group; † significant (P <0.05) as compared to the hypoxic sedentary group; ‡ significant (P <0.05) as compared to hypoxia sedentary + vitamin E (VE) group; # significant (P <0.05) as compared to hypoxia + chronic exercise (CE) group.
 |
Fig. 1. Pearson correlation between MDA and BDNF gene expression. (r=0.7145) (P <0.0001) (n=64). |
Levels of mRNA BDNF (exons 5) in the cortex of both experimental and control animals were measured and analyzed (Figs. 2-5). ΔΔCT between experimental and control samples was calculated and linearized using 2–ΔΔCT for overall change. Chronic exercise significantly increased BDNF gene expression in the cortex of the control animals compared with the control sedentary group (P <0.05) (Figs. 2-4). Moreover, BDNF gene expression in the cortex was significantly increased by hypoxia (P <0.05) with further rise (P <0.05) during the chronic exercise (Figs. 2, 3 and 5). Vitamin E administration significantly decreased BDNF gene expression in the cortex of the control or hypoxic rats subjected to chronic exercise as well as hypoxic sedentary group compared with the corresponding untreated group (P <0.05) (Figs. 2-5), but no changes were observed in control sedentary rats given vitamin E (P >0.05).
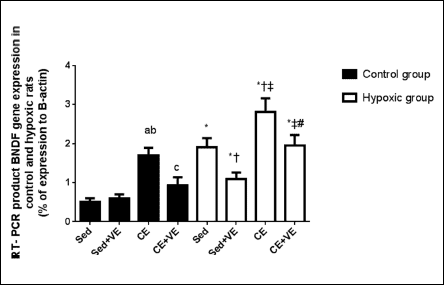 |
Fig. 2. Percent of expression RT-PCR product BDNF gene expression in rats of the control and hypoxic groups. The values are expressed as the mean ± S.D. for 8 rats in each group to the house keeping gene. Sed, sedentary; AE, acute exercise; CE, chronic exercise; VE, vitamin E. a significant (P <0.05) as compared to control sedentary group; b significant (P <0.05) as compared to control sedentary + vitamin E (VE) group; c significant (P <0.05) as compared to control + chronic exercise (CE) group; * significant (P <0.05) as compared to the corresponding control group; † significant (P <0.05) as compared to the hypoxic sedentary group; ‡ significant (P <0.05) as compared to hypoxia sedentary + vitamin E (VE) group; #: significant (P <0.05) as compared to hypoxia + chronic exercise (CE) group. |
 |
Fig. 3. Brain-derived neurotrophic factor (BDNF) gene expression in different experimental groups (overall change relative to sedentary control group 2-ΔΔCT). The values are expressed as the mean ± S.D. for 8 rats in each group to the house keeping gene. Sed, sedentary; CE, chronic exercise; VE, vitamin E. a significant (P <0.05) as compared to control sedentary group; b significant (P <0.05) as compared to control sedentary + vitamin E (VE) group; c significant (P <0.05) as compared to control + chronic exercise (CE) group; * significant (P <0.05) as compared to the corresponding control group; † significant (P <0.05) as compared to the hypoxic sedentary group; ‡ significant (P <0.05) as compared to hypoxic sedentary + vitamin E (VE) group; # significant (P <0.05) as compared to hypoxia + chronic exercise (CE) group. |
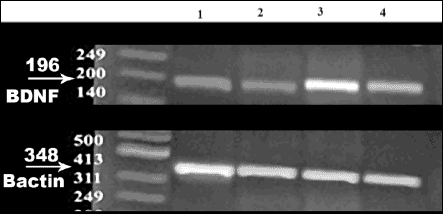 |
Fig. 4. Agarose gel electrophoresis of semi-quantitative RT-PCR product of brain-derived neurotrophic factor (BDNF) gene expression in rats of the control groups. Lane 1: RT-PCR product of BDNF gene in the Sed group; Lane 2: RT-PCR product of BDNF gene expression in the sed + VE group; Lane 3: RT-PCR product of BDNF gene expression in the CE group; Lane 4: RT-PCR product of BDNF gene expression in the CE+ VE group. Sed, sedentary; CE, chronic exercise; VE, vitamin E. |
 |
Fig. 5. Agarose gel electrophoresis of semi-quantitative RT-PCR product of brain-derived neurotrophic factor (BDNF) gene expression in rats from the hypoxic groups. Lane 1: RT-PCR product of BDNF gene in the Sed group; Lane 2: RT-PCR product of BDNF gene expression in the Sed + VE group; Lane 3: RT-PCR product of BDNF gene expression in the CE group; Lane 4: RT-PCR product of BDNF gene expression in the CE+ VE group. Sed, sedentary; CE, chronic exercise; VE, vitamin E. |
DISCUSSION
In the current study, chronic hypoxia was induced in male Sprague-Dawley rats by subjecting the animals to 12% O2 for 8 weeks. The state of hypoxia was confirmed by arterial PO2 analysis. Both the control and hypoxic rats were subjected chronic exercise (swimming 1 hour/day, 5 times/week, for 8 weeks with an increase in swimming duration by 30 min each week up to a maximum of 4.5 h during the last week). Forced-swimming, a relatively mild physiological stress that causes no damage to the body, was used to shape the current animal model. In addition, vitamin E was given to the control and hypoxic rats experiencing chronic exercise. Thus, this study investigated the effects of chronic exercise on hypoxia-induced oxidative stress in the brain cortex of rats with and without vitamin E treatment. In addition, the changes in cortical BDNF gene expression during exercise and hypoxia, with and without vitamin E treatment, were also evaluated. The present results demonstrated that hypoxia induced oxidative stress and increased BDNF gene expression in the cortex of rats as confirmed by biochemical assays and real time-PCR, respectively. Moreover, oxidative stress and BDNF gene expression were increased in the cortex of control or hypoxic rats by chronic exercise. In addition, vitamin E treatment attenuated the oxidative stress and decreased BDNF gene expression in the cortex of hypoxic sedentary rats as well as control or hypoxic animals subjected to chronic exercise, whereas it had no effects on BDNF gene expression in control sedentary rats.
BDNF is a neurotrophic factor involved in critical CNS functions, as well as synaptic transmission and plasticity, and it plays an important role in the survival, maintenance, and growth of neurons (1). The effect of exercise, either voluntary or forced, on BDNF is poorly documented. Furthermore, most studies on BDNF after exercise have focused on the hippocampus, a region which is more involved in learning/memory than in motor function (33). In addition, many recent studies reported an increase in BDNF in the hippocampus of rodents subjected to exercise (33). In contrast, our present study focused on BDNF gene expression in the brain cortex of normal and hypoxic rats with chronic swimming exercise. The results of the current work showed that chronic exercise significantly increased BDNF gene expression in the cortex of control rats (Figs. 2-4). Moreover, hypoxia enhanced BDNF gene expression in the rat cortex with further increase in hypoxic rats subjected to chronic exercise (Figs 2, 3 and 5). In agreement with our results, Rasmussen et al. (34) suggest that the brain has significant BDNF production both at rest and during prolonged exercise, and this may be a major source of the increased plasma BDNF observed during exercise in healthy subjects. An exercise-induced increase in BDNF mRNA expression in the mouse hippocampus and cortex occurs in response to a single exercise bout (34). Almost three quarters of the BDNF present in the venous circulation originated from brain structures, which indicate that brain tissue is the main origin of the circulating BDNF (34). Under normal conditions, BDNF is mainly synthesised and released by neurons and astrocytes, and activated and proliferating astrocytes release much more BDNF during cerebral ischemia or hypoxia (35). Additionally, Liu and Dai 36 showed that a high level of BDNF is released from hypoxic astrocytes. There are other possible explanations for the release of BDNF. BDNF is released from the cerebral vascular endothelium following hypoxic stress (37). While such stress may not be present at rest, exercise could result in cerebral hypoxic stress because cerebral oxygen tension decreases during strenuous exercise (38). These results were confirmed by our study which demonstrated a significant increase in cerebral cortical BDNF expression in hypoxic sedentary rats and following chronic exercise in control and hypoxic rats. The transient up-regulation of BDNF mRNA expression in the mouse hippocampus and cortex in response to exercise emphasises the likely importance of specific parts of the brain as a source of BDNF at rest and during exercise (34).
The synthesis of BDNF may be part of the adaptation to a new stimulus such as exercising to exhaustion, or may be involved in coping with a new or stressful environment, such as the exercise laboratory (34). It may be that a threshold must be surpassed before an increase in BDNF mRNA is exhibited (34), and so cortical BDNF mRNA increased in our study because the animals were highly active during exercise. In a study by Shi et al. (39), acute stress was observed to induce a rapid increase in the expression of BDNF and BDNF mRNA in the hippocampus, in both young and aged animals. Therefore, unexpected and acute stress tends to provide an excited organism with a degree of protection for a short time (39). This suggests that the augmented expression of BDNF mRNA and protein might, either directly or indirectly, contribute to stress protection. On the other hand, in chronically stressed rats, the prevention of neuronal death by exercise was suggested to be mediated through increased BDNF levels (40). Therefore, exercising protects neurons from many types of insults because BDNF promotes neurogenesis in adults and increases synaptic efficiency (41). In support of these studies, the results of the current work showed a significant increase in cortical BDNF expression in normal rats subjected to chronic exercise. Furthermore, hypoxia enhanced cortical BDNF gene expression, with further increase by chronic exercise (Figs. 2, 3 and 5). The increase in cortical BDNF gene expression following physical activity may result from increased neural activation or altered activity patterns during exercise.
Exercise results in an increase in the level of BDNF in the hippocampus, suggesting a role for BDNF in the beneficial effects of exercise on brain function and plasticity (42). In addition, BDNF had been shown to be driven by physical activity (43) and suppressed by immobilization (44) in the rat brain. Exercise also increases levels of BDNF and neurotrophin-4, and high-affinity receptor trkB in neurons and oligodendrocytes in the spinal cord (45). Furthermore, physical activity can benefit the brain after injury (46). For example, exercise improved outcome after traumatic brain injury in rats. Furthermore, exercise has been shown to increase hippocampal BDNF expression (47). Given the protective effects known of BDNF, increasing its expression would potentially protect against stress-induced damage (47). These observations were confirmed by our results which demonstrated a significant increase in cortical BDNF gene expression in the control or hypoxic groups experiencing chronic exercise. Various mechanisms explain increased BDNF expression during exercise and the beneficial effects of exercise on neurons. Exercise imposes a mild stress on neurons, which results from increased activity in neuronal circuits and/or metabolic stress. The cellular stress involves increased levels of intracellular calcium and reactive oxygen species which, in turn, activate kinases and transcription factors. The transcription factors induce the expression of genes that encode neurotrophic factors such as BDNF leading to neurogenesis, synaptic plasticity and cell survival (46).
BDNF can protect neurons against oxidative damage resulting from diverse neuropathologic insults (48). The protective effects of BDNF against neuronal cell death is mediated by activating intracellular signaling cascades via tropomyosin-related kinase B (TrkB), a high affinity receptor for BDNF (2). For example, treatment with exogenous BDNF can markedly attenuate the loss of dopaminergic substantia nigra neurons resulting from oxyradical damage following exposure to 6-hydroxydopamine (49). Furthermore, BDNF protects cultured cortical neurons from NMDA- or H2O2-induced cell death via suppressing the MAPK pathway (50). Consistent with this hypothesis, BDNF expression increases in ischemic brain tissue (51). In agreement with these studies, the results of the present work showed increased cortical BDNF expression and oxidative stress (increased MDA and decreased SOD and CAT activity and levels of GSH and vitamin E) (Table 3) in hypoxic groups with further enhancement following chronic exercise.
Brain-derived neurotrophic factor (BDNF), a member of the neurotrophin family, is an important protein in the regulation of the maintenance, growth and survival of neurons (52). Numerous studies have demonstrated that BDNF expression in the brain is increased in response to hypoxic/ischemic damage (53) or intermittent hypoxia (54). Kim et al. (55) found evidence of increased BDNF expression in brain microvascular endothelial cells in rat pups reared from birth in hypoxia. Therefore it seems likely that BDNF is released from brain microvascular endothelial cells in response to hypoxic stimuli in vivo. Also, Hubold et al. (56) reported that human serum BDNF concentrations are maintained at higher levels upon acute hypoxia as compared to a normoxic control condition. This hypoxia-induced difference in BDNF content results from preserved BDNF concentrations during the hypoxic condition, whereas BDNF levels conversely drop in a time-dependent manner during the normoxic control condition (56). Interestingly, robust production of new neocortical neurons after perinatal hypoxic-ischemic damage occurs and this neuronogenesis has been observed to be accompanied by an increase of BDNF (57). Moreover, in order to prevent fetal brain damage after maternal hypoxia, maternal treatment with MgSO4 is effective which indeed leads to BDNF changes in the hippocampus (58). These data are also compatible with the former hypothesis that BDNF attenuates neurotoxicity associated with hypoxic neurotoxicity (59). In addition, BDNF concentrations in serum, platelets, and plasma were significantly increased in human subjects with asthma as compared to age- and sex-matched control subjects (60). The increased BDNF concentrations in this study were related to potential hypoxic conditions, airflow limitation, and bronchial hyperresponsiveness. In consistent with these observations, our results demonstrated a significant increase in cortical BDNF expression in control exercised rats as well as in hypoxic sedentary group with more elevation following chronic exercise. In addition, a significant positive correlation was demonstrated between MDA level and BDNF gene expression. These results suggest that BDNF gene expression may be related to the oxidative stress induced in control group subjected to chronic exercise as well as in hypoxic sedentary rats with further amplification by chronic exercise (Table 3). Also, the significant decrease in BDNF gene expression in hypoxic and control rats experiencing chronic exercise with vitamin E administration further confirm the oxidative stress-induced stimulation of BDNF expression in those rats. Also exercise increased serum BDNF level (61).
The observed hypoxia-induced rise in BDNF levels (Fig. 2) may be mediated by various mechanisms. Hypoxia can activate the sympathetic nervous system which can partially be confirmed by increased heart rate and epinephrine levels (55). Catecholamines in turn have been previously demonstrated to increase BDNF production in different experimental settings (62). Thus, it appears plausible that a certain stimulating effect of epinephrine on BDNF may contribute to the higher BDNF content upon hypoxia (55). In a study by Wang et al. (63), they reported that BDNF secretion from neuronally differentiated rat pheochromocytoma cells (PC12 cells) is markedly increased by exposure to hypoxic stimuli. Thus, these data support the hypothesis that oxidative stress can increase BDNF availability by stimulating BDNF release (63). Mechanisms underlying this release, including the requirement for (i) sodium influx through tetrodotoxin (TTX) sensitive channels, (ii) Ca2+ influx through voltage-gated channels and (iii) Ca2+ release from inositol triphosphate (IP3)- and ryanodine-sensitive stores (64). The fact that regulated secretion of BDNF from PC12 cells and hippocampal neurons requires activation of both Ca2+ influx and Ca2+ mobilization from internal stores suggests a role for Ca2+- induced Ca2+ release, a mechanism by which cytoplasmic Ca2+ levels can be amplified and prolonged by Ca2+ release from internal stores (65). Furthermore, the effect of hypoxia on BDNF release is largely indirect and requires autocrine or paracrine signaling by endogenous dopamine (63).
In the literature, few studies have investigated the parameters of the lipid peroxidation and antioxidant defence systems of the rat brain during hypoxia. Rauchova et al. (66) reported hypoxia-induced lipid peroxidation in the rat brain. Maiti et al. (67) found a significant increase in lipid peroxidation and a decrease in antioxidants (glutathione peroxidase, glutathione reductase, glutathione) in the cortex, hippocampus and striatum of rat brain exposed to hypoxia for 3 and 7 days. Consistent with these findings, the results of the present study revealed that hypoxia significantly increased MDA, decreased SOD and CAT activity and reduced the levels of GSH and vitamin E (P <0.05) in the cortex of sedentary rats compared with the control sedentary group (Table 3), indicating the development of oxidative stress. There are many events leading to hypoxia-induced oxidative stress. Due to low levels of O2, free electrons in the cell increase, causing the formation of superoxide anions. Through a chain reaction, superoxide anions form free radicals, especially H2O2 and the hydroxyl radical (OH•–), that directly attack the lipid membrane and cause lipid peroxidation. Moreover, the brain is especially susceptible to oxidative damage because it participates in a high degree of oxygen-dependent mitochondrial activity, which is associated with high levels of free iron and polyunsaturated fatty acids and low levels of antioxidant enzymes (SOD, catalase, GSH-peroxidase) and GSH (16). These factors lead to lipid peroxidation of the brain, and this lipid peroxidation increases with the duration of the hypoxic exposure (16). In agreement with these studies, our results demonstrate that the lipid peroxidation product (MDA) increased in the cortex of hypoxic rats compared to the control sedentary group (Table 3).
An elevated generation of lipid peroxidation products such as MDA in the brain cortex of hypoxic rats (Table 3) may also be related to changes in GSH levels and the activity of enzymatic antioxidants. In our study, lower activity of the main antioxidants (CAT, SOD) and decreased GSH levels were detected in the cortex of hypoxic rats (Table 3). The reduced glutathione (GSH) is the main antioxidant in the cell, which directly scavenges free radical and protects biomolecules from free radical attack. The significant decrease in GSHs level following hypoxia in the present study (Table 3) may indicate higher utilisation of GSH for detoxification of hypoxia-induced free radicals. In another study, Maiti et al. (67) observed a decrease in glutathione peroxidase and glutathione reductase activity which led to lower GSH levels. This might have been triggered by low levels of NADPH, which is needed for glutathione reductase to convert oxidised glutathione (GSSG) to GSH (67). Moreover, the first line of defence against hypoxia-induced ROS in the cell is provided by SOD, which catalyses superoxide anions to hydrogen peroxide, and a high level of H2O2 inhibits SOD activity (68). As hypoxia-induced free radical production increased, the formation of H2O2 also increased. This may have caused the inhibition of SOD activity (69) and may be one of the reasons why SOD activity decreases in hypoxia when compared to the control sedentary group in our results (Table 3). Moreover, in the current work, vitamin E levels significantly decreased in the cortex of the hypoxic rats relative to the control sedentary group (Table 3).
The literature provides controversial data regarding oxidative stress in various tissues during exercise. A number of studies report an increase in free radicals during muscular exercise, suggesting exercise-induced oxidative stress (70). Direct evidence for increased rates of ROS production during intensive physical exercise is still scarce, but this notion is supported by other putative manifestations of oxidative stress, such as changes in the thiol/disulphide redox state in the blood plasma and erythrocytes in the context of intensive physical exercise. A decrease in intracellular GSH/GSSG ratios was found in the skeletal muscle of rats after intense muscular exercise; a similar decrease in GSH/ GSSG ratios was detected in the blood of human volunteers after strenuous exercise (71). The thiol compound N-acetylcysteine was found to ameliorate muscle fatigue in humans (72). On the other hand, other studies reported no significant change in TBARS (73) or anti-oxidants (GSH and glutathione peroxidase) (74) in the brains of normal rats during exercise. However, Liu et al. (75) observed a significant decrease in rat brain MDA during chronic exercise but no change with acute exercise in normal rats. Moreover, they detected no significant change in anti-oxidants (GSH, vitamin E) in the brains of normal rats with acute or chronic exercise. In the present study, chronic exercise caused a significant increase in MDA levels, a decrease in GSH and vitamin E levels and a reduction in SOD and CAT enzyme activity in the cortex of control and hypoxic rats compared to the corresponding sedentary group (Table 3). The increase in free radical production with exercise can be explained by various mechanisms. These mechanisms may act synergistically (6). The rate of free radical or oxidant generation in biological tissue is closely related to oxygen consumption. Thus, a substantial increase in ROS generation is to be expected during exercise, as oxygen flux through active muscle may increase by as much as ~100 times the resting value to meet increased energy demands (76). Under physiological conditions, the majority of oxidants are produced in mitochondria. Moreover, the decrease in mitochondrial PO2 rather than increased oxygen flux could be the cause of the exercise-induced increase in ROS production (77). It seems likely that mitochondria, in addition to being the sources of oxidant production, should also be the targets of oxidants. By increasing the oxygen consumption rate, exercise may result in oxidative stress in mitochondria. This results in an increased production of oxidants, which could be detrimental to the tissue (78). Oxidants cause damage to mitochondrial membranes and cytoplasmic structures through the peroxidation of phospholipids, proteins, and nucleotides (75). Previous animal experiments suggest that the mitochondrial membrane-bound glycerol-3-phosphate dehydrogenase is a more important source of free radicals than the complexes of the electron transport chain (79). An alternative mechanism for the increase in free radical production with exercise is that intense exercise is associated with transient tissue hypoxia in several organs, as blood flow is redistributed to cover the increased blood supply in active skeletal muscles and the skin. Exercise can result in microvascular dysfunction, edema and cell damage through mechanical shear forces or through a disturbance of normal cellular metabolism (80). As a result, exercise triggers an inflammatory response, characterized by infiltration of the affected areas by neutrophils and other phagocytic cells, followed by a respiratory burst involving production of superoxide, hydrogen peroxide and other ROS (80). In addition, xanthine oxidase may also contribute to the production of superoxide in the context of intensive physical exercise (81). Our results confirm these findings. In our study, chronic exercise significantly increased MDA level, decreased GSH and vitamin E levels and reduced SOD and CAT enzyme activities in brain cortex of control or hypoxic rats in comparison with corresponding sedentary group (Table 3).
Vitamin E, a nonenzymatic antioxidant, is an important lipid-soluble antioxidant which plays an important role in protecting polyunsaturated fatty acids in the membrane from oxidation involving ROS by terminating free-radical chain reactions (82, 83). Recent studies have suggested that vitamin E analogs were effective on the neuronal cells (84). In addition, vitamin E has been shown to exert beneficial effects against neurodegenerative diseases (85). Also, positive effects of the antioxidant vitamin E on oxidative stress-mediated toxicity in vitro (27) and in vivo (86) have been reported. The interaction between physical training and vitamin E supplementation is neuroprotective against age-related decreases in the antioxidant enzymes and also counteracts increases in lipid peroxidation in the brain (87). Therefore, vitamin E may be expected to play an important role in protecting lipid-rich structures, such as the brain, from free-radical damage. In a study by Khanna et al. (26), they have shown that a-tocopherol (100 nM) blocks the glutamate-induced cell death in cultured rat cortical neurons. Moreover, a tocopherol (10 µM) inhibits the oxidative stress-mediated cytotoxicity induced by (O2•) or NO donor in cultured rat striatal neurons (87). These survival effects could be elicited by scavenging oxygen radical species, as vitamin E analogs were added to the cultured neurons simultaneously with (8) or 5 min before or after (26) the oxidants stimulation. Consistent with these findings, the results of the current work showed that vitamin E significantly decreased MDA, increased SOD and CAT activity and raised the levels of GSH and vitamin E in hypoxic sedentary rats and following chronic exercise in control and hypoxic rats (Table 3), indicating a marked improvement of oxidative stress. In addition, vitamin E significantly decreased BDNF gene expression in the control or hypoxic rats experiencing chronic exercise and in hypoxic sedentary group (Figs. 2-4). These results suggest that cortical BDNF gene expression may be caused by the oxidative stress induced in control and hypoxic rats subjected to chronic exercise and in hypoxic sedentary group.
In conclusion, hypoxia and/or exercise increased BDNF gene expression in the brain cortex of rats as a result of oxidative stress. Vitamin E decreased BDNF gene expression in both the control and hypoxic rats experiencing chronic exercise, as well as in hypoxic sedentary rats by attenuating oxidative stress which confirms the oxidative stress-induced stimulation of BDNF gene expression.
Conflict of interests: None declared.
REFERENCES
- Mattson MP, Maudsley S, Martin B. BDNF and 5-HT: a dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci 2004; 27: 589-594.
- Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H. BDNF function and intracellular signaling in neurons. Histol Histopathol 2010; 25: 237-258.
- Yamada K, Mizuno M, Nabeshima T. Role of brain-derived neurotrophic factor in learning and memory. Life Sci 2002; 70: 735-744.
- Gorski JA, Balogh SA, Wehner JM, Jones KR. Learning deficits in forebrain-restricted brain- derived neurotrophic factor mutant mice. Neuroscience 2003; 121: 341-354.
- Karege F, Perret G, Bondolfi G, Schwald M, Bertschy G, Aubry J-M. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res 2002; 109: 143-148.
- Neeper SA, Gomez-Pinilla F, Choi J, Cotman C. Exercise and brain neurotrophins. Nature 1995; 373: 109.
- Schaaf MJ, Hoetelmans RWM, de Kloet ER, Vreugdenhil E. Corticosterone regulates expression of BDNF and trkB but not NT-3 and trkC mRNA in the rat hippocampus. J Neurosci Res 1997; 48: 334-341.
- Johnson RA, Mitchell GS. Exercise-induced changes in hippocampal brain-derived neurotrophic factor and neurotrophin-3: effects of rat strain. Brain Res 2003; 983: 108-114.
- Oliff HS, Berchtold NC, Isackson P, Cotman CW. Exercise-induced regulation of brain-derived neurotrophic factor (BDNF) transcripts in the rat hippocampus. Brain Res Mol Brain Res 1998; 61: 147-153.
- Nelson DL, Cox MM. Lehninger Principle of Biochemistry. Macmillan Worth Publishers, 2004.
- Magalhaes J, Ascensao A, Soares JM, et al. Skeletal muscle ultrastructural and plasma biochemical signs of endothelium dysfunction induced by a high-altitude expedition (Pumori. 7161 m). Basic Appl Myol 2005; 15: 29-35.
- Jayalakshmi K, Sairam M, Singh SB, Sharma SK, Ilavazhagan G, Banerjee PK. Neuroprotective effect of N-acetyl cysteine on hypoxia induced oxidative stress in primary hippocampal culture. Brain Res 2005; 1046: 97-104.
- Ramanathan L, Gozal D, Siegel JM. Antioxidant response to chronic hypoxia in the rat cerebellum and pons. J. Neurochem 2005; 93: 47-52.
- Buttefield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide associated free radical oxidative stress. Free Radic Biol Med 2002; 32: 1050-1060.
- Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nat Med 2004; 10: S18-S25.
- Reed TT. Lipid peroxidation and neurodegenerative disease. Free Radic Biol Med 2011; 51: 1302-1319.
- Erecinska M, Silver IA. Calcium handling by hippocampal neurons under physiologic and pathologic conditions. Adv Neurol 1996; 71: 119-136.
- Molteni R, Zheng JQ, Ying Z, Gomez-Pinilla F, Twiss JL. Voluntary exercise increases axonal regeneration from sensory neurons. Proc Natl Acad Sci USA 2004; 101: 8473-8478.
- Cotman, CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci 2002; 25: 295-301.
- Radak Z, Asano K, Inoue M, et al. Acute bout of exercise does not alter the antioxidant enzyme status and lipid peroxidation in rat hippocampus and cerebellum. Pathophysiology 1995: 2: 243-245.
- Radak Z, Kaneko T, Tahara S, et al. Regular exercise improves cognitive function and decreases oxidative damage in rat brain. Neurochem Int 2001; 38: 17-23.
- Sano M. Noncholinergic treatment options for Alzheimer’s disease. J Clin Psychiatry 2003; 64 (Suppl. 9): 23-28.
- Fariss MW, Zhang JG. Vitamin E therapy in Parkinson’s disease. Toxicology 2003; 189: 129-146.
- Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem 1994; 62: 376-379.
- Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 1995; 92: 7162-7166.
- Khanna S, Roy S, Ryu H, Bahadduri P, Swaan PW, Ratan RR, Sen CK. Molecular basis of vitamin E action: tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J Biol Chem 2003; 278: 43508-43515.
- Osakada F, Hashino A, Kume T, Katsuki H, Kaneko S, Akaike A. Alpha-tocotrienol provides the most potent neuroprotection among vitamin E analogs on cultured striatal neurons. Neuropharmacology 2004; 47: 904-915.
- Mian R, Marshall JM. The behaviour of muscle microcirculation in chronically hypoxic rats: the role of adenosine. J Physiol 1996; 491: 489-498.
- Ulas M, Cay M. The effects of 17beta-estradiol and vitamin E treatments on oxidative stress and antioxidant levels in brain cortex of diabetic ovariectomized rats. Acta Physiol Hung 2010; 97: 208-215.
- de Lemos ET, Reis F, Baptista S, et al. Exercise training is associated with improved levels of C-reactive protein and adiponectin in ZDF (type 2) diabetic rats. Med Sci Monit 2007; 13: BR168-BR74.
- Tsukinoki K, Saruta J, Sasaguri K, et al. Immobilization stress induces BDNF in rat submandibular glands. J Dent Res 2006; 85: 844-848.
- Tsukinoki K, Saruta J, Muto N, et al. Submandibular glands contribute to increases in plasma BDNF levels. J Dent Res 2007; 86: 260-264.
- Ding Q, Ying Z, Gomez-Pinilla F. Exercise influences hippocampal plasticity by modulating brain-derived neurotrophic factor processing. Neuroscience 2011; 192: 773-780.
- Rasmussen P, Brassard P, Adser H, et al. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp Physiol 2009; 94: 1062-1069.
- Wang JP, Qu WS, Zhang Q. Effect of hypoxia on BDNF release from astrocytes. Zhengzhou Da Xue Xue Bao Yi Xue Ban 2008; 43: 480-482.
- Liu H, Dai T. Effect of brain-derived neurotropic factor released from hypoxic astrocytes on γ-aminobutyric acid type A receptor function in normal hippocampal neurons. Neural Regen Res 2011; 6: 1954-1959.
- Guo S, Kim WJ, Lok J, et al. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci USA 2008; 105: 7582-7587.
- Nybo L, Rasmussen P. Inadequate cerebral oxygen delivery and central fatigue during strenuous exercise. Exerc Sport Sci Rev 2007; 35: 110-118.
- Shi SS, Shao SH, Yuan BP, Pan F, Li ZL. Acute stress and chronic stress change brain-derived neurotrophic factor (BDNF) and tyrosine kinase-coupled receptor (TrkB) expression in both young and aged rat hippocampus. Yonsei Med J 2010; 51: 661-671.
- Marais L, Stein DJ, Daniels WM. Exercise increases BDNF levels in the striatum and decreases depressive-like behavior in chronically stressed rats. Metab Brain Dis. 2009; 24: 587-597.
- Poo M. Neurotrophins as synaptic modulators. Nat Rev Neurosci 2001; 2: 24-32.
- Russo-Neustadt AA, Beard RC, Huang YM, Cotman CW. Physical activity and antidepressant treatment potentiate the expression of specific brain-derived neurotrophic factor transcripts in the rat hippocampus. Neuroscience 2000; 101: 305-312.
- Gold SM, Schulz KH, Hartmann S, et al. Basal serum levels and reactivity of nerve growth factor and brain-derived neurotrophic factor to standardized acute exercise in multiple sclerosis and controls. J Neuroimmunol 2003; 138: 99-105.
- Ueyama T, Kawai Y, Nemoto K, Sekimoto M, Tone S, Senba E. Immobilization stress reduced the expression of neuro- trophins and their receptors in the rat brain. Neurosci Res 1997; 28: 103-110.
- Skup M, Dwornik A, Macias M, Sulejczak D, Wiater M, Czarkowska-Bauch J. Long-term locomotor training up-regulates TrkB (FL) receptor-like proteins, brain-derived neurotrophic factor, and neurotrophin 4 with different topographies of expression in oligodendroglia and neurons in the spinal cord. Exp Neurol 2002; 176: 289-307.
- Mattson MP, Duan W, Wan R, Guo Z. Prophylactic activation of neuroprotective stress response pathways by dietary and behavioral manipulations. NeuroRx 2004; 1: 111-116.
- Maniam J, Morris MJ. Voluntary exercise and palatable high-fat diet both improve behavioural profile and stress responses in male rats exposed to early life stress: role of hippocampus. Psychoneuroendocrinology, 2010; 35: 1553-1564.
- Mattson MP, Duan W, Maswood N. How does the brain control lifespan? Ageing Res Rev 2002; 1: 155-165.
- Altar CA, Boylan CB, Fritsche M, et al. Efficacy of brain- derived neurotrophic factor and neurotrophin-3 on neurochemical and behavioral deficits associated with partial nigrostriatal dopamine lesions. J Neurochem 1994; 63: 1021-1032.
- Boutahar N, Reynaud E, Lassabliere F, Borg J. Brain-derived neurotrophic factor inhibits cell cycle reentry but not endoplasmic reticulum stress in cultured neurons following oxidative or excitotoxic stress. J Neurosci Res 2010; 88: 2263-2271.
- Walton M, Connor B, Lawlor P, et al. Neuronal death and survival in two models of hypoxic-ischemic brain damage. Brain Res Brain Res Rev 1999; 29: 137-168.
- Sakr HF, Khalil KI, Hussein AM, Zaki MS, Eid RA, Alkhateeb M. Effect of dehydroepiandrosterone (DHEA) on memory and brain derived neurotrophic factor (BDNF) in a rat model of vascular dementia. J Physiol Pharmacol 2014; 65: 41-53.
- Kokaia Z, Nawa H, Uchino H, et al. Regional brain-derived neurotrophic factor mRNA and protein levels following transient forebrain ischemia in the rat. Brain Res Mol Brain Res 1996; 38: 139-144.
- Peiris TS, Machaalani R, Waters KA. Brain-derived neurotrophic factor mRNA and protein in the piglet brainstem and effects of intermittent hypercapnic hypoxia. Brain Res 2004; 1029: 11-23.
- Kim H, Li Q, Hempstead BL, Madri JA. Paracrine and autocrine functions of brain-derived neurotrophic factor (BDNF) and nerve growth factor (NGF) in brain-derived endothelial cells. J Biol Chem 2004; 279: 33538-33546.
- Hubold C, Lang UE, Gehring H, et al. Increased serum brain-derived neurotrophic factor protein upon hypoxia in healthy young men. J Neural Transm 2009; 116: 1221-1225.
- Yang Z, Covey MV, Bitel CL, Ni L, Jonakait GM, Levison SW. Sustained neocortical neurogenesis after neonatal hypoxic/ischemic injury. Ann Neurol 2007; 61: 199-208.
- Golan H, Kashtuzki I, Hallak M, Sorokin Y, Huleihel M. Maternal hypoxia during pregnancy induces fetal neurodevelop- mental brain damage: partial protection by magnesium sulfate. J Neurosci Res 2004; 78: 430-441.
- Mitchell JJ, Paiva M, Walker DW, Heaton MB. BDNF and NGF afford in vitro neuroprotection against ethanol combined with acute ischemia and chronic hypoglycemia. Dev Neurosci 1999; 21: 68-75.
- Lommatzsch M, Schloetcke K, Klotz J, et al. Brain-derived neurotrophic factor in platelets and airflow limitation in asthma. Am J Respir Crit Care Med 2005; 171: 115-120.
- Zoladz JA, Majerczak J, Zeligowska E, et al. Moderate-intensity interval training increases serum brain-derived neurotrophic factor level and decreases inflammation in Parkinson’s disease patients. J Physiol Pharmacol 2014; 65: 441-448.
- Chen MJ, Nguyen TV, Pike CJ, Russo-Neustadt AA. Norepinephrine induces BDNF and activates the PI-3K and MAPK cascades in embryonic hippocampal neurons. Cell Signal 2007; 19: 114-128.
- Wang H, Yuan G, Prabhakar NR, Boswell M, Katz DM. Secretion of brain-derived neurotrophic factor from PC12 cells in response to oxidative stress requires autocrine dopamine signaling. J Neurochem 2006; 96: 694-705.
- Balkowiec A, Katz DM. Cellular mechanisms regulating activity-dependent release of native brain-derived neurotrophic factor from hippocampal neurons. J Neurosci 2002; 22: 399-407.
- Albrecht MA, Colegrove SL, Friel DD. Differential regulation of ER Ca2+ uptake and release rates accounts for mul- tiple modes of Ca2+-induced Ca2+ release. J Gen Physiol 2002; 119: 211-233.
- Rauchova H, Koudelova J, Drahota Z, Mourek J. Hypoxia-induced lipid peroxidation in rat brain and protective effect of carnitine and phosphocreatine. Neurochem Res 2002; 27: 899-904.
- Maiti P, Singh SB, Sharma AK, Muthuraju S, Banerjee PK, Ilavazhagan G. Hypobaric hypoxia induces oxidative stress in rat brain. Neurochem Int 2006; 49: 709-716.
- Yunoki M, Kawanchi M, Ukita N, et al. Effect of lecithinized SOD and sequential change in SOD activity after cerebral perfusion. Acta Neurochir Suppl 1998; 71: 142-145.
- Nieber K. Hypoxia and neuronal function under in vitro condition. Pharmacol Ther 1999; 82: 71-86.
- Bailey DM, Young IS, McEneny J, et al. Regulation of free radical outflow from an isolated muscle bed in exercising humans. Am J Physiol Heart Circ Physiol 2004; 287: H1689-H1699.
- Sen CK, Atalay M, Hanninen O. Exercise-induced oxidative stress: glutathione supplementation and deficiency. J Appl Physiol (1985) 1994; 77: 2177-2187.
- Reid MB, Stokic DS, Koch SM, Khawli FA, Leis AA. N-acetyl-cysteine inhibits muscle fatigue in humans. J Clin Invest 1994: 94: 2468-2474.
- Ogonovszky H, Berkes I, Kumagai S, et al. The effects of moderate-, strenuous- and over-training on oxidative stress markers, DNA repair, and memory, in rat brain. Neurochem Int 2005; 46: 635-640.
- Somani SM, Ravi R, Rybak LP. Effect of exercise training on antioxidant system in brain regions of rat. Pharmacol Biochem Behav 1995; 50: 635-639.
- Liu J, Yeo HC, Overvik-Douki E, et al. Chronically and acutely exercised rats: biomarkers of oxidative stress and endogenous antioxidants. J Appl Physiol (1985) 2000; 89: 21-28.
- Vollaard NB, Shearman JP, Cooper CE. Exercise-induced oxidative stress myths, realities and physiological relevance. Sports Med 2005; 35: 1045-1062.
- Bailey DM. What regulates exercise-induced reactive oxidant generation: mitochondrial O2 flux or PO2? Med Sci Sports Exerc 2001; 33: 681-682.
- Ji LL. Exercise, oxidative stress, and antioxidants. Am J Sports Med 1996; 24: S20-S24.
- Miwa S, St-Pierre J, Partridge L, Brand MD. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic Biol Med 2003; 35: 938-948.
- Pyne DB. Exercise-induced muscle damage and inflammation: a review. Aust J Sci Med Sport 1994; 26: 49-58.
- Volek JS, Kraemer WJ, Rubin MR, Gomez AL, Ratamess NA, Gaynor P. L-carnitine L-tartrate supplementation favorably affects markers of recovery from exercise stress. Am J Physiol Endocrinol Metab 2002; 282: E474-E482.
- Mazor D, Brill G, Shorer Z, Moses S, Meyerstein N. Oxidative damage in red cells of vitamin E deficient patients. Clin Chim Acta 1997; 265: 131-137.
- Krawczynska A, Olczak E, Rembiszewska A, Herman AP, Gromadzka-Ostrowska J. Time-dependent supplementation of vitamin E influences leptin expression in the aortic layers of rats fed atherogenic diet. J Physiol Pharmacol 2014; 65: 33-39.
- Numakawa Y, Numakawa T, Matsumoto T, et al. Vitamin E protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. J Neurochem 2006; 97: 1191-1202.
- Pratico D. Evidence of oxidative stress in Alzheimer’s disease brain and antioxidant therapy: lights and shadows. Ann N Y Acad Sci 2008; 1147: 70-78.
- Nakashima H, Ishihara T, Yokota O, et al. Effects of a-tocopherol on an animal model of tauopathies. Free Radic Biol Med 2004; 37: 176-186.
- Jolitha AB, Subramanyam MV, Asha Devi S. Modification by vitamin E and exercise of oxidative stress in regions of aging rat brain: studies on superoxide dismutase isoenzymes and protein oxidation status. Exp Gerontol 2006; 41: 753-763.
- Osakada F, Hashino A, Kume T, Katsuki H, Kaneko S, Akaike A. Neuroprotective effects of a-tocopherol on oxidative stress in rat striatal cultures. Eur J Pharmacol 2003; 465: 15-22.
A c c e p t e d : February 9, 2015