DRUG DELIVERY SYSTEMS IMPROVING CHEMICAL AND PHYSICAL PROPERTIES
OF ANTICANCER DRUGS CURRENTLY INVESTIGATED FOR TREATMENT
OF SOLID TUMORS
INTRODUCTION
Cancer is the second leading cause of death, exceeded only by heart disease. According to the United States Cancer Statistics Incidence and Mortality Web-based Report, every minute someone dies of cancer in the United States. Annual cancer mortality has slightly decreased, however this is mainly related to an improvement in early diagnosis.
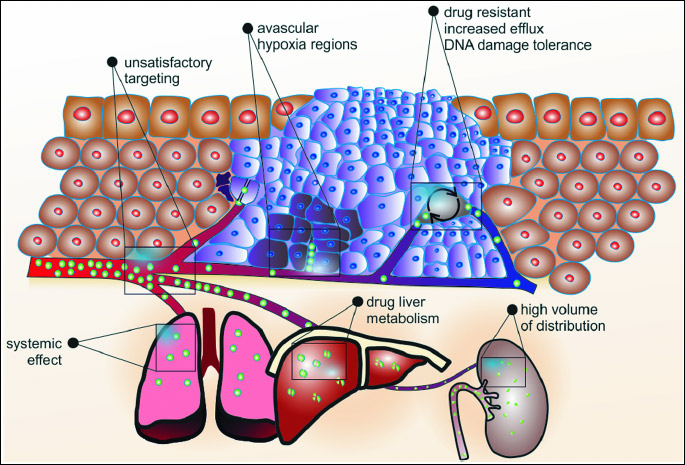
Cytotoxic drugs are still the major treatment modality. These drugs interfere with DNA inhibiting cancer cell division or directly causing cell death. However, results of therapy are still unsatisfactory (1). Firstly, only up to 1 – 2% of the administered drug reaches the solid tumor, while the rest goes to other tissues causing side effects (Fig. 1). Drug distribution in tumor tissue is poor due to characteristics of its microenvironments that limit drug penetration, thereby exposing the tumor to lower than efficacious concentrations of drugs (2). The most important feature of the tumor microenvironment is inadequate vasculature. Perfused vessels are necessary for ensuring adequate drug distribution (Fig. 1). However, due to rapid tumor cell proliferation that outpace the rate of new blood vessel growth, their architecture and, subsequently, their function; perfusion is lower than that in the peritumor normal tissue. Moreover, fast proliferating cancer cells as well as stromal cells in solid tumors press the walls of blood vessels causing their compression and often completely closing their lumen and blocking blood flow. It impairs not only perfusion, but also diffusion of the drug towards tumor mass leading to hypoxia, metabolite accumulation, and inadequate drug delivery (3, 4). Vessel architecture could be improved using antiangiogenic agents. However, this strategy should be rather called 'vascular normalization' because it doesn`t inhibit blood vessel growth but it improves their structure increasing tumor perfusion, drug delivery and treatment efficacy (5). Secondly, even if the drug is distributed in the tumor tissue, cancer has the ability to become resistant to many different types of compounds (Fig. 1). Increased efflux of drug, increased tolerance to DNA damage, and enzymatic deactivation allow cancer cell survive chemotherapy (6). These 'survival' cells might develop multidrug resistance or just might be located in hypoxic avascular areas unreachable for anticancer agents (7-10). Thirdly, anticancer drugs show the same mode of action for all cells. They are not selective for cancer cells and therefore they damage all targeted tissues, especially fast proliferating cells (1). Fourthly, the most important issues of the majority of anticancer drugs are their unsatisfactory pharmacokinetic and chemical limitations. Due to their poor aqueous solubility they are formulated with co-solubilizers leading to severe side effects and decreased efficacy of the therapy (11). Moreover, anticancer drugs have high volume of distribution, being rapidly cleared by liver and kidneys. A rapid increase and subsequent decay of their concentration in blood makes the proper dosage difficult, being one of the limitations of cancer therapy. Additionally, these drugs are not protected from enzymatic degradation (11). Due to above issues in cancer therapy, numerous drug delivery systems are approved or are in developmental stage. These systems aim to improve tumor targeting, overcome lack of specificity and improve pharmacologic and therapeutic properties of classical drugs (Fig. 2).
LIPOSOMES AS A PROMISING DRUG VEHICLE TO SOLID TUMORS
Liposomes are spherical single or bilayer structures described for the first time by Bangham in 1964 (12). They are able to enclose different biologically active compounds and macromolecules (e.g. imaging or therapeutics agents: DNA, peptides, various proteins) both in their lipid bilayer (i.e. hydrophobic molecules) or in their lumen (i.e. hydrophilic molecules) and therefore they are widely investigated in various applications (Fig. 3). Liposomal formulations improve pharmacokinetic and pharmacodynamic profiles of enclosed drugs, ensure controlled and sustained release of drugs and decrease systemic toxicity compared with a free drug formulation. Liposomes can be modified by changing their physicochemical features or adding external elements to enhance their functionality i.e. modifying systemic circulation, increasing accumulation at the target tissue or cellular internalization.
Liposomes are classified according to their size or structure into two types: (a) unilamellar vesicles and (b) multilamellar vesicles (MLV). Unilamellar vesicles can subsequently be classified into two subgroups: small unilamellar vesicles (SUV) and large unilamellar vesicles (LUV). Unilamellar liposomes have a single phospholipid bilayer surrounding the aqueous solution. Multilamellar liposomes are built in an onion-like structure. The size of therapeutically used liposomes vary between 50 and 1000 nm. It enables them to extravasate and penetrate tumor tissue, as a size of the gaps between endothelial cells lining the tumor capillaries ranges from 100 to about 800 nm depending on the cancer type. In contrary, in a typical normal endothelium gaps size is usually between 5 to 10 nm (13). The best extravasation properties have liposomes smaller than 200 nm. In addition, liposomal formulation show enhanced permeability and retention effect (EPR). Moreover, because solid tumors lack adequate lymphatic drainage, there is limited circulatory recovery of the extravasated compounds, leading to their accumulation within the tumor mass.
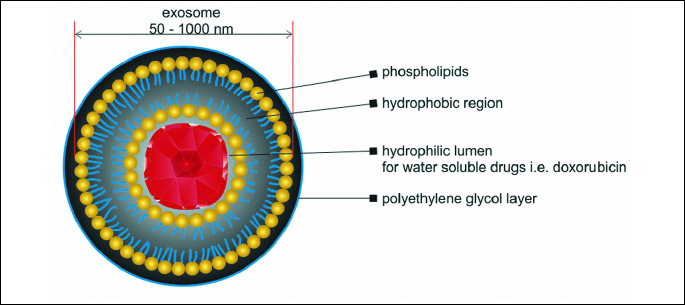
Unmodified liposomes are rapidly cleared by phagocytic cells of the reticuloendothelial system (RES) in blood circulation. To overcome this issue, surface of liposomes is coated with a polyethylene glycol (PEG) which prevents them from rapid clearance by the RES (14).
Liposome tumor targeting
Despite liposomes show better permeability to the tumor mass, they generally lack specificity in tumor targeting. That is why many methods have been developed in order to improve this element. One of them is attaching to their surface various ligands that specifically bind to the receptors overexpressed on the surface of cancer cells (15). Transferrin receptors (TfRs) and folate receptors (FRs) are greatly overexpressed by many cancer cell types in response to their increased metabolic demand. Therefore, they seem to be a natural target (16). Low et al. showed that doxorubicin-loaded PEGylated liposomes targeting FR had 45-fold higher uptake by FR+ epithelial cancer cells than the unmodified liposomes and therefore 85-times higher cytotoxicity (17). Zhai et al. showed that TfR-targeted liposomal formulation of docetaxel was an effective delivery system for the treatment of breast, colon, ovarian, head, neck and non-small-cell lung cancer (18).
Sharma et al. developed a bifunctional liposomal system containing the combination of transferin (Tf) and poly-L-arginine. This smart system was very effective because the Tf-modified liposomes demonstrated tumor targeting and poly-L-arginine promoted cell penetration, leading to drug transport across the endothelium of the blood-brain barrier (19).
Development of the monoclonal antibodies opened new perspectives also for liposome targeting. Antibody binding to the surface of PEGylated liposome should lead to the specific link of the formulation with antigen. Such combination has been evaluated against EGFR, HER2 and matrix metalloproteinases (MMPs) (20, 21). MMPs play role in tumor invasiveness, metastasis and tissue remodeling. Administration of doxorubicin-loaded anti-MT1-MMP liposomes to mice with fibrosarcoma (HT-1080) significantly reduced tumor growth compared with plain liposomes containing doxorubicin (22). This approach faces several challenges, including ligand/target affinity, the quantity of receptors on the cell surface and PEGylation acting as a barrier against interaction with receptors. Nevertheless the use of immunoliposomes enables delivery of a large number of drug molecules per liposome to the targeted cells and multivalent presentation increases uptake of antibody fragments.
Other targeting methods include binding of RGD peptide having high affinity to integrins. Chen et al. developed an integrin-targeted liposomal system for the delivery of doxorubicin. The RGD-coupled liposomal system had a 2.5-fold higher cellular uptake of doxorubicin compared with the unmodified liposomes by integrin-overexpressing human glioma cell line U87MG (23).
Another approach is the development of liposomes sensitive to tumor microenvironment. Suzuki et al. showed that slightly acidic pH-sensitive peptide (SAPSp-lipo) can penetrate well in vivo tumor tissue and in vitro spheroids comprised of cancer cells and extracellular matrix (ECM). The SAPSp-lipo could be delivered to deeper regions within both spheroids and tumor tissues than classic liposomes (24).
Liposomes may be also conjugated to aptamers: single strand (ss) DNA or RNA oligonucleotides that impart high affinity and specific recognition of the target molecules (25). Beak et al. proposed a novel liposome conjugated with an RNA aptamer recognizing the prostate specific membrane antigen (PSMA). They demonstrated in vivo anticancer efficacy of such a liposome encapsulating doxorubicin manifested by decrease of tumor size in LNCaP xenograft (26).
Drug release from the liposome
Besides the tumor targeting, a significant attention has been paid to liposome drug unloading. Therefore, various stimuli to unload the vesicles have been tested. For example, difference between pH at the tumor site and normal environment may facilitate liposome unload. This pH-sensitive degradation of a liposomal carrier releases the entrapped drugs in tissues with a low pH, such as tumors (27). This method uses the fact that pH-sensitive copolymers that are quite stable at pH = 7.5 are hydrolyzed relatively rapidly at pH below 6. Further pH-sensitive liposomes might fuse with the vacuolar membrane after endocytosis and release their contents into the cytoplasm or endosomal vesicle (28).
Temperature-sensitive liposomes are yet another tested approach. They can be prepared from thermosensitive lipids or polymers with low critical solution temperature. Above the low critical solution temperature the polymer precipitates disrupting the liposomes to release the entrapped drug. Hyperthermia is associated with increased tumor permeability and enhanced drug uptake due to enlargement of the microvascular pore size and improvement in blood flow. Therefore using thermosensitive liposomes involves local heating of the tumor site to increase extravasation of drug-loaded nanocarriers. Precise heating can be achieved with physical methods. Temperature based liposomal system ThermoDox® (Celsion, USA) is currently in phase 3 clinical trials and already has demonstrated improved efficacy for cancer-targeted drug delivery (29). Further modification of this method is encapsulation of two agents instead of one. An example of this approach was demonstrated by Maples et al. who constructed Echogenic Low Temperature Sensitive Liposomes (E-LTSL) loaded passively with imaging agent perflouropenthane (PFP) and actively with doxorubicin. Doxorubicin release from the liposome was < 5% within 1 hour at control conditions (25°C) or body temperature (37°C), however reached > 99% after heating (30).
Incorporation of magnetite in the structure of liposomes might enable their disruption at the chosen site. Accumulation of the magnetic nanoparticles in the tumor can be enhanced with magnetic field due to EPR effect. It has been shown that the influence of the external magnetic field caused 6 – 10 fold increase in tumor accumulation compared to unmodified carriers (31). Magnetoliposomes (MLUV) could be prepared with hydrophobic superparamagnetic iron oxide nanoparticles (SPIONs) within the bilayer of the liposomes. It leads to overall stiffening of the vesicle, facilitates rupture of the MLUV membrane in a low-frequency alternating magnetic field (AMF) followed by a subsequent drug release from MLUVs. Drug could be released through MLUV rupture induced by mechanical vibration of SPIONs rather than through localized heating like with the thermoliposomes (32).
Multifunctional approach
Recently tested liposomal formulations are structured in the multifunctional way i.e. using more than one enhancing element. Example of this approach is commercially available PEGylated liposomal doxorubicin (Lipidox®) changed by surface taping with a cell-penetrating peptide TATp, conjugated to PEG1000-PE to enhance its uptake by cells. To overcome early degradation its shielded by the long PEG-Hz-PE, however to improve targeting, its linked to antibody forming 2C5-PEG-PE. This enhanced formula was much more effective in mice with drug sensitive and drug resistant ovarian cancer than classical Lipidox (33). Interesting in vitro study showed that the use of cationic PEGylated liposomes loaded with epirubicin and hepcidin decreased expression and function of multidrug resistant transporters (MDR), reversed epithelial-mesenchymal transition (EMT), modulated autophagy and activated apoptosis, namely increased toxicity of epirubicin in HeLa cells, comparing to classic liposomes (34).
NANOSPONGES AS DRUG DELIVERY SYSTEMS
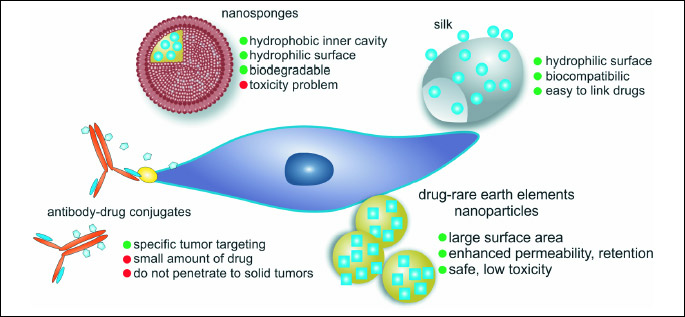
Nanosponges look like a 3D network. They may be built of polyester with many linkers or hyper-cross-linked polymers with cyclodextrin units. They form spherical structures with a hydrophilic outer surface and a hydrophobic inner cavity where the drug may be stored. Therefore these sponges of a size of virus, may be loaded with a drug.
Since these materials are biodegradable, they may be easily broken down inside the body. However, cyclodextrins present low aqueous solubility and are toxic when administered intravenously. Therefore, they may be applied topically (eye administration, cream), orally or directly to the tumor. Recently many various chemical modifications of cyclodextrins have been proposed to overcome their limitations and to facilitate their intravenous administration (35).
An interesting type of nanosponges called pyro-nanosponges, presents free carboxylic groups that can dissociate depending on environmental pH. At acid pH values, the carboxylic groups will be undissociated, whereas at physiological pH they will be dissociated with a net negative charge that can form strong electrostatic interactions with positive groups (36).
Pyro-nanosponges showed a prolonged-release profile over time and slower kinetics than that of carbonate nanosponges. Functionalized nanosponges can be obtained by binding molecules on their surfaces. These attached various 'linkers' ensure their specific binding to cancer cells. These nanosponges are supposed to circulate inside the body until they reach the tumor where they stick to the surface of cancer cell. Then, they may start to release the encapsulated drug in a controllable and predictable fashion (37).
Nanosponges in cancer treatment
Nanosponges may improve one of the greatest limitations of anticancer drugs, namely their low water solubility. Drugs can be molecularly dispersed within the nanosponge carrier structure to avoid crystallization (36).
Cyclodextrins have been proposed as good compounds to deliver paclitaxel. This drug has extremely low water solubility but currently used solubilizers cause serious side effects. On the other hand its common use in patients with breast and ovary cancers makes it the world's major drug on the market (35). The behavior of these paclitaxel-loaded nanoparticles has been investigated after oral and intravenous administration in rats comparing to commercially available taxol. Plasma concentration values markedly increased after oral administration of paclitaxel in nanosponges enhancing threefold the bioavailability of the drug (38).
Nanosponges may also ensure controlled and long release of the drugs. Good example is doxorubicin being one of the most commonly used anticancer drugs. It is a soluble drug, however it cause serious side effects. Therefore, there is a high interest in improving its pharmacokinetics. It shows good encapsulation efficiency in carbonate nanosponges. Doxorubicin release kinetics from a nanosponge in vitro showed a prolonged release profile over time. It was released slowly at very low pH (about 1% after 2 hours), and the percentage increased with pH values (39). This slow release of a drug encapsulated in the cross-linked structure might allow to decrease the required therapeutic dose and limit adverse side effects.
Nanosponges showed the potential to constitute promising tools for the improvement of cancer chemotherapy. However, nowadays structural modification in order to obtain 'targeted nanosponges' ensuring tumor-specific drug accumulation are required.
FERRITIN CAGE-BASED DRUG DELIVERY
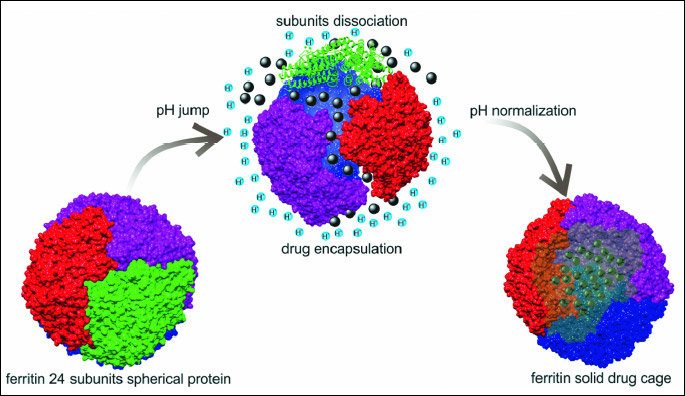
A natural protein-cage architecture of ferritin makes it attractive as a drug delivery tool to various tumors (40, 41). Ferritin is one of the best known proteins capable of maintaining iron in mammalian cells in non-toxic but bioavailable form. It is abundant inside the cells, but it also appears in the serum in larger amounts during liver injury (42). Ferritin is a small protein (20 kDa) composed of 24 subunits, each comprised of two different proteins, a heavy chain (H) and light chain (L) forming a 450 kDa hollow nano-cage (outside diameter 12 – 13 nm; inside diameter 8 nm) capable of incorporating up to 4500 iron atoms (43). However, ferritins may be found as H- or L- monomers. H-ferritin is characteristic for heart tissue, however L-ferritin is typical for liver. Ferritin has been shown to bind transferrin receptor TfR1 (CD71) and subsequently internalize within an endocytic pathway. However, H-ferritin may bind also scavenger receptor (CD163) and T-cell immunoglobulin and mucin domain 2 (TIM-2). A specific receptor for L-ferritin is Scara5 (44). Literature data indicate differences between various cells/tissues and various ferritin forms (monomer: H, L; or dimer) and the receptors by which ferritin may enter the cell. These two forms of ferritins are distinct in their functionality, as H form possess ferroxidase activity while L-ferritins stably incorporate iron at increased cellular iron levels. Therefore, H-chain ferritin confers solid protection against oxidative stress (45). Interestingly, in tumor-bearing patients or during the inflammation, when free radicals are generated at the large scale, blood level of ferritin is significantly elevated. Ferritin is even described as immunomodulator in the four clinical disorders (characterized by hyperferritinemia): macrophage activation syndrome, Still`s disease, septic shock and catastrophic antiphospholipid syndrome (46).
Complexes of ferritin with various compounds
Important feature of ferritin is that the cage can be dissembled at low pH and reassembled in neutral pH (Fig. 4). This pH jump could be used in order to load various compounds inside ferritin cavity (47). Examples are as follow: gadolinium contrast agents, desferrioxamine, metal ions and inorganic nanoparticles, ruthenium carbonyl complexes, magnetite nanoparticles, beta-carotene, folic acid, doxorubicin and few photosensitizers (48-61). However, Pantillo et al. in their recently published paper described that drug encapsulation might be obtained using high pH. They induced L-ferritin dissociation at pH = 13 and reconstituted the cage-like shape at normal pH in the presence of cisplatin. The same group used similar approach to encapsulate Auoxo3, a potential gold(III) based drug (62).
The ability of ferritins to encapsulate metals makes them an ideal tool for cellular imaging, as labelled heavy atoms can be easily sequestered within its core. Furthermore, the ability to modify ferritin through chemical reactions or protein engineering gives the opportunity to use them as contrast agents. Iron-loaded ferritin has also been used as a contrast agent in both electron microscopy and MRI (63-65).
Incorporation of non-metals or non-metal-containing drugs within ferritin is challenging. So, to deal with this problem, drugs may be combined with metals, such as Cu(II), prior to their internalization. Another approach is the addition of accessory molecules to optimise loading of ferritin with drugs (66). However, various compounds may be easily linked to the surface of the ferritin. Therefore, ferritin can be specifically targeted to particular cell types for efficient delivery of therapeutic agents. Kanekiyo et al. showed completely different approach fusing the entire protein of influenza virus hemagglutinin to the surface of ferritin. It shows the potential benefits of implementing synthetic nanoparticle vaccines but also it shows the multipotency of ferritins in medicine.
Ferritins as drug delivery systems to solid tumors
Several ferritin constructs have been used in order to deliver anticancer drugs to tumor tissues after i.v. injection (40, 41, 67). According to current knowledge, malignant cells uptake ferritins to address the need of iron and in many cases they overexpress the TfR1 receptor (68). The key advantages of these protein nanocage constructs over traditional, liposome-based delivery systems include the high stability and solubility of ferritin encapsulated drugs, easy protein surface functionalization and preferential binding to cancer cells overexpressing TfR. However, circulating ferritin is rapidly cleared by expressing TfR and CD163 macrophages (mostly by these located in the spleen and liver) but also hepatocytes and kidney cells, where they accumulate at high yield and thus may have some toxic effect (69).
Liang et al. described encapsulation of H-ferritin with doxorubicin (HFn-Dox) and treatment of HT29 colon tumor-bearing mice. HT29 cells internalized this complex via TfR1 and released doxorubicin in lysosomes. The final effect was 10-fold increase in doxorubicin intracellular concentration in comparison with its plain form. Moreover doxorubicin concentration was 3-16-fold lower when administered inside ferritin cage, comparing with plain drug administration (70). Interesting anticancer approach that has been recently published, it is to use ferritin nanocages in photothermal therapy. NIR (near infrared) dye IR820 was loaded to ferritin and used in multimodal imaging-guided photothermal therapy (PTT) with increased absorbance and lower toxicity when compared to plain dye (71, 72).
A group of Ceci demonstrated that genetically modified variant of H-ferritin, carrying several copies of a-melanocyte-stimulating hormone peptide and loaded with highly monodisperse cobalt Co(II) doped iron oxide NPs was effective against melanoma B16 cells (73).
Very promising approach in anticancer research is coupling ferritin cages with receptor binding peptides. Examples of this approach are three recombinant ferritins: hFTH-affibody, hFTH-4CRGD, and hFTH-affibody-4CRGD binding to EGFR or integrin. Mice with EGFR-overexpressing adenocarcinoma (MDA-MB-468) and integrin-overexpressing glioblastoma (U87MG) ware treated with each of the ferritins listed above. The best results were obtained in U87MG-bearing mice treated with hFTH-affibody-4CRGD (74).
In conclusion, ferritin cages represent interesting scenario for diagnostic guided and stand-alone therapy, enabling delivery of different compounds and making targeting more specific using antibody complexing approach.
CONJUGATES OR DRUGS WITH METAL
NANOPARTICLES OR RARE EARTH ELEMENTS
Metal nanoparticles have unique properties comparing with their solid bulk materials and therefore they have been utilized in many applications, for example: electrochemistry, photochemistry, and biomedicine (75). Currently, in cancer therapy the most commonly investigated compounds are: gold nanoparticles and silver nanoparticles. Recently, rare earth elements seem to be interesting tool for drug delivery as well.
Gold nanoparticles
Gold nanoparticles are widely used in biotechnology and biomedical field because of their large surface area, and interaction with cells (76). Enhanced permeability and retention ensure their accumulation in the tumor mass and interaction with cancer cells. The gold nanoparticles proved to be the safest and much less toxic agents for drug delivery than other inorganic materials (77). Drug vectorization by gold nanoparticles is very important in the context of multidrug resistance of tumors, which is a significant cause of chemotherapy failures (78). Our own study showed that in case of feline fibrosarcoma cells, gold nanoparticle-doxorubicin complex may be a potent new therapeutic agent to increase the efficacy of the drug by overcoming the resistance to doxorubicin. Moreover, as doxorubicin is non-covalently attached to glutathione coated nanoparticles the synthesized system is potentially suitable to a wealth of different drug molecules and not only doxorubicin (78, 79).
Gold nanoparticles enter cancer cell by endocytosis and are diffused through the lipid bilayer of the cell membrane. These gold nanoparticle-conjugated drugs are testes mostly in vitro or in vivo when administered locally. Nowadays, there are some ideas developed to use nanoparticles conjugated with antibodies against cancer cell surface-specific receptors that could specifically bind with malignant cells. These functionalized compounds are used for targeted therapy, however also as a locally applied therapy.
Very good ratio of surface to volume of gold nanoparticles offers a large number of drug molecules being carried by the one metal particle (80). However, these systems usually have complex, multilayer structure that significantly increases production costs and decreases applicative potential (78).
Rare earth elements
The characterization and possibility to exploit of rare earth elements (REE) depend on their morphologies, structure, and chemical compositions (81).
REE show strong fluorescence and therefore they may be used for imaging of organ or particular structures of interest. Han et al. used Eu3+ for labeling of Bel-7402 human liver cancer cells (82) showing their potential as a valuable biocompatible fluorescent labeling material in biological studies. Other group went a step further monitoring strontium fluorapatite with Tb3+ or Eu3+ samples after the loading of drug molecules. They suggested possibility of these compounds to be monitored or tracked during the drug release and whole therapeutic process (83).
Nowadays, special attention is paid on hydroxyapatite (HAP) due to its unique attributes such as interconnecting porosity of bulk material, good biocompatibility, resistance to mechanical force, and sustained release capacity. These make the HAP nanoparticles good candidates for drug, antigen, protein, gene and siRNA delivery and immunoadjuvant therapy (81). HAPs may deliver these compounds without degradation and can ensure slow release in proper place. For example, Chen et al. (84) developed Eu3+ and Gd3+ dual doped calcium phosphate in the presence of amphiphilic block copolymer that posed imaging capacity as well as drug loading and sufficiently long drug release abilities.
Since this topic is relatively new, more experiments conducted to check cytotoxicity of REE at the cellular level should be conducted as well as biodistribution of these particles. So, there is still number of challenges, before practical applications of REE will be possible, e.g. compatibility and stability within biological systems.
ANTIBODY-DRUG CONJUGATES (ADC) AS PROMISING TOOL IN CANCER THERAPY
The concept of linking of the antibody to the toxin and using such a conjugate in the targeted treatment started to be real together with the development of the monoclonal antibodies range. Potential ADC consists of monoclonal antibody responsible for specific tumor targeting and the toxin causing cell apoptosis both linked with the chemical linker. Such complex should be specific enough to avoid toxic influence on the healthy cells and strong enough to kill cancer cells. Linker should be stable while product is transported through the circulation, but able to be broken while endocytosed inside the cell. The whole conjugate should be neutral to immune system recognition to avoid early clearance from the blood. In solid tumors important problem is that their uptake by the external layer of antigen-positive cells may be high but the penetration and distribution throughout a tumor mass may be reduced. To solve this difficulty, different linker strategies are adopted. One of them is usage of cleavable instead of non- cleavable linkers, the first enabling drug after release to escape and influence the neighboring cells. This mechanism is called 'bystander killing', causes apoptosis of non-targeted neighboring carcinoma cells. That improves efficacy in nonresponding tumors, while not showing systemic activity (85).
In the early ADCs, the most commonly used drugs were doxorubicin (86) or methotrexate (87). The reasons are that these agents are readily available and their toxicological properties are well known. A problem with first ADCs is the small amount of the drug, which can be linked to one antibody molecule and that these standard substances are only moderately potent and generally not enough cytotoxic for the targeted tumor cells. To have higher impact on the carcinoma cells much more potent toxic agents are currently used. Among these are inhibitors of tubulin polymerization such as the maytansinoids, dolastatins, auristatin drug analogues and cryptophycin. They are inhibiting cell division by binding tubulin, which arrest the target cell in the G2/M stage of the cell cycle resulting in apoptosis (88). Alkylating agents include the duocarmycin derivatives (89), antibiotics including calicheamicin, which catalyze DNA double-strand breaks (90) and pyrolobenodiazepine (PBD), which is a DNA minor groove binding agent (91). The number of attached drug molecules per antibody is also important for the overall cytotoxicity. The lower number of molecules will not have enough effect, while too many molecules can elicit increased circulatory clearance. It was shown that ADCs loaded with 2 – 4 drugs per mAb achieved the best balance between slow clearance and maximal potency (92).
First ADC product on the market was Mylotarg® (gemtuzumab ozogamycin) launched in 2001 for AML, but subsequently withdrawn in 2010 due to the increased death rate (93). Two other products currently marketed are brentuximab vedotin (Adcetris®) for relapsed HL and relapsed sALCL and trastuzumab emtansine (Kadcyla®) for HER2-positive metastatic breast cancer (mBC) after prior treatment with trastuzumab and taxane. A number of new ADC for tumor treatments are in phase II trials - f.e. lifastuzumab vedotin for non-small lung cancer and ovarian tumor, lorvotuzumab mertansine for small cell lung cancer, labetuzumab-SN-38 for colorectal cancer.
Like with the other cancer technologies resistance to targeted therapy occurs (94). Small number of expressed antigens limits the amount of the antibodies specifically binding to the cells and further amount of the conjugates that is internalized. Moreover the same antigens can be expressed on tumor cells as well as in healthy tissues. So to achieve better effect, toxic agents are linked to bispecific antibody, enhancing the potential affinity to cancer cells only. For example it was shown that HER2xPRLR bispecific ADCs (targeting cancer cells expressing HER2 and prolactin receptor) kill more effectively than HER2 ADC in breast cancer cells (95). Another problem is connected with large size of antibodies, as they cannot easily penetrate solid tumors. To solve this constrain research is focused on using diabodies or minibodies - monoclonal antibodies fragments, that have smaller size and easier penetrate, however at the sake of shorter half-life (96, 97).
SILK AS A DRUG DELIVERY VEHICLE FOR SOLID TUMORS
Silk fibroin is the protein produced by the Bombyx mori. Due to its highly controllable stimuli-responsive self-assembly features in aqueous solution, silk fibroin displays interesting properties desirable for drug delivery applications (98). The primary structure of silk fibroin is prevalently composed of glycine, alanine and serine forming a heterodimeric protein with a heavy chain (325 kDa) and a light chain (25 kDa). The fibroin heavy chain self-assembly results in the formation of micellar structures. In these structures, the hydrophobic crystallizable domains are encapsulated inside a hydrophilic shell of a spherical morphology. A very important value of the silk is its biocompatibility and relatively low immunogenic potential. A number of chemical modifications, like coupling, side chain modifications, covalent fusion of various functional groups and biomacromolecules makes silk the platform for sustained drug delivery (99).
Conjugates of silk and anticancer drugs
In cancer studies, silk has been utilized as a vehicle to deliver a wide range of bioactive molecules including genes, small molecules, and drugs. For each class of compounds, various silk modifications have been used for the delivery and release of the drugs. Conjugates of silk and anticancer drugs have been extensively tested but mostly in various basic and pre-clinical studies. These complexes showed significant activity against breast cancer (silk and doxorubicin, metotrexate, emodin, curcumin), gastric cancer (silk and paclitaxel) and melanoma (silk and curcumin, emodin). The silk particles were able to enter the cancer cells in vitro to end up in lysosomes releasing the drug in a pH dependent manner. The silk nanoparticles showed unique abilities e.g. of the drug release over 9 days, or when blended with albumin were successfully applied to load and release a poorly water-soluble anticancer drug, methotrexate (100). Interesting approach used Cheema et al. coating with silk liposomes loaded with a hydrophobic anticancer drug, emodin. This new formulation allowed to decrease the emodin release rate, therefore enhancing uptake of the drug-loaded liposomes (98). However, only in case of a complex of silk and plasmid DNA the route of administration was intravenous, in contrary to other complexes studied in vitro or administered intratumorally (100). The adenoviral gene therapy as silk-elastin-like protein polymer hydrogels loaded with adenovirus administered i.v. has been particularly effective demonstrated significant reduction in tumor volume (99).
In summary, the majority of the currently approved anticancer drugs have limitations in their chemical and physical properties as well as in the abilities to effectively target the tumor tissue. Therefore development of special drug delivery systems is crucial to improve outcomes of chemotherapy. Despite some of them have been approved and used in cancer therapy (e.g. discussed in this review liposomes, or ADCs), they do not address all medical needs. That is why investigators and big pharma companies are still inventing and testing innovative approaches to drug delivery for cancer therapy.
Acknowledgments: This manuscript has been supported by the grant SONATA BIS from the Polish National Science Centre (NCN) no. UMO-2015/18/E/NZ6/00642.
Conflict of interests: None declared.
REFERENCES
- Duc P. Drug delivery systems for cancer therapeutics. US Pharm 2011; 36: 12-15.
- Hobbs SK, Monsky WL, Yuan F, et al. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA 1998; 95: 4607-4612.
- Kizaka-Kondoh S, Inoue M, Harada H, Hiraoka M. Tumor hypoxia: a target for selective cancer therapy. Cancer Sci 2003; 94: 1021-1028.
- Lewis C, Murdoch C. Macrophage responses to hypoxia. Am J Pathol 2005; 167: 627-635.
- Stylianopoulos T, Jainb RK. Combining two strategies to improve perfusion and drug delivery in solid tumors. Proc Natl Acad Sci USA 2013; 110: 18632-18637.
- Krol M, Pawlowski KM, Majchrzak K, Szyszko K, Motyl T. Why chemotherapy can fail? Pol J Vet Sci 2010; 13: 399-406.
- Muthana M, Guannoudis A, Scott SD, et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res 2011; 71: 1805-1815.
- Leppert W, Zajaczkowska R, Wordliczek J, Dobrogowski J, Woron J, Krzakowski M. Patophysiology and clinical characteristics of pain in most common locations in cancer patients. J Physiol Pharmacol 2016; 67: 787-799.
- Kotowski M, Bogacz A, Bartkowiak-Wieczorek J, et al. The influence of the tumor necrosis factor-alpa-308G>A polymorphism on the efficacy of immunosuppressive therapy in patients after kidney transplantation. J Physiol Pharmacol 2016; 67: 819-826.
- Skidan I, Steiniger SC. In vivo models for cancer stem cell research: a practical guide for frequently used animal models and available biomarkers. J Physiol Pharmacol 2014; 65: 157-169.
- Unal H, Ozturk N, Bilensoy E. Formulation development, stability and anticancer efficacy of core-shell cyclodextrin nanocapsules for oral chemotherapy with camptothecin. Beilstein J Org Chem 2015; 11: 204-212.
- Bangham AD, Horne RW. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J Mol Biol 1964; 8: 660-668.
- Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol 2008; 26: 57-64.
- Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv Drug Deliv Rev 2016; 99: 28-51.
- Allen TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer 2002; 2: 750-763.
- Xu L, Huang CC, Huang W, et al. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol Cancer Ther 2002; 1: 337-346.
- Low PS, Henne WA, Doorneweerd DD. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc Chem Res 2007; 41: 120-129.
- Zhai G, Wu J, Yu B, Guo C, Yang X, Lee RJ. A transferrin receptor-targeted liposomal formulation for docetaxel. J Nanosci Nanotechnol 2010; 10: 5129-5136.
- Sharma G, Modgil A, Sun C, Singh J. Grafting of cell-penetrating peptide to receptor-targeted liposomes improves their transfection efficiency and transport across blood-brain barrier model. J Pharm Sci 2012; 101: 2468-2478.
- Kim SK, Huang L. Nanoparticle delivery of a peptide targeting EGFR signaling. J Control Release 2012; 157: 279-286.
- Shmeeda H, Tzemach D, Mak L, Gabizon A. HER-2-targeted pegylated liposomal doxorubicin: retention of target-specific binding and cytotoxicity after in vivo passage. J Control Release 2009; 136: 155-160.
- Hatakeyama H, Akita H, Ishida E, et al. Tumor targeting of doxorubicin by anti-MT1-MMP antibody-modified PEG liposomes. Int J Pharm 2007; 342: 194-200.
- Chen Z, Deng J, Zhao Y, Tao T. Cyclic RGD peptide-modified liposomal drug delivery system: enhanced cellular uptake in vitro and improved pharmacokinetics in rats. Int J Nanomed 2012; 7: 3803. doi: 10.2147/IJN.S33541
- Suzuki S, Itakura S, Matsui R, et al. Tumor microenvironment-sensitive liposomes penetrate tumor tissue via attenuated interaction of the extracellular matrix and tumor cells and accompanying actin depolymerization. Biomacromolecules 2017; 18: 535-543.
- Kang H, O'Donoghue MB, Liu H, Tan W. A liposome-based nanostructure for aptamer directed delivery. Chem Commun 2010; 46: 249-251.
- Baek SE, Lee KH, Park YS, et al. RNA aptamer-conjugated liposome as an efficient anticancer drug delivery vehicle targeting cancer cells in vivo. J Control Release 2014; 196: 234-242.
- Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anticancer drug delivery. J Control Release 2010; 148: 135-146.
- Torchilin VP. Targeted pharmaceutical nanocarriers for cancer therapy and imaging. AAPS J 2007; 9 :128-147.
- Grull H, Langereis S. Hyperthermia-triggered drug delivery from temperature-sensitive liposomes using MRI-guided high intensity focused ultrasound. J Control Release 2012; 161: 317-327.
- Maples D, McLean K, Sahoo K, et al. Synthesis and characterisation of ultrasound imageable heat-sensitive liposomes for HIFU therapy. Int J Hyperthermia 2015; 31: 674-685.
- Mejias R, Perez-Yague S, Gutierrez L, et al. Dimercaptosuccinic acid-coated magnetite nanoparticles for magnetically guided in vivo delivery of interferon gamma for cancer immunotherapy. Biomaterials 2011; 32: 2938-2952.
- Joniec A, Sek S, Krysinski P. Magnetoliposomes as potential carriers of doxorubicin to tumors. Chemistry 2016; 22: 17715-17724.
- Apte A, Koren E, Koshkaryev A, Torchilin VP. Doxorubicin in TAT peptide-modified multifunctional immunoliposomes demonstrates increased activity against both drug-sensitive and drug-resistant ovarian cancer models. Cancer Biol Ther 2014; 15: 69-80.
- Juang V, Lee HP, Lin AM, Lo YL. Cationic PEGylated liposomes incorporating an antimicrobial peptide tilapia hepcidin 2-3: an adjuvant of epirubicin to overcome multidrug resistance in cervical cancer cells. Int J Nanomedicine 2016; 11: 6047-6064.
- Trotta F, Zanetti M, Cavalli R. Cyclodextrin-based nanospongues as drug carriers. Beilstein J Org Chem 2012; 8: 2091-2099.
- Trotta F, Dianzani C, Caldera F, Mognetti B, Cavali R. The application of nanospongues to cancer drug delivery. Expert Opin Drug Deliv 2014; 11: 931-941.
- Bolmal UB, Manvi FV, Rajkumar K, Palla SS, Paladugu A, Reddy KR. Recent advances in nanospongues as drug delivery system. Int J Pharm Sci Nanotechnol 2013; 6: 1934-1944.
- Torne S, Ansari K, Vavia P, et al. Enhanced oral paclitaxel bioavailability after administration of paclitaxel-loaded nanospongues. Drug Deliv 2010; 17: 419-425.
- Cavalli R, Trotta F, Tumiatti WJ. Cyclodextrin-based nanospongues for drug delivery. J Incl Phenom Macrocycl Chem 2006; 56: 209-213.
- Vanucci L, Falvo E, Formara M, et al. Selective targeting of melanoma by PEG-masked protein-based multifunctional nanoparticles. Int J Nanomed 2012; 7: 1489-1509.
- Falvo E, Tremante E, Fraioli R, et al. Antibody-drug conjugates: targeting melanoma with cisplatin encapsulated in protein-cage nanoparticles based on human ferritin. Nanoscale 2013; 5: 12278-12285.
- Reissmann KR, Dietrich MR. On the presence of ferritin in the peripheral blood of patients with hepatocellular disease. J Clin Invest 1956; 35: 588-595.
- Harrison PM, Arosio P. The ferritins: molecular properties, iron storage function and cellular regulation. Biochim Biophys Acta 1996; 1275: 161-203.
- Li JY, Paragas N, Ned RM, et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev Cell 2009; 16: 35-46.
- Bresgen N, Eckl PM. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015; 5: 808-847.
- Rosario C, Zandman-Goddard G, Meyron-Holtz EG, D'Cruz DP, Shoenfeld Y. The hyperferritinemic syndrome: macrophage activation syndrome, Still's disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med 2013; 11: 185. doi: 10.1186/1741-7015-11-185.
- Pontillo N, Pane F, Messori L, Amoresano A, Merlino A. Cisplatin encapsulation within a ferritin nanocage: a high-resolution crystallographic study. Chem Commun (Camb) 2016; 52: 4136-4139.
- Aime S, Frullano L, Crich SG. Compartmentalization of a gadolinium complex in the apoferritin cavity: a route to obtain high relaxivity contrast agents for magnetic resonance imaging. Angew Chem Int 2002; 41: 1017-1019.
- Cutrin JC, Crich SG, Burghelea D, Dastru W, Aime S. Curcumin/Gd loaded apoferritin: a novel 'theranostic' agent to prevent hepatocellular damage in toxic induced acute hepatitis. Mol Pharmaceutics 2013; 10: 2079-2085.
- Dominguez-Vera JM, Colacio E. Nanoparticles of prussian blue ferritin: a new route for obtaining nanomaterials. Inorg Chem 2003; 42: 6983-6985.
- Ueno T, Suzuki M, Goto T, Matsumoto T, Nagayama K, Watanabe Y. Size selective olefin hydrogenation by a Pd nanocluster provided in the Apo-ferritin cage. Angew Chem Int Ed Eng 2004; 43: 2527-2530.
- Li M, Viravaidya C, Mann S. Polymer-mediated synthesis of ferritin-encapsulated inorganic nanoparticles. Small 2007; 3: 1477-1481.
- Iwahori K, Yoshizawa K, Muraoka M, Yamashita I. Fabrication of ZnSe nanoparticles in the apoferritin cavity by designing a slow chemical reaction system. Inorg Chem 2005; 44: 6393-6400.
- Pulsipher KW, Dmochovski IJ. Ferritin encapsulation and templated synthesis of inorganic nanoparticles. Methods Mol Biol 2015; 1252: 27-37.
- Fujita K, Tanaka Y, Sho T, et al. Intracellular CO release from composite of ferritin and ruthenium carbonyl complexes. J Am Chem Soc 2014; 136: 16902-16908.
- Polanams J, Ray AD, Watt RK. Nanophase iron phosphate, iron arsenate, iron vanadate, and iron molybdate minerals synthesized within the protein cage of ferritin. Inorg Chem 2005; 44: 3203- 3209
- Kilic MA, Ozlu E, Callis S. A novel protein-based anticancer drug encapsulating nanosphere: apoferritin-doxorubicin complex. J Biomed Nanotechnol 2012; 8: 508-514.
- Uchida M, Flenniken ML, Allen M, et al. Targeting of cancer cells with ferrimagnetic ferritin cage nanoparticles. J Am Chem Soc 2006; 128: 16626-16633.
- Chen L, Bai G, Yang R, Zang J, Zhou T, Zhao G. Encapsulation of b-carotene within ferritin nanocages greatly increases its water-solubility and thermal stability. Food Chem 2014; 149: 307-312.
- Zhen Z, Tang W, Zhang W, Xie J. Folic acid conjugated ferritins as photosensitizer carriers for photodynamic therapy. Nanoscale 2015; 7: 10330-10333.
- Zhen Z, Tang W, Guo C, et al. Ferritin nanocages to encapsulate and deliver photosensitizers for efficient photodynamic therapy against cancer. ACS Nano 2013; 7: 6988-6996.
- Ferraro G, Monti DM, Amoresano A, et al. Gold-based drug encapsulation within a ferritin nanocage: X-ray structure and biological evaluation as a potential anticancer agent of the Auoxo3-loaded protein. Chem Commun (Camb) 2016; 52: 9518-9521.
- Wang Q, Mercogliano CP, Lowe JA. Ferritin-based label for cellular electron cryotomography. Structure 2011; 19: 147-154.
- Jin R, Lin B, Li D, Ai H. Superparamagnetic iron oxide nanoparticles for MR imaging and therapy: design considerations and clinical applications. Curr Opin Pharmacol 2014; 18C: 18-27.
- Vande Velde G, Rangarajan JR, Toelen J, Dresselaers T., Ibrahimi A, Krylychkina O. Evaluation of the specificity and sensitivity of ferritin as an MRI reporter gene in the mouse brain using lentiviral and adeno-associated viral vectors. Gene Ther 2011; 18: 594-605.
- He D, Marles-Wright J. Ferritin family proteins and their use in bionanotechnology. N Biotechnol 2015; 32: 651-657.
- Zhen Z, Tang W, Chuang YJ, et al. Tumor vasculature targeted photodynamic therapy for enhanced delivery of nanoparticles. ACS Nano 2014; 8: 6004-6013.
- Fan K, Cao C, Pan Y, et al. Magnetoferritin nanoparticles for targeting and visualizing tumor tissues. Nat Nanotechnol 2012; 7: 459-464.
- Kitagawa T, Kosuge H, Uchida M, et al. RGD-Conjugated human efrritin nanoparticles for imaging vascular inflammation and angiogenesis in experimental carotid and aortic disease. Mol Imaging Biol 2012; 14: 315-324.
- Liang M, Fan K, Zhou M, et al. H-ferritin-nanocaged doxorubicin nanoparticles specifically target and kill tumors with a single-dose injection. Proc Natl Acad Sci USA 2014; 111: 14900-14905.
- Gao FP, Lin YX, Li LL, et al. Supramolecular adducts of squaraine and protein for noninvasive tumor imaging and photothermal therapy in vivo. Biomaterials 2014; 35: 1004-1014.
- Huang P, Rong P, Jia A, et al. Dye-loaded ferritin nanocages for multimodal imaging and photothermal therapy. Adv Mater 2014; 26: 6401-6408.
- Fantechi E, Innocenti C, Zanardelli M, et al. A smart platform for hyperthermia application in cancer treatment: cobalt-doped ferrite nanoparticles mineralized in human ferritin cages. ACS Nano 2014; 8: 4705-4719.
- Kwon KC, Ko HK, Lee J, Lee EJ, Kim K, Lee J. Enhanced in vivo tumor detection by active tumor cell targeting using multiple tumor receptor-binding peptides presented on genetically engineered human ferritin nanoparticles. Small 2016; 12: 4241-4253.
- Di Guglielmo C, Lopez DR, De Lapuente J, Mallafre JM, Suarez MB. Embryotoxicity of cobalt ferrite and gold nanoparticles: a first in vitro approach. Reprod Toxicol 2010; 30: 271-276.
- Mendoza KC, McLane VD, Kim S, Griffin JD. In vitro application of gold nanoprobes in live neurons for phenotypical classification, connectivity assessment, and electrophysiological recording. Brain Res 2010; 1325: 19-27.
- Lukianova-Hleb EY, Wagner DS, Brenner MK, Lapotko DO. Cell-specific transmembrane injection of molecular cargo with gold nanoparticle-generated transient plasmonic nanobubbles. Biomaterials 2012; 33: 5441-5450.
- Wojcik M, Lewandowski W, Krol M, et al. Enhancing anti-tumor efficacy of doxorubicin by non-covalent conjugation to gold nanoparticles - in vitro studies on feline fibrosarcoma cell lines. PLoS One 2015; 10: e0124955. doi: 101371/journal.pone.0124955.
- Zabielska-Koczywas K, Dolka I, Krol M, et al. Doxorubicin conjugated to glutathione stabilized gold nanoparticles (Au-GSH-Dox) as an effective therapeutic agent for feline injection-site sarcomas - chick embryo chorioallantoic membrane study. Molecules 2017; 22: 253. doi: 10.3390/molecules22020253
- Alaqad K, Saleh TA. Gold and silver nanoparticles: synthesis methods, characterization routes and applications towards drugs. J Environ Anal Toxicol 2016; 6: 384.
- Perera TS, Han Y, Lu X, Wang X, Dai H, Li S. Rare earth doped apatite nanomaterials for biological application. J Nanomater 2015; Article ID 705390.
- Han Y, Wang X, Li S. Biocompatible europium doped hydroxyapatite nanoparticles as a biological fluorescent probe. Curr Nanosci 2010; 6: 178-183.
- Niu N, Wang D, Huang S et al. Controlled synthesis of luminescent F-substituted strontium hydroxyapatite with hierarchical structures for drug delivery. Cryst Eng Comm 2012; 14: 1744-1752.
- Chen F, Huang P, Zhu YJ, Wu J, Cui DX. Multifunctional Eu3+/Gd3+ dual doped calcium phosphate vesicle like nanosperes for sustained drug release and imaging. Biomaterials 2012; 33: 6447-6455.
- Ogitani Y, Hagihara K, Oitate M, Naito H, Agatsuma T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci 2016; 107: 1039-1046.
- Shih LB, Goldenberg DM, Xuan H, Lu HW, Mattes MJ, Hall TC. Internalization of an intact doxorubicin immunoconjugate. Cancer Immunol Immunother 1994; 38: 92-98.
- Smyth MJ, Pietersz GA, McKenzie IF. The mode of action of methotrexate-monoclonal antibody conjugates. Immunol Cell Biol 1987; 65: 189-200.
- Klute K, Nackos E, Tasaki S, Nguyen DP, Bander NH, Tagawa ST. Microtubule inhibitor-based antibody-drug conjugates for cancer therapy. Onco Targets Ther 2014; 7: 2227-2236.
- Lee van der MM, Groothuis PG, Ubink R, et al. The preclinical profile of the duocarmycin-based HER2-targeting ADC SYD985 predicts for clinical benefit in low HER2-expressing breast cancers. Mol Cancer Ther 2015; 14: 692-703.
- Shao RG. Pharmacology and therapeutic applications of enediyne antitumor antibiotics. Curr Mol Pharmacol 2008; 1: 50-60.
- Mantaj J, Jackson PJ, Rahman KM, Thurston DE. From anthramycin to pyrrolobenzodiazepine (PBD)-containing antibody-drug conjugates (ADCs). Angew Chem Int Ed Engl 2017; 56: 462-488.
- Hamblett, KJ, Senter PD, Chace DF, et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 2004; 10: 7063-7070.
- FDA News Release For Immediate Release: June 21, 2010.
- Ellis LM, Hicklin DJ. Resistance to targeted therapies: refining anticancer therapy in the era of molecular oncology. Clin Cancer Res 2009; 15: 7471-7478
- Andreev J, Thambi N, Perez Bay AE, et al. Bispecific antibodies and antibody-drug conjugates (ADCs) bridging HER2 and prolactin receptor improve efficacy of HER2 ADCs. Mol Cancer Ther 2017; 16: 681-693.
- Kim KM, McDonagh CF, Westendorf L, et al. Anti-CD30 diabody-drug conjugates with potent antitumor activity. Mol Cancer Ther 2008; 7: 2486-2497.
- Asano R, Shimomura I, Konno S, et al. Rearranging the domain order of a diabody-based IgG-like bispecific antibody enhances its antitumor activity and improves its degradation resistance and pharmacokinetics. MAbs 2014; 6: 1243-1254.
- Yucel T, Lovett ML, Kaplan DL. Silk-based biomaterials for sustained drug delivery. J Control Release 2014; 190: 381-397.
- Zhao Z, Li Y, Xie MB. Silk fibroin-based nanoparticles for drug delivery. Int J Mol Sci 2015; 16: 4880-4903.
- Jastrzebska K, Kucharczyk K, Florczak A. Silk as an innovative biomaterial for cancer therapy. Rep Pract Oncol Radiother 2015; 20: 87-98.
- Cheema SK, Gobin AS, Rhea R, Lopez-Berestein G, Newman RA, Mathur AB. Silk fibroin mediated delivery of liposomal emodin to breast cancer cells. Int J Pharm 2007; 341: 221-229.
A c c e p t e d : April 28, 2017