THE EFFECT OF THE NITROXIDE PIROLIN ON OXIDATIVE STRESS
INDUCED BY DOXORUBICIN AND TAXANES IN THE RAT BRAIN
INTRODUCTION
Anticancer therapy employing doxorubicin (DOX) and the taxanes paclitaxel (PTX) and docetaxel (DTX) is burdened by serious side-effects, e.g. neurotoxicity (1) and reactive oxygen species (ROS) have been shown to play an indisputable role in the development of these side-effects (2, 3). Oxidative stress (OS), induced by DOX, is considered to underlie the cognitive impairment (CI) experienced by up to 75% of cancer survivors treated with DOX (4, 5). Oxidative stress-induced depletion of reduced glutathione (GSH) and impairment in the activities of GSH-related enzymes have been suggested to be responsible for CI in DOX-treated mice (6). Doxorubicin-induced OS, neuroinflammation and apoptosis have been indicated as causative for depressive-like rat behaviors (7). Cognitive impairment, evoked by chemotherapy, including DTX or PTX, has been also reported in clinical investigations (8) and confirmed in rodents (9)), suggesting the involvement of OS and oxidative DNA damage and repair in this neurotoxic effect (3, 10).
Recent data indicate that DOX, PTX and DTX can cross the blood-brain barrier (BBB) in limited amounts, which are sufficient to adversely affect the brain and induce toxic effects on the central nervous system (CNS) (9, 11, 12). Increased BBB permeabilization and the disturbance of its functions have been demonstrated under OS conditions (13), and ROS production has been shown to participate in DOX-, PTX- and DTX-induced neuronal degeneration (2, 3). An increase in 4-hydroxy-2-nonenal (4-HNE), one of the main products of lipid peroxidation (LPO), has been discovered in the brains of mice receiving DOX (14). The generation of ROS has been also suggested to be a key mechanism responsible for the genotoxic effects of DOX (e.g., in rat hearts (15)).
Doxorubicin by itself weakly induces cellular defense against OS (16), but ROS and products of their reactions are relatively strong inducers of various signaling pathways. DOX can generate ROS, deplete GSH and activate nuclear factor kB (NF-κB) in isolated rat neurons (17). The primary cell-protective pathways involve the nuclear factor E2-related protein 2 (Nrf-2), the antioxidant responsive element (ARE) (18), p53, NF-κB and Forkhead transcription factors (FOXO) (19). ROS, produced by drugs, activate p53 and result in additional ROS production and the induction of apoptosis. Under OS conditions, FOXO upregulate antioxidant enzymes catalase (CAT) and manganese superoxide dismutase (MnSOD), contributing to a decrease in ROS (20). During the inflammatory response, NF-κB regulates genes for cytokines, e.g. tumor necrosis factor-α (TNF-α). Furthermore, its effects include MnSOD and B Cell Lymphoma 2 (Bcl-2) regulation (19).
TNF-α has been suggested to be a major mediator inducing further cytokine production, BBB damage and brain inflammation via the activation of microglia. TNF-α can dually influence the CNS (21), initiating both cell survival or apoptotic pathways via TRADD and FADD domains and the activation of caspase-8, NF-κB and JNK (c-Jun N-terminal kinase) (22). DOX has been shown to oxidize apolipoprotein A1 (APOA1) in mice and human blood plasma, which stimulates TNF-α release by macrophages and increases its level in plasma (23). TNF-α is transported into the brain, where it induces inflammation and apoptosis (5).
One of the crucial signaling ROS is NO, which is produced by nitric oxide synthases (NOS). The dual role of neuronal NOS (nNOS), expressed through inhibition of oxidases (24, 25), has been demonstrated for the regulation of the myocardial redox environment and the attenuation of unfavorable heart remodeling or the contribution to an increase in ventricular wall thickness (26). NO has also been shown to inhibit cytochrome oxidase in the brain tissue (27), but the role of nNOS in the neuronal toxicity of anticancer drugs remains unknown.
An important target of ROS attack is DNA, whose damage activates processes and signaling pathways leading to cell cycle arrest and apoptosis. Poly(ADP-ribosylation) of nuclear acceptors by DNA-binding poly (ADP-ribose) polymerase-1 (PARP-1) accompanies the formation of DNA lesions. Binding of cleaved PARP-1 fragments to DNA strand breaks inhibits repair enzymes and conserves ATP (28).
Increased BBB permeabilization under conditions of OS, the influx of anticancer drugs into the brain and CNS oxidative damage induced by DOX, DTX and PTX indicate the need for the application of effective antioxidative neuroprotector(s) (29). This approach may be more beneficial than reducing drug dosages and effectiveness. The rationale for this hypothesis is based on the results of studies showing that 4-HNE causes inactivation of neuronal enzymes, DNA lesions (30), interruption of BBB integrity (13, 31) and apoptosis in mouse and human hearts through apoptosis-inducing factor, mitochondria associated 2 (AIFm2), which can be attenuated by superoxide dismutase (SOD) mimics (32). Antioxidant 2-mercaptoethane sulfonate (Mesna) reduced plasma levels of TNF-α and its soluble receptors in patients receiving DOX-based chemotherapy (33). Therefore, we assume that non-immunogenic, BBB-permeable (34) and non-toxic nitroxides could play a neuroprotective role, the more that they perform as SOD mimics (35) and have been shown to prevent or attenuate oxidative damage in various models (36, 37). Despite some studies demonstrating in vivo protection of brain cells by nitroxides (38, 39), their capability to attenuate the CNS toxicity of DOX, DTX and PTX has not yet been widely investigated. Therefore, the aim of our preliminary study was to verify whether these chemotherapeutics induce oxidative damage in rat brains in vivo and whether the five-membered pyrroline nitroxide Pirolin (PL) can act as a neuroprotector in the rat CNS. In our animal model, Wistar rats were treated with DOX, PTX or DTX, applied alone or in combination with PL. We analyzed the brain homogenates for the representative OS biomarkers: lipid hydroperoxides, thiobarbituric acid-reactive substances (TBARS), thiol groups (-SH) and the activity of antioxidant enzymes SODs and CAT. To gain deeper insights into the mechanisms of OS-induced CNS damage and its protection by PL, we also investigated the genotoxic effects of DOX, DTX, PTX and PL by measuring DNA damage as well as the expression of some of the key components of ROS and protective signaling pathways: nNOS, TNF-α and PARP-1. To the best of our knowledge, there have been no studies focusing on the neuroprotective properties of PL against OS-induced CNS damage evoked by DOX and taxanes; revealing these properties is the novelty of our study.
MATERIALS AND METHODS
Materials
Sequoia Research Products Ltd., Pangbourne, United Kingdom was a provider of doxorubicin, paclitaxel and docetaxel. The Rozantsev protocol was used to synthesize Pirolin from 3-carbamoyl-2,2,5,5-tetramethylpyrroline (40). The nitroxide crystals were recrystallized from ethanol and purity of PL was checked by the measurement of its melting point (203 – 204°C). Acrylamide, N,N’-methylenebis(acrylamide), butylated hydroxytoluene (BHT; 2,6-di-tert-butyl-4-hydroxytoluene), bovine serum albumin (BSA), ferrous ammonium sulfate, collagenase, 4’,6-diamidino-2-phenylindole (DAPI), D-sorbitol, Ponceau S, DL-dithiothreitol, Hanks’ balanced salt solution (HBSS), low melting point (LMP) agarose, normal melting point (NMP) agarose and phosphate buffered saline (PBS) were purchased from Sigma-Aldrich. Bromophenol blue, Coomassie Brilliant Blue R 250, TEMED, Tween® 80, sodium dodecyl sulfate, Roti®-Mark 10-150 were from Carl Roth GmbH, Germany. The adrenaline and xylenol orange (XO) were obtained from MP Biomedicals. Ammonium persulfate, hydrogen peroxide (H2O2), 2-thiobarbituric acid (TBA), Tris(hydroxymethyl)aminomethane and glycine were obtained from POCH, Poland. All other reagents were of the highest purity available. Deionized Q water was used in the preparation of all solutions (Millipore Corp.).
We used a UV-VIS spectrophotometer (Pharmacia Bio-Tech) to measure the corresponding absorbances, specific for each of the methods that we employed to determine the content of hydroperoxides, TBARS, thiol groups and protein concentration. We performed kinetic measurements in enzyme assays using a CARY 50 spectrophotometer (Varian Inc., Australia).
The comet analysis was performed using an Eclipse fluorescence microscope (Nikon, Tokyo, Japan) equipped with a COHU-4910 video camera (Cohu, San Diego, CA) and a UV-1 filter block (an excitation filter of 359 nm and a barrier filter of 461 nm). The percentage of DNA in the comet tail (% tail-DNA) was calculated using image analysis software Lucia-Comet v. 4.51 (Laboratory Imaging, Prague, Czech Republic).
Animals and experimental design
The study was carried out on adult (2-month-old) male Wistar rats weighing 180 – 220 g. The animals were housed with free access to water and fed a standard diet. The rats were divided into one control (vehicle-treated) and seven experimental (anticancer drug/Pirolin-treated) groups with five animals in each group. A vehicle (1 ml of 5% glucose) and a single dose of 10 mg in 1 ml of 5% glucose of each compound/kg body weight were injected i.p. once on the first day of the experiment (Fig. 1). Four days after injection (Fig. 1), all of the rats were sacrificed.

The experimental setting included the following groups: vehicle-treated rats (group 1, control); compound-treated rats: PL (group 2), DOX (group 3), DTX (group 4), PTX (group 5), DOX + PL (group 6), DTX + PL (group 7) and PTX + PL (group 8). Doses of DOX, DTX and PTX were calculated as corresponding to those applied in chemotherapy for human breast cancer (41) and based on the formula that made it possible to estimate a dose appropriate for animal studies (42). The 10 mg/kg b.w. dose of PL and the four-day period of time (Fig. 1) between the injection of the compounds and the sacrifice of the rats were selected on the basis of our earlier study, which investigated the effect of this nitroxide against OS induced by the same dose of DOX in heart myocytes in vivo and indicated protective properties of PL (37). No reactions between PL and DOX occurred in solution, as checked by the electron paramagnetic resonance (EPR) technique (43).
We adhered to the rules described in the European Communities Council Directive (86/609/EEC from 24 November 1986) and the National Institutes of Health guide for the care and use of laboratory animals (NIH Publications No. 8023, revised 1978). The experimental procedures were designed to minimize animal suffering, and we reduced the number of animals that we used considering that this study is preliminary. The experiments were carried out under the approval of the appropriate institutional local ethics committee.
Preparation of brain tissue samples for biochemical analyses
On the fourth day after injection (Fig. 1), all of the rats were anesthetized and sacrificed by cervical dislocation. The whole brains were excised, washed with 0.9% NaCl and weighed. Part of the brain (fresh tissue) was taken for the comet assay analysis and the remaining part was frozen on solid CO2 and then stored at –80°C until analysis. Immediately before the biochemical analyses, each of the frozen brains was mixed with cold (4°C) 1.15% KCl (9 ml of KCl per gram of tissue) and homogenized with the mechanical rotor-stator homogenizer. The homogenate was centrifuged at 3,000g for 10 min at 4°C and the supernatant was used for the biochemical assays. All of the biochemical measurements were performed in triplicate.
Biochemical analyses
1. Lipid peroxidation - hydroperoxide content
We used the ferrous oxidation-xylenol orange (FOX) assay of Gay and Gebicki (44) to measure the hydroperoxide concentration. In brief, the appropriate volumes of reagents at the final concentrations of 25 mM H2SO4, 125 mM xylenol orange, 100 mM D-sorbitol, and 250 mM ferrous iron were mixed with the brain tissue homogenate in a volume of 1 ml. After 30 min incubation in the dark, the samples were centrifuged (3,000g, 5 min, 20°C) and absorbance of the supernatant was read at 560 nm against blank probe (water and XO/Fe2+). The standard curve was constructed for fixed concentrations of t-butyl hydroperoxide and the content of hydroperoxides was calculated and expressed in nanomoles per milligram of protein.
2. Lipid peroxidation - thiobarbituric acid-reactive substances (TBARS) level
The amount of TBARS in the brain tissue was assayed using the protocol of Rice-Evans et al. (45). Briefly, tissue homogenate and 15% trichloroacetic acid in 0.25 M hydrochloric acid (HCl) were mixed in the 1:1 ratio and incubated for 10 min on ice. Then, the samples were centrifuged for 5 min at 3,000g and 4°C in order to remove the proteins. Equal volumes of supernatant and 0.37% TBA in 0.25 M HCl containing 2% BHT were mixed, and the reaction mixture was incubated at 100°C for 10 min. After cooling and a short centrifugation (3,000g, 5 min, 20°C), the absorbance of supernatant was measured at 535 nm against the reaction blank (chemicals without tissue homogenate). The amount of TBARS in the samples was calculated from the molar absorption coefficient e = 1.56 × 105 M–1cm–1 and expressed as nanomoles of TBARS per milligram of protein.
3. Thiol group content
The content of thiol groups was measured spectrophotometrically at 412 nm according to Ellman’s method (46) and calculated as nanomoles of -SH groups per milligram of protein, using a molar extinction coefficient of 1.36 × 104 M–1cm–1.
4. Superoxide dismutase activity
We applied the Misra and Fridovich (47) assay to measure SOD activity. The rate of adrenaline autooxidation in a sample containing adrenaline alone and the rate of adrenaline oxidation in samples containing adrenaline and brain tissue homogenate were measured spectrophotometrically at 480 nm. The maximum rate of adrenaline oxidation (0.025 A/min) was taken to be 100% (0% inhibition of adrenaline oxidation). The amount of homogenate resulting in 50% inhibition of adrenaline oxidation was considered to be one unit of SOD. Five mM sodium cyanide, which inhibits copper, zinc superoxide dismutase (CuZnSOD), was used to distinguish between MnSOD and CuZnSOD activities. We calculated CuZnSOD activity by subtracting MnSOD activity from the total SOD activity. All of the enzyme activities were expressed in international units per milligram of protein.
5. Catalase activity
The spectrophotometric method of Aebi (48) was used to measure the activity of catalase. The decomposition rate of hydrogen peroxide by catalase was measured at 240 nm. The amount of brain homogenate that resulted in a decrease rate of H2O2 absorbance equal to 0.036 A/min was regarded as being one unit of catalase activity. The activity of catalase was expressed in international units per milligram of protein.
6. Protein concentration
The protein assay was carried out according to the standard Lowry method (49). After development of the color reaction, the absorbance was read at 750 nm. The protein concentration was calculated from the standard curve prepared for bovine serum albumin.
7. Western blot analysis
The procedure was carried out as described previously (50). Total proteins, isolated from the brain homogenized in ice-cold buffer (50 mM Tris, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 0.1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA), 10% glycerol, 150 mM NaCl) and containing protease inhibitors, were electrophoresed and electrotransferred to polyvinylidene fluoride (PVDF) sheets. The blots were stained with Ponceau S to determine equal loading. After blocking in 5% non-fat milk for 1 hour, the PVDF sheets were washed with Tris buffered saline with 10% Tween 20 (TBST) buffer and treated with specific antibodies against TNF-α (1:1000; Abcam), nNOS (1:1000; Acris), PARP-1 (1:1000; Sigma-Aldrich) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:3000; Sigma-Adrich) for 1.5 h. Then, the PVDF sheets were washed again with TBST, and polyclonal, rabbit-antimouse secondary antibody (1:5000; Abcam), conjugated with horseradish peroxidase, was added and incubation was continued for the next 1 hour. The washed blot was treated with enhanced chemiluminescence Western blot detection solution (Thermo Scientific) and exposed to AGFA X-ray film. The densitometric analysis was performed using Image J software, NIH, USA (51).
8. Comet assay
Brain tissue samples (about 1 mm3) were suspended in 1.5 ml of HBSS containing 100 µg/ml of collagenase and 20 µM CaCl2 (pH 7.4) and incubated for 1 h at 37°C. Then, the samples were centrifuged for 10 min at 1500g and 4°C and washed twice with PBS. The cells were suspended in PBS in order to obtain a suspension containing 1 – 2 × 105 cells/ml. The viability of the cells was determined using the trypan-blue exclusion method.
The comet assay was performed under alkaline conditions according to the procedure of Singh et al. (52). We also used the modifications of Klaude et al. (53) to identify DNA damage (single- and double-strand breaks). A suspension of cells in 0.75% LMP agarose, dissolved in PBS, was spread onto microscope slides precoated with 0.5% NMP agarose. The slides were submerged in a pre-cooled lysis solution for 1 hour at 4°C in a buffer consisting of 2.5 M NaCl, 100 mM EDTA, 1% Triton X-100, 10 mM Tris, pH 10 and then placed in an electrophoresis unit. The DNA was allowed to unwind for 20 min in the electrophoretic buffer, which consisted of 300 mM NaOH and 1 mM EDTA, pH > 13. The electrophoresis was conducted at an ambient temperature of 4°C (the temperature of the running buffer did not exceed 12°C) for 20 min at an electric field strength of 0.73 V/cm (28 mA). Then, the slides were washed in water, drained, stained with 2 µg/ml DAPI and covered with cover slips. All of the steps were conducted under dimmed light or in the dark to prevent additional DNA damage.
The comets were observed at 200 × magnification, and 50 images were randomly selected for analysis from each sample. Two parallel tests with aliquots of the same sample were performed for a total of 100 cells. The percentage of DNA in the comet tail, positively correlated with the level of DNA breakage in a cell, was considered to be an index of DNA damage in each sample.
Statistical analysis
We used the Shapiro-Wilk test to assess the normality of the data distribution. We evaluated the homogeneity of variance using Levene’s test. The statistical significance between groups was tested using ANOVA and the a posteriori Tukey-Kramer test. In the case of non-homogeneity of variance, the Kruskal-Wallis test, followed by the Conover-Inman test, was applied. ‘Nested’ ANOVA was used in the case of interactions between compounds (the impact of DOX, DTX and PTX was more important than the impact of PL). Differences were considered to be statistically significant at least at P < 0.05 (54). For the statistical evaluations, we used Statistica (StatSoft Inc., Tulsa, OK, USA) and StatsDirect software (StatsDirect Ltd., England).
RESULTS
Lipid peroxidation
We evaluated the significance of differences between the experimental groups for the primary LPO biomarkers (hydroperoxides and TBARS) on the basis of a non-parametric Kruskal-Wallis test (P < 0.0001). The post-hoc Conover-Inman test for all-pairwise comparisons was applied to validate the significance of changes between the experimental and control groups. The significance of PL inclusion in the drug treatment (differences between the drug + PL groups and the drug alone) was assessed using the Kruskal-Wallis and Conover-Inman tests.
Our results revealed that DOX, DTX and PTX could induce LPO in the rat brain, demonstrated by an increase in the amount of the hydroperoxides and TBARS (Fig. 2). Compared with the control group, DOX increased both hydroperoxides and TBARS by approximately two-fold. DTX and PTX exhibited different effects: DTX induced a two-fold increase in hydroperoxides comparable to DOX and a smaller, 1.4-fold increase in TBARS compared with the control group. PTX showed an opposite effect and caused an approximately 1.3-fold increase in hydroperoxides and a two-fold increase in TBARS (Fig. 2).
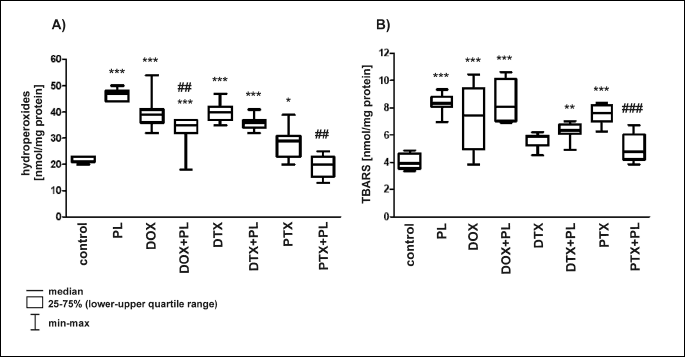
A: *P < 0.05 vs. control; ***P < 0.001 vs. control; DOX + PL vs. DOX, ##P < 0.01; PTX + PL vs. PTX, ##P < 0.01.
B: **P < 0.01 vs. control; ***P < 0.001 vs. control; PTX + PL vs. PTX, ###P < 0.001.
We observed a dual-nature activity of PL. Used alone, PL performed like a prooxidant and caused an increase in hydroperoxides and TBARS compared with the control group. Moreover, the changes were larger than those induced by DOX, DTX or PTX. In combination with two of the anticancer drugs, PL behaved as an antioxidant and ensured nearly full (PTX + PL) or partial (DOX + PL) protection of rat brains against PTX- and DOX-induced LPO. No changes in LPO products, compared with the control group, were noted in rats receiving the PTX + PL combination (Fig. 2). In the DOX + PL group, only hydroperoxides were significantly decreased compared with DOX alone, and there was an accompanying small increase in TBARS (Fig. 2). We can accordingly conclude that PL, at the administered dose, was not able to effectively protect rat brains against LPO induced by DOX and DTX.
Changes in the amount of thiol groups
We found statistically significant changes in the content of the thiol groups after treatment of rats with anticancer drugs, which caused a decrease (DOX, DTX) or approximately a 1.5-fold increase (PTX) in their amount (Fig. 3). PL by itself did not induce significant changes. The addition of PL to the PTX treatment moderately increased the level of -SH groups compared with PTX alone. PL acted synergistically with DTX and enhanced the effect of the drug, causing an additional 25% decrease in the thiol pool (Fig. 3). When PL was applied with DOX, we noted significant restoration of the thiols to the level of the control group.
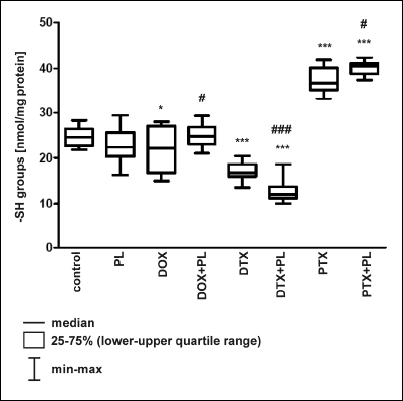 |
Fig. 3. The effect of Pirolin (PL) on the amount of thiol groups (-SH) in the brain of rats injected i.p. with doxorubicin (DOX), docetaxel (DTX) and paclitaxel (PTX). The content of -SH groups was measured spectrophotometrically using the Ellman’s method and expressed in nanomoles of -SH groups per milligram of protein. The results are presented as a median, lower-upper quartile range (25 – 75%) and min-max range. Kruskal-Wallis test (P < 0.0001) showed that there was a statistically significant difference in -SH groups level between the different treatments. The post-hoc, all-pairwise comparisons Conover-Inman test was used to determine which group(s) differed from the controls with the probability denoted on the graph (*P < 0.05, ***P < 0.001). The significance of PL administration (the drug + PL vs. the drug alone) was also checked using the Kruskal-Wallis and Conover-Inman tests; only those cases where the impact of PL was significant are shown below and are denoted on the graph: *P < 0.05 vs. control; ***P < 0.001 vs. control; DOX + PL vs. DOX, #P < 0.05; PTX + PL vs. PTX, #P < 0.05; DTX + PL vs. DTX, ###P < 0.001. |
The significance of changes was statistically evaluated, as described in the legend of Fig. 3.
Changes in the activity of the antioxidant enzymes
The significance of changes in the activities of SOD, MnSOD, CuZnSOD and CAT was statistically evaluated, as described in the legend of Fig. 4.
1. SOD, MnSOD and CuZnSOD activity
Treatment with any of the anticancer drugs caused a significant decrease in the activities of total SOD and CuZnSOD. DOX and DTX resulted in the most profound changes: about a 40% decrease in the total activity of SOD and more than a two-fold decrease in the activity of CuZnSOD. PTX induced about a 10% decrease in total SOD activity and about a 40% decrease in CuZnSOD activity (Fig. 4A and 4C). It is interesting that DOX and DTX, which compared with PTX caused considerably larger changes in the total activity of SOD and CuZnSOD, did not influence significantly the activity of MnSOD. In turn, PTX caused more than a two-fold increase in the activity of MnSOD (Fig. 4B).
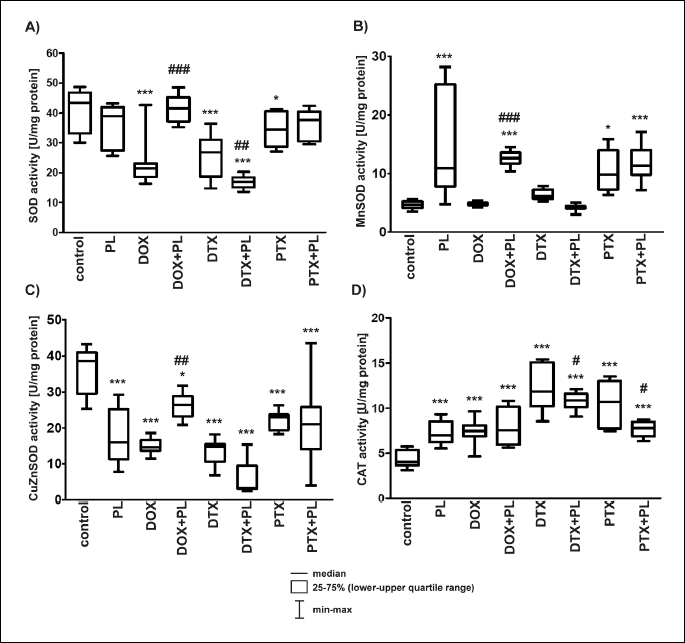
A: (SOD) *P < 0.05 vs. control, ***P < 0.001 vs. control; DTX + PL vs. DTX, ##P < 0.01; DOX + PL vs. DOX, ###P < 0.001.
B: (MnSOD) *P < 0.05 vs. control, ***P < 0.001 vs. control; DOX + PL vs. DOX, ###P < 0.001.
C: (CuZnSOD) *P < 0.05 vs. control, ***P < 0.001 vs. control, DOX + PL vs. DOX, ##P < 0.01.
D: (CAT) ***P < 0.001 vs. control, DTX + PL vs. DTX, #P < 0.05, PTX + PL vs. PTX, #P < 0.05.
Pirolin used alone did not affect significantly the total SOD, but it increased by more than three-fold the activity of MnSOD and decreased by about two-fold the activity of CuZnSOD (Fig. 4A, 4B and 4C). The performance of PL differed when it was used in combination with anticancer drugs. PL fully (total SOD) or partially (CuZnSOD) restored the activity of SOD enzymes impaired by DOX. Compared with DOX and the control group, more than a two-fold increase in the activity of MnSOD was observed after treatment with DOX + PL. PL acted synergistically with DTX and enhanced its effects, which resulted in an additional 1.5-fold decrease in the total activity of SOD and a 2.5-fold decrease in the activity of CuZnSOD compared with DTX alone. It should be noted that MnSOD activity in the DTX + PL group was comparable to that of the control group (Fig. 4B). No significant differences in the activities of SOD enzymes between the PTX + PL and PTX groups were found (Fig. 4A, 4B and 4C).
2. Catalase activity
In contrast with the various effects of DOX, DTX, PTX and PL on SOD activity, all of these compounds significantly increased CAT activity compared with the control group (Fig. 4D). Taxanes, particularly DTX, induced the largest changes.
Pirolin moderately modified the effects of DTX and PTX. We observed a decrease in the CAT activity for DTX + PL and PTX + PL combinations compared with DTX or PTX alone. The enzyme activity remained, however, higher than in the control group. No significant differences were found between the rats treated with the DOX + PL combination and the rats treated with DOX alone (Fig. 4D).
Expression of neuronal nitric oxide synthase
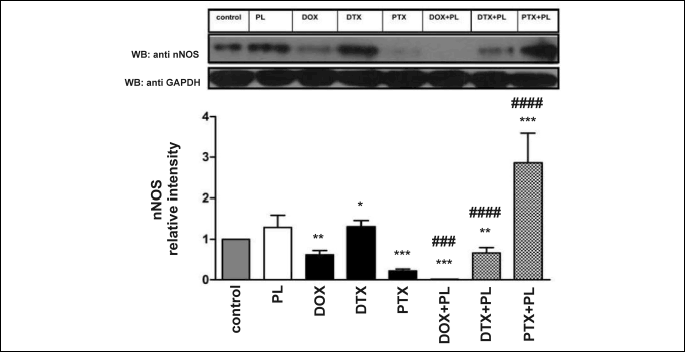
Doxorubicin, DTX, PTX and PL influenced nNOS expression in different ways (Fig. 5). DOX and PTX suppressed enzyme expression (PTX to a considerably larger extent), and DTX resulted in an increase in enzyme expression. The administration of PL alone induced an insignificant increase in nNOS expression, but PL used in combination with DOX or DTX suppressed nNOS expression. We observed an approximately 8-fold decrease in enzyme expression in the DOX + PL group compared with the group injected with DOX alone. The expression of nNOS increased more than 10-fold after PTX + PL treatment compared with the effect of PTX alone.
The statistical significance of changes in nNOS expression was evaluated, as described in the legend of Fig. 5.
Tumor necrosis factor-α expression
Pirolin, DTX and PTX alone suppressed the expression of TNF-α in the rat brain. DOX, in contrast, induced its overexpression (Fig. 6). The addition of PL to the DOX treatment did not result in any significant changes compared with the effects observed for DOX alone. We did note, however, a large increase in TNF-α expression after treatment with combinations of PL with taxanes (more profound in the case of PTX), compared with DTX or PTX alone.
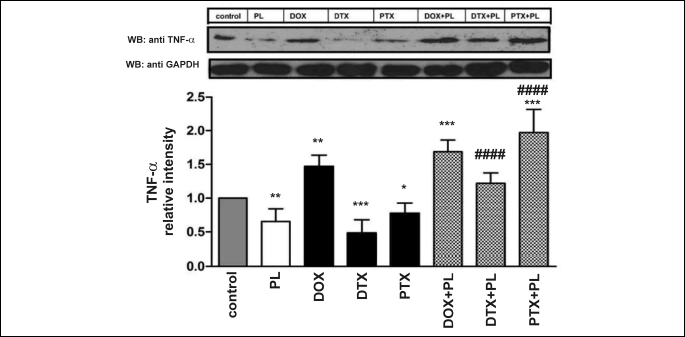
The statistical significance of changes between the experimental groups was evaluated using an ANOVA test (P = 0.0013). We used the post-hoc, all-pairwise comparisons Tukey test to assess the significance of changes between the results obtained for experimental groups and the control group. We checked the significance of PL administration (comparison of drug + PL versus drug) using a nested ANOVA test.
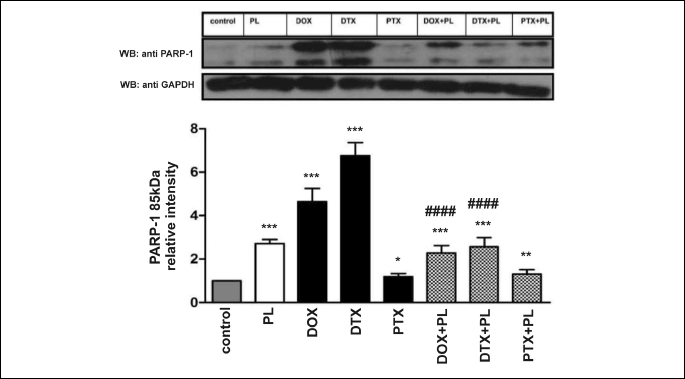
Poly (ADP-ribose) polymerase-1
Extensive PARP-1 (85 kDa) cleavage was observed after treatment with DOX or DTX, which indicates apoptotic changes. The most pronounced increase (i.e., approximately 7-fold) in the cleaved form of PARP-1 was noted after DTX administration. PTX, in contrast to DTX, exhibited a smaller effect. PL induced an increase in PARP-1 cleavage that was smaller than the effects associated with DOX or DTX but larger than those triggered by PTX.
Pirolin, used concurrently with DOX and DTX, attenuated apoptotic changes caused by these two drugs, and it suppressed an increase in PARP-1 expression and significantly inhibited the formation of its cleaved form compared with DOX or DTX alone. At the same time, we did not observe any effects of PL on changes induced by PTX (Fig. 7).
The same statistical tests as in the case of the TNF-α expression data were used to assess the significance of the results for PARP-1 (Fig. 7).
Comet assay
Docetaxel was the most genotoxic and resulted in the largest DNA damage, expressed as a 3.5-fold increase in the tail DNA (Fig. 8). DOX and PTX caused less DNA damage, and PL did not show any significant effect on DNA. The addition of PL to the chemotherapy protocol fully protected DNA from damage induced by PTX, attenuated changes caused by DOX, but did not alter the effect of DTX.
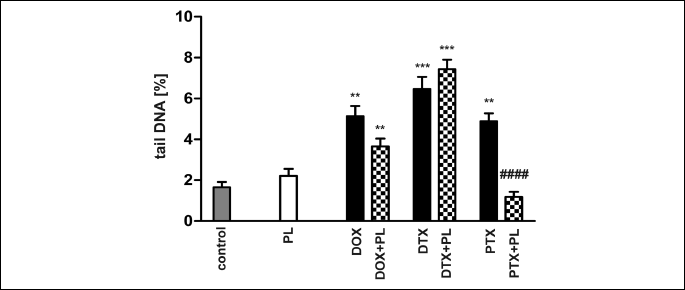
The statistical evaluation of the results was performed, as described in the legend of Fig. 8.
DISCUSSION
Doxorubicin, DTX and PTX induced OS in the CNS, increasing LPO, CAT activity and DNA damage. They affected the -SH group level and SOD activity, as well as TNF-α, nNOS and PARP-1 expression.
Pirolin exhibited different effects when administered alone or in combination with anticancer drugs. It disturbed some OS biomarkers, enhanced the prooxidative DTX effect, but decreased DOX-induced oxidative damage and protected DNA against DOX- and PTX-induced damage. PL enhanced the DOX-induced decrease of nNOS expression and reversed the effects of DTX and PTX on nNOS and TNF-α expression. PL also reduced PARP-1 cleavage evoked by DOX and DTX.
Oxidative damage generated by doxorubicin and taxanes in the rat brain
Doxorubicin induced an increase in LPO in rat brains, consistent with the reported increase in 4-HNE in the brain of DOX-treated mice (14). PTX induced more extensive LPO than DTX, consistent with the various effects of PTX and DTX on rat hearts, where PTX happened to be more cardiotoxic (55). The differences in the effects of DTX and PTX might arise from their diverse pharmacokinetics and pharmacodynamics, which necessitates additional studies.
Both taxanes increased MnSOD activity, which might be related to their influence on the MnSOD expression (56). DOX and taxanes increased CAT activity, consistent with the reported generation of H2O2 (57, 58). H2O2 forms a hydroxyl radical binding to the active site of the enzyme (59), which can reduce CuZnSOD activity, as observed in our study.
Since the crucial role of ROS and 4-HNE in BBB impairment and its permeabilization during OS has been demonstrated (13), we suggest that LPO, DNA damage and PARP cleavage in the brain may be due to the detrimental actions of DOX, DTX and PTX on the blood vessel endothelium and the permeation of their small amounts via an impaired BBB. Moreover, the depletion of GSH (e.g., as a result of the transport of GSH-4-HNE conjugates (31)) might enhance the impairment of the BBB integrity. An increased LPO, demonstrated as an elevated level of MDA, has been found by Sakr et al., (60) in the brain cortex of rats subjected to hypoxic conditions and has been suggested by these authors as related to a decreased level of GSH and a decreased activity of the main antioxidant enzymes CAT and SOD. Under the same conditions, these parameters were markedly improved by vitamin E, well known antioxidant which has been shown to protect cell membranes rich in lipids, such as those in the brain cells, against damage induced by ROS.
In our study, DOX and DTX decreased the amounts of -SH groups and induced extensive LPO, which could contribute to increased BBB permeability. PTX evoked LPO but simultaneously raised the thiol level, which might contribute to its reduced permeation through the BBB compared with that of DOX or DTX.
Based on earlier studies (61), we suggest that the genotoxic effects of DOX, PTX and DTX relate to the OS generated by these drugs. The differences in DTX and PTX performance imply potentially distinct mechanisms of their proapoptotic activity in the CNS. DTX and PTX affected DNA integrity differently, which might be attributed to differences in the transport of even small amounts of these drugs across the BBB in the conditions of extensive OS. PTX is a better substrate for p-glycoprotein than DTX, which can result in a lesser intracellular accumulation of PTX (62).
More DNA oxidative damage caused by DTX and particularly PARP-1 cleavage triggered by DOX and DTX, could be attributed to increased BBB permeability resulting from -SH depletion. DOX and DTX caused both PARP-1 cleavage and substantial decreases in -SH groups, and PTX caused an increase in thiols, which corresponds well with the lesser PARP-1 cleavage.
The dose-dependent PARP-1 cleavage accompanied by a significant increase in DNA damage measured by comet assay has been demonstrated in HT29 human colon cancer cells treated with novel synthetic pyrazoles belonging to DNA-intercalating drugs: tospyrquin and tosind (63). These changes were suggested to be associated with an activation of pro-apoptotic effector caspase-3.
Changes in tumor necrosis factor-α and neuronal nitric oxide synthase expression induced by doxorubicin, docetaxel and paclitaxel
TNF-α, a possible mediator of indirect outcomes of action of anticancer drugs (e.g., LPO in the brain (64)), passes the BBB (65) and elicits the local TNF-α production (66). We noted the overexpression of TNF-α in the brain of DOX-treated rats, suggesting that it might mediate OS generated by DOX. PTX and DTX decreased TNF-α expression. DTX evoked a more pronounced effect, consistent with results (67) of patients with advanced breast cancer. Decreased TNF-α expression rather excludes TNF-α involvement in the OS induction by PTX and DTX in the CNS.
The effect of DOX on nNOS expression in the CNS remains elusive. In our study, a single dose of 10 mg DOX/kg b.w. reduced almost twice nNOS expression, which is contradictory to a report demonstrating that 1 mg of DOX/kg b.w., injected for 12 weeks, did not alter nNOS expression in rat hearts (68). In rat neurons, nNOS takes part in the redox cycling of DOX (17), however, DOX and ROS can also inhibit nNOS activity (69).
No information about the influence of taxanes on nNOS in the brain is available in the literature. Here, we report for the first time that PTX and DTX influence nNOS expression in the rat brain. PTX suppressed nNOS expression to a greater degree than DOX, and DTX induced a slight increase. This finding is important and novel since nNOS inhibition or overactivation may disorder physiological functions and contribute to chemotherapy-induced side-effects.
Does Pirolin protect rat brains from oxidative damage induced by anticancer drugs?
Used alone, PL induced a slight decrease in -SH groups, consistent with earlier studies in which PL, compared with other nitroxides, was reduced more slowly in cells and resulted in less GSH loss (70).
Increased LPO after PL treatment might be due to its SOD-mimic properties and the production of H2O2 (71). Nitroxide performance depends on the presence of other oxidants/reductants (72). In oxygenated conditions, nitroxides have exhibited slower reduction (73). In brains with a good blood flow, PL might undergo slower reduction than in less-oxygenated tissues and perform longer as a SOD-mimetic. Since nitroxides pass the BBB (34) and enter the mitochondria (74), PL possibly might increase CAT, MnSOD activities and LPO, driven by H2O2. The effect of PL on MnSOD activity might be indirect via its influence on protein expression. PL reduced TNF-α level, which excludes TNF-α involvement in PL action. Another nitroxide, Tempol, reduced TNF-α levels in rat plasma and prevented mitochondrial OS and cardiac dysfunction (75). In another study on rat closed head injury, Tempol attenuated TNF-α toxicity (76).
Nitroxides can also display prooxidative activity, producing H2O2, which oxidizes GSH (71). At high concentrations, as reducing agents during the detoxification of ROS (e.g., hydroxyl radicals), nitroxides could form prooxidant oxoammonium cations (77).
It has been shown that LPO markers should not be solely used to describe OS since they may also increase under different pathological conditions and physiological states (78). It has been shown that endurance physical activity can increase the level of TBARS in the cerebellum of adolescent rats (79). Furthermore, different effects on the activity of antioxidant enzymes and the level of total glutathione were observed depending on the brain region. For instance, the highest increase in the activity of CAT and elevated level of total glutathione were found in the cerebellum. These changes, however, were insufficient to effectively protect this brain region from an increased amount of TBARS (79). Similarly, anticancer drugs probably could influence various brain regions to a different extent. Thus, based on the results of the present work, we can suggest that further studies are needed to elucidate, if PL is able to efficiently protect particular brain regions against CI induced by anticancer drug-treatment.
In this study, PL displayed dual activity: it influenced some OS markers unfavorably, but it decreased TNF-α levels and did not damage DNA despite increased PARP-1 cleavage. Therefore, the overall effect of PL on brain tissue was not harmful.
Pirolin injected with DOX decreased the level of hydroperoxides, consistent with studies demonstrating protective PL activity toward the damage of plasma membrane lipids evoked by anthracyclines (36). Restored SOD activity in the brains of rats receiving DOX in combination with PL was mainly due to an increase in MnSOD activity. This effect indicates that PL strongly influences mitochondria and suggests the intensification of SOD-mimic activity of PL in the presence of DOX and partial participation of PL in overall SOD activity. PL, combined with DTX, augmented OS. Apparently, DTX caused changes in redox conditions, which triggered the prooxidative action of PL.
Pirolin enhancement of DOX-induced inhibition of nNOS expression might have partially resulted from enzyme inhibition due to exposure to H2O2 (80), produced via intensified SOD activity. The opposite effect was observed for the combination of PL with DTX: PL reversed the effect of DTX, and PL with PTX increased nNOS expression. This finding, however, necessitates verification in future studies.
Pirolin attenuated the proapoptotic effects of DOX and DTX by a significant decrease in PARP-1 cleavage, which might be beneficial. Oxidative activation of PARP-1 by DOX accounts for DOX cardiotoxicity (81). Moreover, overactivation of PARP-1 causes cell death. Therefore, the attenuation of PARP-1 expression and cleavage by PL might prevent apoptosis of the brain cells triggered by DOX and taxanes. PL protected rat brains against DOX toxicity and demonstrated an effect similar to PARP-1 inhibitors. Previously, inhibitory activity had been confirmed for the nitroxide-conjugated derivatives of PARP-1 inhibitors (82).
Pirolin also effectively reduced DNA damage caused by DOX and PTX. PTX, administered with PL, moderately increased PARP-1 expression with no enhancement of its cleavage. Mild expression of PARP enhances DNA repair, which corresponds to a decrease in the comet tail DNA in the PTX + PL group compared with PTX alone. These results indicate that PL can be protective toward DOX- and taxane-induced DNA damage. We suggest that PL does not act as a PARP-1 inhibitor but rather as an antioxidant, resulting in decreased DNA damage and PARP-1 expression. Since caspase-3 produces an 85 kDa PARP-1 fragment, we assume that PL used with DOX and taxanes exhibits antiapoptotic activity in the brain via PARP-1 cleavage reduction.
The doses of anticancer drugs used in our study, comparable to clinically effective doses used in chemotherapy of human breast cancer, induced systemic toxicities. In these conditions, PL shielded against DOX cardiotoxicity (37) and protected partially rat blood plasma against oxidative damage caused by DOX + DTX chemotherapy (43). Therefore, the effects of PL alone appear to be less important than its influence on the toxicity of anticancer drugs. This finding highlights the potential usefulness of adding PL to DOX and taxane chemotherapy rather than using PL by itself.
Conclusions
We have demonstrated that DOX, DTX and PTX induce OS and damage to rat brain tissue. Our study confirmed the complex dual performance of PL, which depends on the properties of oxidants in its environment. Our work has also highlighted the possibility of applying pyrroline nitroxides as new CNS cytoprotectors against DOX-, DTX- and PTX-induced OS.
Acknowledgements: The authors thank Mrs. Anna Selmi, MSc, and Mr. Maciej Konka, MSc for excellent technical assistance in some of the experiments.
Financial support for this work was provided by Grant N 401 2337 33 of the Ministry of Science and Higher Education (Poland) and, in part, by grant no. 506/980 of the University of Lodz, Poland.
Conflict of interest: None declared.
REFERENCES
- Mielke S, Sparreboom A, Mross K. Peripheral neuropathy: a persisting challenge in paclitaxel-based regimes. Eur J Cancer 2006; 42: 24-30.
- Chen Y, Jungsuwadee P, Vore M, Butterfield DA, St. Clair DK. Collateral damage in cancer chemotherapy: oxidative stress in nontargeted tissues. Mol Interv 2007; 7: 147-156.
- Mir O, Alexandre J, Tran A, et al. Relationship between GSTP1 Ile(105)Val polymorphism and docetaxel-induced peripheral neuropathy: clinical evidence of a role of oxidative stress in taxane toxicity. Ann Oncol 2009; 20: 736-740.
- Ahles TA, Saykin AJ, Furstenberg CT, et al. Neuropsychologic impact of standard-dose systemic chemotherapy in long-term survivors of breast cancer and lymphoma. J Clin Oncol 2002; 20: 485-493.
- Gaman AM, Uzoni A, Popa-Wagner A, Andrei A, Petcu EB. The role of oxidative stress in etiopathogenesis of chemotherapy induced cognitive impairment (CICI)-“chemobrain”. Aging Dis 2016; 7: 307-317.
- Joshi G, Aluise CD, Cole MP, et al. Alterations in brain antioxidant enzymes and redox proteomic identification of oxidized brain proteins induced by the anti-cancer drug adriamycin: implications for oxidative stress-mediated chemobrain. Neuroscience 2010; 166: 796-807.
- Wu YQ, Dang RL, Tang MM, et al. Long chain omega-3 polyunsaturated fatty acid supplementation alleviates doxorubicin-induced depressive-like behaviors and neurotoxicity in rats: involvement of oxidative stress and neuroinflammation. Nutrients 2016; 8: 243. doi:10.3390/nu8040243
- Wefel JS, Saleeba AK, Buzdar AU, Meyers CA. Acute and late onset cognitive dysfunction associated with chemotherapy in women with breast cancer. Cancer 2010; 116: 3348-3356.
- Fardell JE, Zhang J, De Souza R, et al. The impact of sustained and intermittent docetaxel chemotherapy regimens on cognition and neural morphology in healthy mice. Psychopharmacology 2014; 231: 841-852.
- Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don’t divide what’s to repair? Mutat Res 2007; 614: 24-36.
- Christie LA, Acharya MM, Parihar VK, Nguyen A, Martirosian V, Limoli CL. Impaired cognitive function and hippocampal neurogenesis following cancer chemotherapy. Clin Cancer Res 2012; 18: 1954-1965.
- Li P, Albrecht BJ, Yan X, Gao M, Weng HR, Bartlett MG. A rapid analytical method for the quantification of paclitaxel in rat plasma and brain tissue by high-performance liquid chromatography and tandem mass spectrometry. Rapid Commun Mass Spectrom 2013; 27: 2127-2134.
- Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res 2009; 43: 348-364.
- Joshi G, Sultana R, Tangpong J, et al. Free radical mediated oxidative stress and toxic side effects in brain induced by the anti cancer drug adriamycin: insight into chemobrain. Free Radic Res 2005; 39: 1147-1154.
- Manjanatha MG, Bishop ME, Pearce MG, Kulkarni R, Lyn-Cook LE, Ding W. Genotoxicity of doxorubicin in F344 rats by combining the comet assay, flow-cytometric peripheral blood micronucleus test, and pathway-focused gene expression profiling. Environ Mol Mutagen 2014; 55: 24-34.
- Satoh T, McKercher SR, Lipton SA. Nrf2/ARE-mediated antioxidant actions of pro-electrophilic drugs. Free Radic Biol Med 2013; 65: 645-657.
- Lopes MA, Meisel A, Carvalho FD, Bastos M de L. Neuronal nitric oxide synthase is a key factor in doxorubicin-induced toxicity to rat-isolated cortical neurons. Neurotox Res 2011; 19: 14-22.
- Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol 2004; 37: 139-143.
- Ma Q. Transcriptional responses to oxidative stress: Pathological and toxicological implications. Pharmacol Ther 2010; 125: 376-393.
- Kops GJ, Dansen TB, Polderman PE, et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 2002; 419: 316-321.
- Kraft AD, McPherson CA, Harry GJ. Heterogeneity of microglia and TNF signaling as determinants for neuronal death or survival. NeuroToxicology 2009; 30: 785-793.
- Park KM, Bowers WJ. Tumor necrosis factor-αlpha mediated signaling in neuronal homeostasis and dysfunction. Cell Signal 2010; 22: 977-983.
- Aluise CD, Miriyala S, Noel T, et al. 2-Mercaptoethane sulfonate prevents doxorubicin-induced plasma protein oxidation and TNF-α release: implications for the reactive oxygen species-mediated mechanisms of chemobrain. Free Radic Biol Med 2011; 50: 1630-1638.
- Khan SA, Lee K, Minhas KM, et al. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation–contraction coupling. Proc Natl Acad Sci USA 2004; 101: 15944-15948.
- Jin CZ, Jang JH, Wang Y, et al. Neuronal nitric oxide synthase is up-regulated by angiotensin II and attenuates NADPH oxidase activity and facilitates relaxation in murine left ventricular myocytes. J Mol Cell Cardiol 2012; 52: 1274-1281.
- Zhang YH, Casadei B. Sub-cellular targeting of constitutive NOS in health and disease. J Mol Cell Cardiol 2012; 52: 341-350.
- Bustamante J, Czerniczyniec A, Lores-Arnaiz S. Brain nitric oxide synthases and mitochondrial function. Front Biosci 2007; 12: 1034-1040.
- D’Amours D, Sallmann FR, Dixit VM, Poirier GG. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J Cell Sci 2001; 114: 3771-3778.
- Tangpong J, Miriyala S, Noel T, Sinthupibulyakit C, Jungsuwadee P, St Clair DK. Doxorubicin-induced central nervous system toxicity and protection by xanthone derivative of Garcinia mangostana. Neuroscience 2011; 175: 292-299.
- Ong WY, Lu XR, Hu CY, Halliwell B. Distribution of hydroxynonenal-modified proteins in the kainate-lesioned rat hippocampus: evidence that hydroxynonenal formation precedes neuronal cell death. Free Radic Biol Med 2000; 28: 1214-1221.
- Mertsch K, Blasig I, Grune T. M4-Hydroxynonenal impairs the permeability of an in vitro rat blood-brain barrier. Neurosci Lett 2001; 314: 135-138.
- Miriyala S, Thippakorn C, Chaiswing L, et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic Biol Med 2016; 91: 68-80.
- Hayslip J, Dressler EV, Weiss H, et al. Plasma TNF-α and soluble TNF receptor levels after doxorubicin with or without co-administration of Mesna - a randomized, cross-over clinical study. PLoS One 2015; 10: e0124988. doi:10.1371/journal.pone.0124988
- Shen J, Liu S, Miyake M, et al. Use of 3-acetoxymethoxycarbonyl-2,2,5,5-tetramethyl-1-pyrrolidiny-loxyl as an EPR oximetry probe: potential for in vivo measurement of tissue oxygenation in mouse brain. Magn Reson Med 2006; 55: 1433-1440.
- Soule BP, Hyodo F, Matsumoto K, et al. The chemistry and biology of nitroxide compounds. Free Radic Biol Med 2007; 42: 1632-1650.
- Koceva-Chyla A, Sokal A, Kania K, Gwozdzinski K, Jozwiak Z. The nitroxides pirolin and pirolid protect the plasma membranes of rat cardiomyocytes against damage induced by anthracyclines. Cell Mol Biol Lett 2003; 8: 171-177.
- Koceva-Chyla A, Gwozdzinski K, Kochman A, Stolarska A, Jozwiak Z. Effects of pyrroline and pyrrolidine nitroxides on lipid peroxidation in heart tissue of rats treated with doxorubicin. Cell Mol Biol Lett 2003; 8: 179-183.
- Kato N, Yanaka K, Hyodo K, Homma K, Nagase S, Nose T. Stable nitroxide Tempol ameliorates brain injury by inhibiting lipid peroxidation in a rat model of transient focal cerebral ischemia. Brain Res 2003; 979: 188-193.
- Zhang R, Shohami E, Beit-Yannai E, Bass R, Trembovler V, Samuni A. Mechanism of brain protection by nitroxide radicals in experimental model of closed-head injury. Free Radic Biol Med 1998; 24: 332-340.
- Rozantsev EG. Svobodnyje Iminoksylnyje Radicaly, Chimia. [in Russian] Moscow 1970.
- Misset JL, Dieras V, Gruia G, et al. Dose-finding study of docetaxel and doxorubicin in first-line treatment of patients with metastatic breast cancer. Ann Oncol 1999; 10: 553-560.
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J 2008; 22: 659-661.
- Tabaczar S, Koceva-Chyla A, Czepas J, Pieniazek A, Piasecka-Zelga J, Gwozdzinski K. Nitroxide pirolin reduces oxidative stress generated by doxorubicin and docetaxel in blood plasma of rats bearing mammary tumor. J Physiol Pharmacol 2012; 63: 153-163.
- Gay C, Gebicki JM. A critical evaluation of the effect of sorbitol on the ferric-xylenol orange hydroperoxide assay. Anal Biochem 2000; 284: 217-220.
- Rice-Evans CA, Diplock AT, Symons MC. Techniques in free radicals research. In: Laboratory Techniques in Biochemistry and Molecular Biology, RH Burdon, PH van Knippenberg (eds). Amsterdam, London, New York, Tokyo. Elsevier, 1991.
- Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys 1959; 82: 70-77.
- Misra HP, Fridovich I. Superoxide dismutase and peroxidase: a positive activity stain applicable to polyacrylamide gel electropherograms. Arch Biochem Biophys 1977; 183: 511-515.
- Aebi H. Catalase in vitro. Methods Enzymol 1984; 105: 121-126.
- Lowry OJ, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem 1951; 193: 265-275.
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227: 680-685.
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012; 9: 671-675.
- Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 1988; 175: 184-191.
- Klaude M, Eriksson S, Nygren J, Ahnstrom G. The comet assay mechanisms and technical considerations. Mutat Res 1996; 363: 89-96.
- Zar JH. Biostatistical Analysis. New York, Prentice-Hall, Englewood Cliffs, 1984.
- Minotti G, Saponiero A, Licata S, et al. Paclitaxel and docetaxel enhance the metabolism of doxorubicin to toxic species in human myocardium. Clin Cancer Res 2001; 7: 1511-1515.
- Das KC, Guo X, White CW. Protein kinase Cdelta-dependent induction of manganese superoxide dismutase gene expression by microtubule-active anti-cancer drugs. J Biol Chem 1998; 273: 34639-34645.
- Alexandre J, Batteux F, Nicco C, et al. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int J Cancer 2006; 119: 41-48.
- Rabi T, Bishayee A. d-Limonene sensitizes docetaxel-induced cytotoxicity in human prostate cancer cells: generation of reactive oxygen species and induction of apoptosis. J Carcinog 2009; 8: 9. doi: 10.4103/1477-3163.51368
- Zhang H, Andrekopoulos C, Joseph J, Crow J, Kalyanaraman B. The carbonate radical anion-induced covalent aggregation of human copper, zinc superoxide dismutase, and alpha-synuclein: intermediacy of tryptophan- and tyrosine-derived oxidation products. Free Radic Biol Med 2004; 36: 1355-1365.
- Sakr HF, Abbas AM, El Samanoudy AZ. Effect of vitamin E on cerebral cortical oxidative stress and brain-derived neurotrophic factor gene expression induced by hypoxia and exercise in rats. J Physiol Pharmacol 2015; 66: 191-202.
- Fabbri F, Carloni S, Brigliadori G, Zoli W, Lapalombella R, Marini M. Sequential events of apoptosis involving docetaxel, a microtubule-interfering agent: a cytometric study. BMC Cell Biol 2006; 7: 6.
- Kemper EM, Verheij M, Boogerd W, Beijnen JH, van Tellingen O. Improved penetration of docetaxel into the brain by co-administration of inhibitors of P-glycoprotein. Eur J Cancer 2004; 40: 1269-1274.
- Toton E, Ignatowicz E, Bernard MK, Kujawski J, Rybczynska M. Evaluation of apoptotic activity of new condensed pyrazole derivatives. J Physiol Pharmacol 2013; 64: 115-123.
- Tangpong J, Cole MP, Sultana R, et al. Adriamycin-induced, TNF-α mediated central nervous system toxicity. Neurobiol Dis 2006; 23: 127-139.
- Osburg B, Peiser C, Domling D, et al. Effect of endotoxin on expression of TNF receptors and transport of TNF-αlpha at the blood-brain barrier of the rat. Am J Physiol Endocrinol Metab 2002; 283: E899-E908.
- Szelenyi J. Cytokines and the central nervous system. Brain Res Bull 2001; 54: 329-338.
- Tsavaris N, Kosmas C, Vadiaka M, Kanelopoulos P, Boulamatsis D. Immune changes in patients with advanced breast cancer undergoing chemotherapy with taxanes. Br J Cancer 2002; 87: 21-27.
- Liu B, Li H, Qu H, Sun B. Nitric oxide synthase expressions in ADR-induced cardiomyopathy in rats. J Biochem Mol Biol 2006; 39: 759-765.
- Luo D, Vincent SR. Inhibition of nitric oxide synthase by antineoplastic anthracyclines. Biochem Pharmacol 1994; 47: 2111-2112.
- Bujak S, Gwozdzinski K. Nitroxides lead to reduced level of glutathione in red blood cells. In: Free Radicals and Oxidative Stress: Chemistry, Biochemistry and Pathophysiological Implications, G Galaris (ed). Medimond International Proceedings 2003, pp. 105-108.
- Krishna MC, Russo A, Mitchell JB, Goldstein S, Dafni H, Samuni A. Do nitroxide antioxidants act as scavengers of O2·– or as SOD mimics? J Biol Chem 1996; 271: 26026-26031.
- Aronovitch Y, Godinger D, Israeli A, Krishna MC, Samuni A, Goldstein S. Dual activity of nitroxides as pro- and antioxidants: catalysis of copper-mediated DNA breakage and H2O2 dismutation. Free Radic Biol Med 2007; 42: 1317-1325.
- Glebska J, Gwozdzinski K. Oxygen-dependent reduction of nitroxides by ascorbic acid and glutathione. EPR investigations. Curr Top Biophys 1998; 22 (Suppl. B): 75-82.
- Monti E, Supino R, Colleoni M, Costa B, Ravizza R, Gariboldi MB. Nitroxide TEMPOL impairs mitochondrial function and induces apoptosis in HL60 cells. J Cell Biochem 2001; 82: 271-276.
- Mariappan N, Soorappan RN, Haque M, Sriramula S, Francis J. TNF-α-induced mitochondrial oxidative stress and cardiac dysfunction: restoration by superoxide dismutase mimetic Tempol. Am J Physiol Heart Circ Physiol 2007; 293: H2726-2737.
- Trembovler V, Beit-Yannai E, Younis F, Gallily R, Horowitz M, Shohami E. Antioxidants attenuate acute toxicity of tumor necrosis factor-αlpha induced by brain injury in rat. J Interferon Cytokine Res 1999; 19: 791-795.
- Offer T, Russo A, Samuni A. The pro-oxidative activity of SOD and nitroxide SOD mimics. FASEB J 2000; 14: 1215-1223.
- Dotan Y, Lichtenberg D, Pinchuk I. Lipid peroxidation cannot be used as a universal criterion of oxidative stress. Prog Lipid Res 2004; 43: 200-227.
- Chalimoniuk M, Jagsz S, Sadowska-Krepa E, Chrapusta SJ, Klapcinska B, Langfort J. Diversity of endurance training effects on antioxidant defenses and oxidative damage in different brain regions of adolescent male rats. J Physiol Pharmacol 2015; 66: 539-547.
- Kar R, Kellogg DL III, Roman LJ. Oxidative stress induces phosphorylation of neuronal NOS in cardiomyocytes through AMP-activated protein kinase (AMPK). Biochem Biophys Res Commun 2015; 459: 393-397.
- Pacher P, Liaudet L, Bai P, et al. Activation of poly(ADP-ribose) polymerase contributes to development of doxorubicin-induced heart failure. J Pharmacol Exp Ther 2002; 300: 862-867.
- Kalai T, Balog M, Szabo A, et al. New poly(ADP-ribose) polymerase-1 inhibitors with antioxidant activity based on 4-carboxamidobenzimidazole-2-ylpyrroline and -tetrahydropyridine nitroxides and their precursors. J Med Chem 2009; 52: 1619-1629.
A c c e p t e d : April 3, 2017