NON-ISCHEMIC HEART PRECONDITIONING
INTRODUCTION
Collateral circulation in the coronary arterial bed is very poor. In most cases, it is not able to provide blood to the area of the myocardium supplied by an occluded artery. As a result, blockade of blood flow in the coronary artery results in necrosis of the myocardium down below the occlusion. It is, therefore, obvious that early reconstitution of the blood flow is necessary to maintain the ischemic portion of the myocardium vivid. However, restoration of the blood flow in the ischemic area produces a complex of dangerous consequences, including exacerbation of cardiomyocyte injury and worsening of the heart condition manifested by an increase in the infarct size, impaired contractility, ventricular arrhythmias, microvascular dysfunction, myocyte necrosis, impaired healing of the infarct area, developing heart failure and other symptoms (1, 2). The complex of symptoms is called ‘ischemia/reperfusion (I/R) injury’ (1). Therefore, it is understandable that much effort has been put to developing means to prevent/reduce these changes. The pharmacological heart conditioning was used with no fully satisfactory results (3-5). A new opening was brought out by Murry et al. (6) in experiments performed on dogs. In one group of the animals, 4 cycles of 5 min occlusion of the left circumflex coronary artery separated by 5 min reperfusion were applied. Next, the vessel was occluded either for 40 min or for 3 hours. In the control group, 40 min and 3 h occlusions were also conducted but without the proceeding procedure of 5 min occlusions/reperfusions. Four days later, the infarct size was measured in each group. The pre-infarction procedure reduced the infarct size after 40 min ischemia to only 25% of that seen in the respective control group. It did not affect the infarct size after 3 h occlusion of the artery, as compared to the group not receiving the procedure. The researchers called the phenomenon seen in the first group ‘preconditioning with ischemia’. Now, the procedure is usually referred to as ‘ischemic preconditioning’. In the next years much work has been done to elucidate the phenomenon and its clinical application. It can be induced before I/R (ischemic perconditioning), during evolving infarction (ischemic preconditioning) or after infarction at the onset of reperfusion (ischemic postconditioning) (7, 8). It was further established that the same procedure applied not only to another coronary artery of the same heart (9, 10) but also to distant arteries in other organs induces heart preconditioning (11, 12). The latter procedure was called remote heart preconditioning (12, 13). It can be easily applied in a non-invasive way, e.g. using a blood pressure cuff on an arm or a leg (14). Acute ischemic heart conditioning is a promising, adjunctive clinical procedure in the protection against ischemia/reperfusion injury (15). Recently, a few contributions have been published indicating some beneficial effects of daily, prolonged, repeated remote ischemic conditioning. The procedure has been shown to improve the endothelial function and microcirculation both in healthy subjects (16) and in patients with chronic heart failure (17). It also attenuates the left ventricular remodeling after myocardial infarction (18), reduces the blood pressure, the plasma level of NT-proBNP and tissue plasminogen activator (t-PA) in patients with chronic ischemic heart failure (19, 20). Another study on patients with mild ischemic heart failure demonstrated an elevation in the left ventricular ejection fraction, heart rate variability, 6-minute walk distance and a reduction in the plasma level of BNP after the procedure (21).
Another mode of heart conditioning is non-ischemic conditioning provided by physical exercise or by tachycardia. Exercise induces cardioprotection both indirectly and directly. The indirect action requires a long-term endurance type of training that normalizes plasma lipid level and composition, improves insulin sensitivity, reduces blood pressure and body weight (22-24). Direct action means that exercise preconditions the heart either by the blood-borne/neural factors or through increasing heart workload. Numerous reports have shown that direct cardioprotection may by exerted by different types of exercise: prolonged endurance training, a few day endurance exercise or even one bout of exercise. Some reports have also revealed that resistance or interval training induces heart preconditioning. It has been calculated, basing on the available data, that exercise reduces the infarct size by 34% on average (range 19 – 78%) as compared to non-exercising controls (25). The current study is devoted to direct heart preconditioning by exercise or by tachycardia. Firstly, we present data on the cardioprotective effect of different types of muscular exercise. Next, factors known to mediate the phenomenon are discussed. Finally, sections on cardioprotective effect of tachycardia and on studies in humans are included. It is not a systematic review but rather a descriptive overview of the problem. The literature was searched via PubMed in the electronic MEDLINE databases.
EFFECT OF DIFFERENT TYPE OF EXERCISE ON HEART PRECONDITIONING
The methods used to investigate this problem could be applied exclusively to animal experiments. The results available point at two factors of the preconditioning action of exercise: intensity and duration. The intensity above 55% VO2max was usually employed (26), whereas time of exercise varied a lot. In the literature concerning the problem of exercise-induced preconditioning (or cardioprotection) the word ‘training’ is somewhat misleading since even a three-day exercise is often called ‘training’. Exercise physiologists would oppose using the word ‘training’ in such a case. Training is usually regarded as a procedure which leads to a measurable increase in function as well biochemical and/or structural changes in the body (27, 28). Exercise repeated for a few days does not produce any noticeable effects. It should rather be considered as pre-training adaptation (or habituation) to training conditions. However, since the authors used the word ‘training’ we will follow it as below. Generally, the experimental protocols used in the studies on the cardioprotective action of exercise were similar. The animals were exercised and then after a selected time of post-exercise recovery the hearts were incised, perfused ex vivo, I/R procedure was performed and desired parameters were studied. If the procedure was different it will be indicated. The data about type and duration of training are collected in Table 1.
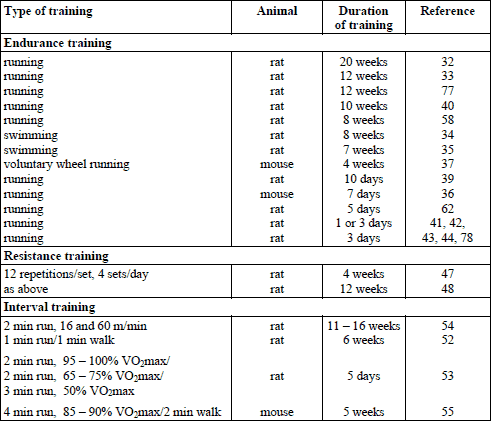
LONG-TERM ENDURANCE TRAINING
According to the above, only results obtained after training lasting more than one week will be discussed in this section. Since many data unanimously indicate that prolonged endurance training exerts a strong cardioprotective effect against I/R injury (26, 29-31), only a few examples are presented here. More information is also included in the section ‘Mechanism of exercise-induced preconditioning’.
The longest training protocol lasted for > 20 weeks in rats. The exercise intensity on a treadmill was increasing gradually up to 35 m/min and up to 20 min daily. The hearts were removed and perfused. The left anterior descending coronary artery (LADA) was occluded for 60 min and then reperfused for 2 hours. The training reduced the infarct size by 25% (32). The same group of authors trained rats for 12 weeks and found the infarct size to be reduced by 34% (33). Swimming training for 8 weeks reduced the infarct size by 24% (34). Interesting data were obtained by (35), who trained rats for 7 weeks (swimming) and thereafter induced myocardial infarction in vivo. Four weeks later, in the trained group the scar area was 1.6-fold smaller, arteriolar density was 1.7-fold higher, and left ventricular shortening fraction was 1.9-fold higher in the trained compared to sedentary animals. In mice, a 7- day treadmill training reduced the infarct size by 34% (36), whereas 4-week voluntary wheel running by 22% (37). Long term endurance exercise was shown to improve cardiac function after I/R in rats, which was manifested by better recovery of contractile function, lower diastolic stiffness, greater efficiency of work, and greater extracellular calcium responsiveness than hearts from sedentary post-I/R rats (38). In another study, rats were trained for 10 days, 60 min daily with increasing speed. Twenty four hours after the last exercise bout, hearts were subjected to I/R (25 min/120 min). Exercise protected against infarction and arrhythmia, and preserved coronary flow (39). Different approach was employed in rats by (40). After a 10-week endurance training program, the animals were anesthetized, mechanically ventilated, and the chest was opened by thoracotomy. Coronary occlusion was achieved by a ligature around the left coronary artery and I/R was evoked (20/10min). The exercise-trained animals maintained higher peak systolic blood pressure throughout I/R compared with the untrained ones.
SHORT-TERM ENDURANCE TRAINING
All studies using short-term endurance training showed reduction in the post I/R infarct size (26, 29-31). Here are some examples of the experiments reporting functional advantages of this type of training. Taylor et al. (41) run rats for 100 min at 20 m/min, 6o upgrade, on 1 or 3 consecutive days. Twenty four hours later they studied cardiac function using isolated perfused heart. Applied global ischemia lasted for 17 min and subsequent reperfusion for 30 min. Both 1- and 3-day exercise accelerated post I/R recovery of cardiac output. Locke et al. (42) exercised rats for 60 min at 30 m/min also on 1 or 3 consecutive days. Twenty four hours after exercise, hearts were subjected to I/R (30/30 min). Animals exercising for 3 days showed recovery of the left ventricular developed pressure after I/R faster than the sedentary ones. Lennon et al. (43) exercised rats for 3 consecutive days, 60 min daily, at workload of 55 or 75% VO2max. Three 2 – 3 min rest periods were introduced during each bout of exercise. Twenty four hours after the third exercise bout, I/R (20/30min) was applied. I/R markedly decreased cardiac output and cardiac work in both groups but the reductions were less pronounced in the trained one. No difference was recorded in the level of cardioprotection between the two exercised groups. However, it should be mentioned that the rats were habituated to running for 5 days at 55% VO2 max, which might have interfered with the final effect of exercise. French et al. (44) exercised rats for 3 days at ~70% VO2max, 60 min/daily, 30 m/min. The exercise reduced the infarct size, apoptosis and necrosis of cardiomyocytes and prevented calpain activation as compared to the sedentary group. Importantly, the cardioprotective effect of short-term endurance training was comparable to that achieved by the long-term one (26, 31).
RESISTANCE EXERCISE
Resistance exercise training is highly recommended in the prevention of metabolic and circulatory disorders (45, 46). Unfortunately, only two studies on the cardioprotective effect of this type of training have been found. In one study (47), rats were adapted to resistance exercise for 4 weeks and in the other study (48) for 12 weeks, according to the same protocol. After the last exercise bout in the training program the hearts were excised and perfused. The LADA was occluded for 40 min and then reperfused for 80 min. The 4-week training had no effect on the infarct size, left ventricular developed pressure and coronary flow compared to the sedentary I/R group (47). The 12-week training reduced the infarct size and allowed maintenance of higher coronary flow and left ventricular developed pressure over the period of ischemia and reperfusion compared to the untrained controls (48). It suggests that only long-term resistance training exerts cardioprotection against I/R.
INTERVAL TRAINING
Generally speaking, in an anaerobic type of interval training, short-term efforts of high intensity are interspersed with short-term efforts of low intensity. This training is mainly used to improve anaerobic capacity. In an aerobic type of interval training, repeated short-term high intensity aerobic exercise bouts are interspersed with very brief rest periods. It shortens the time of training necessary to produce results obtained after long, continuous training (49-51). A few reports are available on the cardioprotective action of the aerobic type of interval training in different species. Rats were trained for 6 weeks. Five 1 min runs at 75 m/min, 15o grade, were interspersed with 1 min walks at 20 m/min. Another group of rats was trained at the same time at 20 m/min. Fourty eight hours after the last exercise, I/R (20/30min) was performed. The training improved the mechanical function of the heart, which was manifested by higher left ventricular developed pressure and lower left ventricular end diastolic pressure during reperfusion than in the sedentary rats (52). Rahimi et al. (53) exercised rats for 5 days, 6 × 2 min at 95 – 100% VO2max. The exercise was interspersed with 5 × 2 min run at 65 – 5% VO2 max and 3 min run at 50% VO2max. The I/R procedure (30/90min) was applied on day 1, 7 and 14 after the last exercise bout. The training reduced the infarct size: by 50% after one day, by 35% after 7 days and by 19% after 14 days (the latter was insignificant). Training did not affect the occurrence of arrhythmia. Bowles at al. (54) compared cardioprotective effects of 11 – 16 week endurance training of low intensity (20 m/min, 0% grade, 60 min/day), moderate intensity (30 m/min, 5o grade, 60 min/day) and interval training (10 bouts of alternating 2 min runs at 16 and 60 m/min, 5o grade). The percent recovery of cardiac output after ex vivo total 25 min ischemia was: 36 in the control group, 61.2 in low intensity trained, 68.1 in moderate intensity trained and 73.2 in interval trained rats. The differences between the trained groups were not significant, indicating that the decisive factor was the training itself and not the type of training. Interval training proved also to be substantially effective in I/R reduction in mice maintained on high-fat diet for 8 weeks. Then, one group underwent 5 week high-intensity interval training (daily, 10 × 4 min at 85 – 90% VO2max, 25° inclination, interspersed by 2 min active rest), whereas the other group was kept sedentary. The training reduced I/R infarct size by 47% as compared to high fat diet sedentary rats (55). These data clearly indicate that the interval training is a potent stimulus in the development of the cardioprotective phenotype.
A SINGLE BOUT OF EXERCISE
Domenech et al. (56) instrumented dogs with a snare on the anterior descending coronary artery. After healing, the animals were exercised acutely, 5 times × 5min. In one group of the animals the aorta was occluded 10 min after exercise, whereas in the other one a day after exercise. The occlusion lasted for 1 hour and subsequent reperfusion for 4 hours. The exercise reduced the infarct size by 78% in the first group and by 46% in the other group, indicating that only one bout of exercise is able to induce preconditioning and that it decreases rather fast. These effects could not be explained by changes in collateral flow to the ischemic region. It should be stressed, however, that it was the interval exercise. In another report by the same group (57), the exercise reduced the infarct size by 56% after 1 hour coronary artery occlusion. Very recently, a study was published in which the effect of a single maximal exercise was compared to an 8-week endurance training in a group of rats. Three days after the last exercise in the training program, I/R procedure (30/60min) was performed. The left ventricle developed pressure was similar in both groups at the basal conditions. However, after I/R the pressure in the trained group was over 70% higher than after a single bout of exercise (58).
ISOLATED CARDIOMYOCYTES
Kang et al. (59) subjected rats to a 6-week endurance training. Thereafter, the cardiomyocytes were isolated and subjected to hypoxia/reoxygenation. The training protected the cells against apoptosis produced by the procedure. This is an important information indicating that the cells themselves acquire preconditioning phenotype during training.
PERSISTENCE OF EXERCISE PRECONDITIONING
Yamashita et al. (60) showed that the exercise-produced cardioprotection is biphasic. They exercised rats acutely for 25 – 30 min on a treadmill moving with the speed of 27 – 30 m/min. Thereafter, in situ LADA was ligated for 20 min and then reperfused. After 0.5, 48 and 60 hours of reperfusion the infarct size was smaller than in sedentary animals, whereas the reduction was not observed after 3, 24, 36 and 72 hours of reperfusion. It was paralleled by changes in the myocardial activity of manganese superoxide dismutase, the reactive oxygen species scavenger. Interestingly, a biphasic cardioprotection was also reported after exercise in dogs (56). In humans, remote ischemic preconditioning protects against ischemia-reperfusion injury of the endothelium also in a biphasic manner (61). In a study by Lennon et al. (62) rats were subjected to a 5-day endurance training and allowed to rest for 1, 3, 9 or 18 days. At those periods of time the hearts were subjected to I/R (20.5/30min). The exercise-trained animals showed higher relative cardiac output and cardiac work at 30 min of reperfusion and on day 1, 3, and 9 after training. It remains an open question how the mechanism inducing cardioprotection could be maintained active for such a long time after the exercise bout.
MECHANISMS OF EXERCISE-INDUCED HEART PRECONDITIONING
Prolonged cardiac ischemia results in several biochemical changes in the myocardium, including a reduction in ATP level, accumulation of hydrogen ions, increased level of free calcium in the cytosol, increased formation of reactive oxygen species (ROS) and activation of calpain (calcium dependent protease). Subsequent reperfusion further increases the production of ROS, overloading with free calcium, activation of caspase-3 and calpain, and opening of the mitochondrial permeability transition pores (mPTP). These complex factors induce mitochondrial injury and in consequence lead to cardiomyocyte death (63, 64). Several factors have been shown to participate in the exercise-induced cardioprotection (Table 2). However, it seems to be a multifactorial phenomenon rather than a consequence of an action of a single factor. Factors contributing to exercise-induced preconditioning could be divided into two groups, namely the extracardiac (neural and blood-borne) and the intracardiac ones.
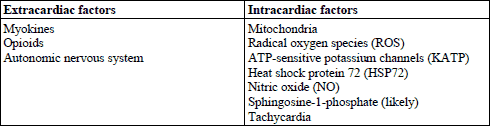
EXTRACARDIAC FACTORS
Neural factors
Some data indicate that in case of remote ischemic preconditioning, stimulation of the sensory nerve endings in the ischemic area participates in the heart conditioning. However, it is not certain which level of the spinal cord is mandatory for the signal transduction. Both parasympathetic and sympathetic efferent pathways to the heart seem to be involved in this reflex (13, 65). We are not aware of any studies on the skeletal muscle originated neural pathways mediating the exercise-induced cardioprotection. However, the contractile activity changes the metabolic environment in the muscles. It could activate sensory receptors within the muscle and the signal would be transferred to the heart as it happens during remote ischemic preconditioning. The fact that higher workloads are needed to produce cardioprotection would support such an assumption.
BLOOD-BORNE FACTORS
Myokines
The skeletal muscles are very active secretory tissue. The bioactive compounds secreted by the muscles are called myokines. In humans, over 300 compounds of likely bioactive properties have been found to be secreted by the muscles but only a few have been examined closely. Myokines exert broad effects both on the muscles themselves and on other organs of the body. According to the available data, acute exercise increases secretion and blood concentration of the following myokines: interlueukin-6 (IL-6), interleukin-7 (IL-7), interleukin-15 (IL-15), decorin, follistatin-like 1(FSTL1), brain-derived neurotrophic factor (BDNF), angiopoietin-like protein 4 (ANGPTL4), and MCP-1 (66, 67). So far, only a role of IL-6 in the acute exercise-induced heart preconditioning has been studied. In wild mice, exercise (running for 3 days, 18 m/min, 60 min daily) protected against I/R induced arrhythmia and necrosis and reduced the infarct size. In IL-6 knockout mice the exercise did not reduce the post I/R infarct size but augmented arrhythmia (68). Serum concentration of IL-6 increased several times after exercise only in wild mice. Serum IL-6 receptor level increased in both groups after exercise but it was much higher in the wild than in the IL-6 knockout animals (68). That strongly points to IL-6 as the principal mediator in exercise-induced cardioprotection. These data are in line with the results showing involvement of IL-6 in ischemic preconditioning (69, 70). FSTL1, another myokine, was shown in mice and swine to accelerate heart infarct healing (71). No data are available on its acute cardioprotective effect. We are certainly only at the beginning of investigating the role of myokines in exercise-induced heart preconditioning. However, it seems to be a very promising area of research.
Opioids
Opioids (opiates) were shown to be involved in ischemic heart preconditioning (72, 73). Some data indicate that opioids are also involved in the exercise-induced cardioprotection, acting both as blood-borne and myocardial factors. In a study by Michelsen et al. (74) healthy young men underwent four cycles of 2 min high-intensity exercise with 3 min intervals of low intensity exercise. Five minutes after the last exercise bout, the blood was collected and plasma dialysate was prepared. Next, rabbit hearts were isolated and perfused with the dialysate for 25 min and washed out for 5 min. The left marginal coronary artery (in rabbit, it is equivalent to LADA in rats and dogs) was occluded for 40 min and it was followed by reperfusion for 120 min. The dialysate reduced infarct size from 60 to 35% of the area at risk and improved the left ventricular developed pressure. In the next experiment, naloxone (a nonspecific opioid receptor antagonist) was added to the dialysate. Naloxone abolished the cardioprotective effects of exercise. This would indicate that opioids released during exercise are present in the dialysate and they could be responsible for the cardioprotective effect of exercise. The source of opioids remains unclear. Unfortunately, the level of plasma opioids was not measured in the study. Dickson et al. (75) investigated this problem in rats. Naltrexone (a nonspecific antagonist of opioid receptors) was administered twice: on day 4 and 5 before the test exercise. Rats exercised for 20 min at 25 m/min. I/R was generated in isolated perfused heart (20 min/3 h). The exercise reduced the infarct size by 50% as compared to nonexercised rats. This effect was absent 5 days after exercise. Blockade of the opioid receptors thoroughly removed exercise-induced cardioprotection. Immediately after exercise, the mRNA expression of all three opioid receptors (δ, κ and µ) and mRNA expressions of their precursors increased several-fold, almost disappearing 8 hours later. The authors concluded that opioids mediate, at least in part, the cardioprotective effect of exercise. In another study (76) rats were exercised for 4 days, 60 min daily at ~70% of VO2max. Twenty four hours after the last exercise bout, I/R was produced in vivo by LADA closing for 30 min and subsequent reperfusion for 60 min. 10 min prior to I/R, selective or nonselective opioid receptor antagonists were administered intravenously. The exercise reduced the infarct size by about 30% and this effect was thoroughly abolished by the blockade of both non-selective or selective δ-opioid receptor antagonist. Neither the blockade of µ- nor κ- opioid receptors affected the infarct size after exercise. These data clearly indicate that the exercise-induced preconditioning was mediated by δ-opioid receptors. A role of opioids in cardioprotection produced by long-term training was also studied (77). Rats were exercised for 12 weeks at 60% VO2 max. Naloxone was administered before each bout of exercise. Fourty eight hours after the last exercise bout the LADA was ligated and rats were recovering for 7 days. The training reduced the infarct size over 3 times, the effect being prevented by naloxone. Treatment with morphine right before I/R also had a cardioprotective effect, but only a mild additive effect of training and morphine was observed. The role of δ-opioid receptors in exercise-induced cardioprotection was at the same time described by Miller et al. (78), who used a 3-day training, 30 min at 30 m/min. Naltrindole, a selective δ-opioid receptor blocker was administered on each day before exercise. Twenty four hours after the last exercise bout the rats were anesthetized and in vivo I/R procedure was performed. The LADA was ligated for 50 min and then reperfused for 120 min. The exercise reduced the infarct area by over 50% as compared to the sedentary group. This effect of exercise was partially prevented by the blockade of δ-opioid receptors. The left ventricle mRNA of proenkephalin was elevated immediately after exercise and 120 min later. The exercise did not affect the ventricular mRNA expression of δ-opioid receptors. Neither the protein expression of Leu-enkephalin nor δ-opioid receptors were affected by the exercise. Also the serum concentration of enkephalin remained stable. The blockade of δ-opioid receptors prevented necrosis but not apoptosis induced by I/R. They hypothesized that both the blood borne and the intracardiac opioids were likely to participate in the exercise-induced preconditioning.
Autonomic nervous system
Muscular exercise increases the activity of the adrenergic nervous system. It induces tachycardia and increases inotropy during exercise (27, 28). It has been shown that the adrenergic system is involved in cardioprotection (25, 37). This is discussed in the ‘Nitric oxide’ section.
INTRACARDIAC FACTORS
Mitochondria
As already indicated, mitochondria play a key role in I/R heart injury (63, 64). The available data clearly indicate that endurance training also induces the development of a mitochondrial phenotype that is resistant to I/R injury. Other major features of the phenotype include decreased reactive oxygen species (ROS) production, increased tolerance to high calcium level, reduced rate of opening of the mitochondrial permeability transition pores (mPTP) and elevation in the content of antiapoptotic proteins (64, 79-82). Endurance training induces changes in the expression of a number of mitochondrial proteins. Many of them participate in ATP production. However, no direct relationship was reported between the latter group of proteins and cardioprotection (64, 83). The mPTP play an important role in cardioprotection afforded by ischemic preconditioning. The pores are present in the inner mitochondrial membrane and are closed. Their opening at the onset of reperfusion results in the appearance of a cascade of events leading finally to cell death. Ischemic conditioning prevents opening of the pores (84, 85). Long-term endurance training was shown to increase resistance of the mitochondrial permeability transition pore opening during I/R in rats (86, 87) and mice (88). Also short-term (5 days) training reduced mPTP opening in I/R in rats. Interestingly, the training did not affect the heart contractile function and coronary flow and LDH release (89), suggesting a specific response of mPTP to the training. Unfortunately, we are not aware of any data regarding a response of mPTP to acute exercise. The data on mitochondrial ROS and ATP-sensitive potassium channels are included in the sections below.
Reactive oxygen species
ROS are claimed to play the major role in I/R injury (90, 91). Therefore, studies were undertaken to elucidate a role of the antioxidative defense system in the exercise-induced cardioprotective phenotype. ROS are produced both in mitochondria and in the cytosol (80, 82, 91). The major components of the antioxidative defense system include such enzymes as superoxide dismutase (SOD), cytosolic form, located also in the mitochondrial intermembrane space (copper-zinc [CuZn] SOD) and mitochondrial matrix form -manganese [Mn] SOD, catalase, glutathione, glutathione peroxidase, glutathione reductase and thioredoxin reductase (82, 91). The data collected by (30) and (91) indicate that both short-term and long-term exercise training increases the activity of the heart MnSOD. Low-intensity exercise does not seem to affect the cardiac activity of MnSOD (82). Most studies failed to show an elevation in the activity of catalase and glutathione peroxidase in the myocardium after exercise (26, 82, 91). The data regarding the effect of training on the activity of myocardial glutathione reductase are not uniform (26, 82). Yamashita et al. (60) used antisense oligodeoxyribonucleotide against MnSOD. The treatment prevented exercise training-induced reduction in the infarct size, which was confirmed by French et al. (44). Hamilton et al. (92) showed that antisense oligodeoxyribonucleotide prevents arrhythmia during I/R. On the other hand, Lennon et al. (93) observed no influence of antisense oligonucleotides on the development of myocardial stunning after I/R. They concluded that different mediators may protect against stunning and infarction. Taylor and Starnes (94) administered MPG, a free radical scavenger, before exercise and preformed I/R procedure 24 hours after exercise. The compound did not affect the heart external work output. It remains to be established whether the cytosolic isoform of SOD participates in the exercise-induced cardioprotection (30).
ATP-sensitive potassium channels
There are two groups of the ATP-sensitive potassium channels: sarcolemmal (sKATP) and inner mitochondrial membrane (mKATP) (95, 96). The channels are closed when the level of intracellular ATP is adequate. The reduction in the level of ATP in the cytoplasm, which takes place during ischemia, results in the opening of sKATP channels and subsequent leakage of potassium ions from cardiomyocytes. This, in turn, prevents entry of calcium ions into the cells, thus preventing calcium overload. Decreased availability of ATP also leads to opening of mKATP channels, which may protect mitochondria against calcium overload and subsequent damage during I/R (97, 98). Brown et al. (33) blocked sKATP channels with a compound termed HMR 1098 and mKATP channels with 5-hydroxydecanoic acid in endurance trained rats. Blockade of sKATP channels completely abolished the training-acquired cardioprotection, whereas blockade of mKATP channels did not provide any effect. The results would strongly indicate that sKATP channels are responsible for cardioprotection, which, however, has not been confirmed yet. As already mentioned, in dogs, acute high intensity exercise both prior to and 24 h before coronary occlusion induced early and late cardiac preconditioning. The early effect was removed by the blockade of mKATP channels (56). In a subsequent study, according to the same protocol, late cardiac preconditioning by acute exercise was also mediated through mKATP channels (99). Very recent data revealed that remote ischemic preconditioning also reduced infarct size and arrhythmia, the effect being abolished by the blockade of mKATP channels (100). Certainly, by far more studies are necessary to elucidate the role of KATP channels in exercise-induced cardioprotection.
Heat shock protein 72 (HSP72)
Stress stimuli increase synthesis of a group of proteins collectively called heat stress proteins (HSPs). One of them is HSP72. Overexpression of HSP72 protects the myocardium against I/R injury (101). Endurance exercise increases the level of myocardial HSP72 (41, 102). Therefore, it may be expected that exercise-induced elevation in the heart HSP72 could be an important protector against I/R. However, the heart HSP72 content was not increased by exercise in cold, but was elevated several-fold after exercise in room temperature. Moreover, the cardioprotective effect of exercise was similar in both conditions (41, 103), thus indicating that the exercise-induced elevation of the heart HSP72 content is not necessary to produce the cardioprotective phenotype. Obviously, HSP72 may be replaced by other factors.
Nitric oxide (NO)
Nitric oxide plays an important role in ischemic heart conditioning (104). Exercise increases shear stress put on the vessels. As a result, the expression and activity of the endothelial isoform of nitric oxide synthase (eNOS) increase (91). Some data indicate that the compound is involved in the exercise-induced cardiac phenotype. Mice were trained for 7 days at 60 – 70% VO2max, 60 min daily, which resulted in eNOS activation and increased expression of the inducible isoform of NOS (iNOS) in the heart. Twenty four hours after exercise, I/R (30 min/2 h) procedure was performed. The infarct size was significantly smaller in the hearts from exercised mice than from sedentary control mice. In eNOS(–/–) knockout mice, the exercise-induced upregulation of iNOS and cardioprotection did not occur. Also, exercise preconditioning did not occur when the iNOS selective inhibitor 1400 W was administered right before coronary artery occlusion or when iNOS(–/–) mice were employed. This would indicate that iNOS was responsible for exercise-induced cardiopotection (36). In another study on mice, a voluntary 4-week exercise training was employed and I/R was produced 1 or 7 days after training accomplishment. The reduction in the infarct size was similar in both cases. Training increased the expression and activity of eNOS, indicating that eNOS was involved in preconditioning at each time period. The blood level and myocardial level of nitrite and nitrosothiols also increased both early and late after training (25, 37). Nitrites are a storage form of NO. During I/R they can be converted to NO by reductases present in the heart. Also nitrosothiols can be NO donors (105, 106). It was further shown that the activation of eNOS and elevation in the production of NO in the myocardium during exercise were partly mediated by the β3 adrenergic receptors (25, 37). The involvement of NO in exercise-induced cardioprotection was also shown in dogs. The animals were subjected to acute exercise of increasing intensity. Twenty four or fourty eight hours later they received intravenously aminguanidine, the inhibitor of iNOS. Thirty min later, the LADA was ligated for 25 min. In the control group, exercise reduced the severity of arrhythmia, increased survival rate and increased the activity of the enzyme in the left ventricle after 24 h but not after 48 h of recovery. The administration of aminoguanidine prevented the cardioprotective effects of exercise (107). In another study on dogs, a selective iNOS inhibitor (AEST) and nonselective NOS inhibitor (L-NAME) were used. Exercise markedly diminished ventricular arrhythmia 24 h after I/R. This effect was substantially reduced by L-NAME administered before exercise as well as by AEST given prior to coronary artery ligation. This would indicate that NO is cardioprotective both early and late after exercise (108). However the results obtained in rats are in odds with the results obtained in dogs (109). It should be added that in trained rats, coronary endothelial cells but not cardiomyocytes were shown to be responsible for the eNOS-dependent cardioprotection (110).
Ceramide and sphingosine-1-phosphate
Ceramide and sphingosine-1-phosphate (S1P) are key bioactive sphingolipids (111, 112). I/R increases the content of ceramide in the perfused rat heart, the effect being partially prevented by ischemic preconditioning (113). In the rat, acute exercise was shown to reduce the heart content of ceramide (114). Ceramide activates apoptosis of cardiomyocytes after I/R (115-117). However, no data regarding the effect of exercise on post-I/R content of ceramide in the myocardium are available. S1P was repeatedly shown to exert a very powerful cardioprotective action against I/R injury. Exogenous S1P increases viability of cardiomyocytes incubated under hypoxic conditions and reduces the infarct size in the isolated, perfused rat heart after I/R. It also mediates the beneficial effect of ischemic pre- and postconditioning in the heart subjected to I/R. Formation of S1P in the heart is catalyzed by the enzyme sphingosine kinase 1 and its catabolism is catalyzed by the enzyme sphingosine-1-phosphate lyase. The reduction in the activity of sphingosine kinase 1 or knocking out its gene eliminates a cardioprotective effect of ischemic preconditioning in mice. Also knocking out the sphingosine-1-phosphate lyase gene has a very potent cardioprotective effect against I/R injury in the mouse heart (116-118). It is important to note that the extracellular S1P binds to S1P receptors present on the plasma membrane both of the mother cells (autocrine action), neighboring cells (paracrine action) and on remote cells, reaching them by the blood plasma (119, 120). Acute exercise was shown to increase plasma S1P in untrained healthy humans (121) and it would increase the exercise-induced cardioprotective potential of the compound. However, the late post-infarction plasma S1P concentration was found to be reduced in humans (122, 123) and rats (124). The plasma concentration of S1P remained unchanged during percutaneous coronary artery intervention (125), but it was elevated during reperfusion after percutaneous artery occlusion (126). However, as in case of ceramide, no data are available regarding the influence of exercise on post-I/R content of S1P in the heart.
HUMAN STUDIES
For obvious reasons, no direct data are available on the cardioprotective effect of acute exercise in humans. It should be mentioned, however, that Michelsen et al. (74) showed in healthy humans that a transferable, cardioprotective factor is released to the plasma during exercise. A few reports are also available indicating the cardioprotective effect of acute muscular activity against cardiac ischemia in patients with coronary heart disease. Bilinska et al. (127) studied patients with stable angina. The patients performed a symptom-limited exercise test, followed by the second exercise test (as above) either on day 1, 2, 3 or 4 of recovery. The patients showed improved exercise time and other heart variables on day 1 and 2 after exercise. The improvements decreased on day 3 and disappeared on day 4 of recovery. This observation clearly showed that the first exercise preconditioned the patients’ hearts to the second exercise bout. These data were confirmed by Lambiase et al. (128) and Crisafulli et al. (129). Lambiase et al. (128) examined two groups of patients with stable angina due to isolated and discrete stenosis within LADA. One group of patients performed 3 maximal exercise tests (basal, 15 and 90 min later) and recovered for 14 days. Then, each patient had elective percutaneous coronary intervention (PCI) comprising two balloon inflations lasting for 180 s. The second group of patients also performed 3 exercise bouts as above but 24 h later had two additional tests separated by 15 min rest, and then 4 h later they underwent PCI. The tests performed 24 h after the first 3 tests showed improved performance and reduction in ventricular ectopic frequency. During the first and the second balloon inflation the peak ST elevation was markedly reduced as compared to the first group. This indicates that in the second group, the first sets of exercise produced late preconditioning as manifested both during exercise and PCI. Crisafulli et al. (129) examined a group of patients with stable angina. First, they performed a maximal exercise-test and then, the subsequent maximal tests 30 min and 2 days later. In these two subsequent tests (30 min and 2 days later) , the onset of ischemia was delayed, and the allowed workload, heart rate and cardiac output were higher as compared to the respective data in the first exercise-test. Only exercise after the 2-day recovery elevated myocardial contractility and stroke volume. However, it should be kept in mind that the exercise test in humans with stable angina lasted until symptoms of ischemia appeared. Therefore, it is difficult to evaluate whether silent ischemia contributed to the preconditioning. Certainly, many more studies are needed to fully investigate the exercise-induced preconditioning in humans and its possible clinical usefulness.
TACHYCARDIA-INDUCED PRECONDITIONING
Some data clearly indicate that tachycardia per se induces heart preconditioning. Koning et al. (130) subjected open-chest pigs to 30 min ventricular pacing at 200 beats/min followed by 15 min normal sinus rhythm and then produced myocardial infarction by a 60-min coronary artery occlusion. The pacing resulted in a reduction in the infarct size by ~30%. 30 min pacing immediately preceding the occlusion without intervening sinus rhythm reduced the infarct size by ~40%. The reductions in the infarct size were abolished by pretreatment with the KATP channel blocker glibenclamide. The results would suggest that tachycardia itself, without ischemia, provides considerable acute preconditioning. That cardioprotective effect of tachycardia was also confirmed in dogs (131). Anesthetized dogs were induced with 5 periods of tachycardia (213 ± 12 cycles/min), each of 5 min duration, and 5 min of intervening periods at control heart rate. Next, the LADA was occluded for 60 min and reperfused for 4.5 hours. The tachycardia reduced the infarct size by 46% with respect to the group without preconditioning. This effect was not due to changes in the collateral flow. During tachycardia, myocardial interstitial adenosine showed an approximately twofold increase. Blockade of adenosine receptors with 8 phenyltheophylline, before or after preconditioning tachycardia, reverted its protecting effect but it did not modify infarct size in non-preconditioned dogs. No differences in cytosolic or particulate protein kinase C activity or translocation of α-, β-, ε-, and ξ- protein kinase C isozyme were observed between preconditioned and non-preconditioned dogs. It was concluded that tachycardia, in the absence of ischemia, mimics the preconditioning effect of ischemia in the dog, the effect being mediated by adenosine. It should be added that several other factors might contribute to tachycardia-induced preconditioning. Tachycardia increases the usage of glucose (132, 133) and free fatty acids by the heart (134). It induces numerous changes in the metabolism of sphingolipids (135), glycerolipids (136) and major components of the lipolytic complex (137) in the ventricles. Endurance training in rats reduces glycolysis and elevates glucose and palmitate oxidation both before and after an insult of ischemia. The latter changes have been collectively called a ‘protective metabolic phenotype’(138). All these changes in the metabolic milieu could affect heart susceptibility to I/R. Moreover, cardiomyocytes belong to a group of cells empowered with the mechanism of mechanotransduction. The elevation in mechanical tension induces the release of bioactive compounds by the cells. They act in either autocrine or paracrine way (139, 140). It is likely that increased mechanical tension during tachycardia also releases the local factor(s) contributing to preconditioning. It is a tenet that the increasing exercise intensity is paralleled by heart rate elevation. As already mentioned, higher exercise intensity is needed to produce cardioprotection. It may be hypothesized that exercise-induced tachycardia may participate in the generation of preconditioning by exercise.
CONCLUSIONS AND PERSPECTIVES
Ischemic heart conditioning is a well-recognized, clinically applicable phenomenon. In its shadow, numerous data have been published indicating that heart preconditioning may be induced in a nonischemic way, namely by muscular exercise or tachycardia. Exercise-induced heart preconditioning is a very interesting phenomenon. It can be produced by different types of exercise, such as long term or short term endurance training, acute endurance exercise, resistance training, interval training and even by one bout of exercise. Cardiac preconditioning reduces the infarct size, arrhythmia and stunning after ischemia/reperfusion. Surprisingly, even cardiomyocytes isolated from the hearts of trained rats show a higher level of resistance against hypoxia/reoxygenation. Exercise-induced preconditioning is a multifactorial process involving both extracardiac and intracardiac factors. The extracardiac factors include interleukin 6, opiates and the adrenergic system. The major intracardiac factors include the antioxidant defense system, heart opiates, nitric oxide, ATP-dependent potassium channels, heat shock protein 72 and sphingosine-1-phosphate. Several reports indicate that removal of only one factor prevents exercise-induced cardioprotection. Therefore, it remains unclear how all the factors cooperate at the same time. Experimental tachycardia has also been found to reduce the infarct size. Tachycardia-induced cardioprotection is probably mediated by adenosine. All information concerning exercise and tachycardia-induced cardioprotection has been obtained in experiments performed on animal models. In healthy humans, the presence of a transferable, cardioprotective factor in the plasma has been observed after exercise. Some data obtained in humans with stable angina indicate that one exercise bout increases the time of allowable exercise workload of next bouts. Ischemic heart conditioning is produced by a non-physiological procedure, whereas exercise and tachycardia-induced preconditioning is generated by physiological stimuli. They usually act along since exercise is accompanied by heart rate acceleration. No acute clinical application of the two stimuli has been reported so far. However, they may represent a considerable potential. It is tempting to claim that in case of exercise-inducing preconditioning, the principal, but yet not recognized stimulus, originates from the contracting skeletal muscles thus ‘taking care’ of the heart. The stimulus would activate all other mediators of cardioprotection. IL-6 would be a candidate to play such a role though it exerts many extra-heart effects. However, the existence of another, yet unidentified, myokine acting specifically on the heart cannot be excluded. Its identification and synthesis would probably enable cardiac preconditioning without exercise before procedures on patients’ coronary arteries.
Acknowledgements: This work was supported by the Medical University of Bialystok, grants no. N/ST/ZB/17/002/1118 and N/ST/ZB/15/005/1153.
Conflict of interests: None declared.
REFERENCES
- Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med 2007; 357: 1121-3135.
- Vinten-Johansen J, Jiang R, Reeves JG, Mykytenko J, Deneve J, Jobe LJ. Inflammation, proinflammatory mediators and myocardial ischemia-reperfusion injury. Hematol Oncol Clin North Am 2007; 21: 123-145.
- Santillo E, Migale M, Postacchini D, Balestrini F, Incalzi RA. Cardioprotection by conditioning mimetic drugs. Antiinflamm Antiallergy Agents Med Chem 2016; 15: 15-30.
- Sivaraman V, Yellon DM. Pharmacologic therapy that simulates conditioning for cardiac ischemic/reperfusion injury. J Cardiovasc Pharmacol Ther 2014; 19: 83-96.
- Schmidt MR, Pryds K, Botker HE. Novel adjunctive treatments of myocardial infarction. World J Cardiol 2014; 6: 434-443.
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74: 1124-1136.
- Heusch G. Molecular basis of cardioprotection. Signal transduction in ischemic pre-, post-, and remote conditioning. Circ Res 2015; 116: 674-699.
- Przyklenk K. Reduction of myocardial infarct size with ischemic ‘conditioning’: physiologic and technical consideration. Anesth Analg 2013; 117: 891-901.
- Przyklenk K, Bauer B, Ovize M, Kloner RA, Whittaker P. Regional ischemic ‘preconditioning’ protects remote virgin myocardium from subsequent sustained coronary occlusion. Circulation 1993; 87: 893-899.
- Whittaker P, Przyklenk K. Reduction of infarct size in vivo with ischemic preconditioning: mathematical evidence for protection via non-ischemic tissue. Basic Res Cardiol 1994; 89: 6-15.
- Gho BC, Schoemaker RG, van den Doel MA, Duncker DJ, Verdouw PD. Myocardial protection by brief ischemia in noncardiac tissue. Circulation 1996; 94: 2193-2200.
- Kleinbongard P, Skyschally A, Heusch G. Cardioprotection by remote ischemic conditioning and its signal transduction. Pflugers Arch 2017; 469: 159-181.
- Heusch G, Botker HE, Przyklenk K, Redington A, Yellon D. Remote ischemic conditioning. J Am Coll Cardiol 2015; 65: 177-195.
- Stokfisz K, Ledakowicz-Polak A, Zagorski M, Zielinska M. Ischaemic preconditioning - Current knowledge and potential future applications after 30 years of experience. Adv Med Sci 2017; 62: 307-316.
- Heusch G, Rassaf T. Time to give up on cardioprotection? A critical appraisal of clinical studies on ischemic pre-, post-, and remote conditioning. Circ Res 2016; 119: 676-695.
- Jones H, Hopkins N, Bailey TG, Green DJ, Cable NT, Thijssen DH. Seven-day remote ischemic preconditioning improves local and systemic endothelial function and microcirculation in healthy humans. Am J Hypertens 2014; 27: 918-925.
- Kono Y, Fukuda S, Hanatani A, et al. Remote ischemic conditioning improves coronary microcirculation in healthy subjects and patients with heart failure. Drug Des Devel Ther 2014; 8: 1175-1181.
- Yamaguchi T, Izumi Y, Nakamura Y, et al. Repeated remote ischemic conditioning attenuates left ventricular remodeling via exosome-mediated intercellular communication on chronic heart failure after myocardial infarction. Int J Cardiol 2015; 178: 239-246.
- Pryds K, Nielsen RR, Jorsal A, et al. Effect of long-term remote ischemic conditioning in patients with chronic ischemic heart failure. Basic Res Cardiol 2017; 112: 67. doi: 10.1007/s00395-017-0658-6
- Pryds K, Kristiansen J, Neergaard-Petersen S, et al. Effect of long-term remote ischaemic conditioning on platelet function and fibrinolysis in patients with chronic ischaemic heart failure. Thromb Res 2017; 153: 40-46.
- Chen L, Zhou Q, Jin H, et al. Effects of remote ischaemic conditioning on heart rate variability and cardiac function in patients with mild ischaemic heart failure. Heart Lung Circ 2018; 27: 477-483.
- Bassuk SS, Manson JE. Physical activity and the prevention of cardiovascular disease. Curr Atheroscler Rep 2003; 5: 299-307.
- Lavie CJ, Arena R, Swift DL, et al. Exercise and the cardiovascular system: clinical science and cardiovascular outcomes. Circ Res 2015; 117: 207-219.
- Moghetti P, Bacchi E, Brangani C, Dona S, Negri C. Metabolic effects of exercise. Front Horm Res 2016; 47: 44-57.
- Calvert JW, Lefer DJ. Role of -adrenergic receptors and nitric oxide signaling in exercise-mediated cardioprotection. Physiology (Bethesda) 2013; 28: 216-224.
- Kavasis AN. Exercise preconditioning of the myocardium. Sports Med 2009; 39: 923-935.
- McArdle WD, Katch FI, Katch VL. Exercise Physiology. Wolters Kluwer, 2015.
- Wilimore JH, Costil DL. Physiology of Sport and Exercise. Human Kinetics, 1999.
- Starnes JW, Taylor RP. Exercise-induced cardioprotection: endogenous mechanisms. Med Sci Sports Exerc 2007; 39: 1537-1543.
- Powers SK. Exercise: teaching myocytes new tricks. J Appl Physiol 2017; 123: 460-472.
- Borges JP, Lessa MA. Mechanisms involved in exercise-induced cardioprotection: a systematic review. Arq Bras Cardiol 2015; 105: 71-81.
- Brown DA, Jew KN, Sparagna GC, Musch TI, Moore RL. Exercise training preserves coronary flow and reduces infarct size after ischemia-reperfusion in rat heart. J Appl Physiol 2003; 95: 2510-2518.
- Brown DA, Chicco AJ, Jew KN, et al. Cardioprotection afforded by chronic exercise is mediated by the sarcolemmal, and not the mitochondrial, isoform of the KATP channel in the rat. J Physiol 2005; 569: 913-924.
- Zhang KR, Liu HT, Zhang HF, et al. Long-term aerobic exercise protects the heart against ischemia/reperfusion injury via PI3 kinase-dependent and Akt-mediated mechanism. Apoptosis 2007; 12: 1579-1588.
- Freimann S, Scheinowitz M, Yekutieli D, Feinberg MS, Eldar M, Kessler-Icekson G. Prior exercise training improves the outcome of acute myocardial infarction in the rat. Heart structure, function, and gene expression. J Am Coll Cardiol 2005; 45: 931-938.
- Akita Y, Otani H, Matsuhisa S, et al. Exercise-induced activation of cardiac sympathetic nerve triggers cardioprotection via redox-sensitive activation of eNOS and upregulation of iNOS. Am J Physiol Heart Circ Physiol 2007; 292: H2051-H2059.
- Calvert JW, Condit ME, Aragon JP, et al. Exercise protects against myocardial ischemia-reperfusion injury via stimulation of (3)-adrenergic receptors and increased nitric oxide signaling: role of nitrite and nitrosothiols. Circ Res 2011; 108: 1448-1458.
- Bowles DK, Starnes JW. Exercise training improves metabolic response after ischemia in isolated working rat heart. J Appl Physiol 1994; 76: 1608-1614.
- Frasier CR, Moukdar F, Patel HD, et al. Redox-dependent increases in glutathione reductase and exercise preconditioning: role of NADPH oxidase and mitochondria. Cardiovasc Res 2013; 98: 47-55.
- Powers SK, Demirel HA, Vincent HK, et al. Exercise training improves myocardial tolerance to in vivo ischemia-reperfusion in the rat. Am J Physiol 1998; 275: R1468-R1477.
- Taylor RP, Harris MB, Starnes JW. Acute exercise can improve cardioprotection without increasing heat shock protein content. Am J Physiol 1999; 276: H1098-H1102.
- Locke M, Tanguay RM, Klabunde RE, Ianuzzo CD. Enhanced postischemic myocardial recovery following exercise induction of HSP72. Am J Physiol 1995; 269: H320-H325.
- Lennon SL, Quindry JC, French JP, Kim S, Mehta JL, Powers SK. Exercise and myocardial tolerance to ischaemia-reperfusion. Acta Physiol Scand 2004; 182: 161-169.
- French JP, Hamilton KL, Quindry JC, Lee Y, Upchurch PA, Powers SK. Exercise-induced protection against myocardial apoptosis and necrosis: MnSOD, calcium-handling proteins, and calpain. FASEB J 2008; 22: 2862-2871.
- Artero EG, Lee DC, Lavie CJ, et al. Effects of muscular strength on cardiovascular risk factors and prognosis. J Cardiopulm Rehabil Prev 2012; 32: 351-358.
- Lemes IR, Ferreira PH, Linares SN, Machado AF, Pastre CM, Netto J. Resistance training reduces systolic blood pressure in metabolic syndrome: a systematic review and meta-analysis of randomised controlled trials. Br J Sports Med 2016; Mar 8: bjsports-2015-094715. doi: 10.1136/bjsports-2015-094715
- Doustar Y, Soufi FG, Jafary A, Saber MM, Ghiassie R. Role of four-week resistance exercise in preserving the heart against ischaemia-reperfusion-induced injury. Cardiovasc J Afr 2012; 23: 451-455.
- Soufi FG, Saber MM, Ghiassie R, Alipour M. Role of 12-week resistance training in preserving the heart against ischemia-reperfusion-induced injury. Cardiol J 2011; 18: 140-145.
- Batacan RB, Duncan MJ, Dalbo VJ, Tucker PS, Fenning AS. Effects of high-intensity interval training on cardiometabolic health: a systematic review and meta-analysis of intervention studies. Br J Sports Med 2017; 51: 494-503.
- Gist NH, Fedewa MV, Dishman RK, Cureton KJ. Sprint interval training effects on aerobic capacity: a systematic review and meta-analysis. Sports Med 2014; 44: 269-279.
- Logan GR, Harris N, Duncan S, Schofield G. A review of adolescent high-intensity interval training. Sports Med 2014; 44: 1071-1085.
- Libonati JR, Kendrick ZV, Houser SR. Sprint training improves postischemic, left ventricular diastolic performance. J Appl Physiol (1985) 2005; 99: 2121-2127.
- Rahimi M, Shekarforoush S, Asgari AR, et al. The effect of high intensity interval training on cardioprotection against ischemia-reperfusion injury in Wistar rats. EXCLI J 2015; 14: 237-346.
- Bowles DK, Farrar RP, Starnes JW. Exercise training improves cardiac function after ischemia in the isolated, working rat heart. Am J Physiol 1992; 263: H804-H809.
- Lund J, Hafstad AD, Boardman NT, et al. Exercise training promotes cardioprotection through oxygen-sparing action in high fat-fed mice. Am J Physiol Heart Circ Physiol 2015; 308: H823-H829.
- Domenech R, Macho P, Schwarze H, Sanchez G. Exercise induces early and late myocardial preconditioning in dogs. Cardiovasc Res 2002; 55: 561-566.
- Parra VN, Macho P, Domenech RJ. Late cardiac preconditioning by exercise in dogs is mediated by mitochondrial potassium channels. J Cardiovasc Pharmacol 2010; 56: 268-274.
- Borges JP, Cruz MD, Lanza M, Lessa MA. Aerobic exercise training induces superior cardioprotection following myocardial ischemia reperfusion injury than a single aerobic exercise session in rats. Motriz, Rio Claro 2017; 23 (spec. iss.): e101620. doi: http://dx.doi.org/10.1590/S1980-6574201700SI0011
- Kang PM, Yue P, Liu Z, Tarnavski O, Bodyak N, Izumo S. Alterations in apoptosis regulatory factors during hypertrophy and heart failure. Am J Physiol Heart Circ Physiol 2004; 287: H72-H80.
- Yamashita N, Hoshida S, Otsu K, Asahi M, Kuzuya T, Hori M. Exercise provides direct biphasic cardioprotection via manganese superoxide dismutase activation. J Exp Med 1999; 189: 1699-1706.
- Loukogeorgakis SP, Panagiotidou AT, Broadhead MW, Donald A, Deanfield JE, MacAllister RJ. Remote ischemic preconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans: role of the autonomic nervous system. Am Coll Cardiol 2005; 46: 450-456.
- Lennon SL, Quindry J, Hamilton KL, et al. Loss of exercise-induced cardioprotection after cessation of exercise. J Appl Physiol 2004; 96:1299-1305.
- Heusch G, Gersh BJ. The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J 2017; 38: 774-784.
- Powers SK, Smuder AJ, Kavazis AN, Quindry JC. Mechanism of exercise-induced cardioprotection. Physiology 2014; 29: 27-38.
- Donato M, Evelson P, Gelpi RJ. Protecting the heart from ischemia/reperfusion injury: an update on remote ischemic preconditioning and postconditioning. Curr Opin Cardiol 2017; 32: 784-790.
- Gorgens SW, Eckardt K, Jensen J, Drevon CA, Eckel J. Exercise and regulation of adipokine and myokine production. Prog Mol Biol Transl Sci 2015; 135: 313-336.
- Giudice J, Taylor JM. Muscle as a paracrine and endocrine organ. Curr Opin Pharmacol 2017; 34: 49-55.
- McGinnis GR, Ballmann C, Peters B, et al. Interleukin-6 mediates exercise preconditioning against myocardial ischemia reperfusion injury. Am J Physiol Heart Circ Physiol 2015; 308: H1423-H1433.
- Dawn B, Xuan YT, Guo Y, et al. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc Res 2004; 64: 61-71.
- Smart N, Mojet MH, Latchman DS, Marber MS, Duchen MR, Heads RJ. IL-6 induces PI 3-kinase and nitric oxide-dependent protection and preserves mitochondrial function in cardiomyocytes. Cardiovasc Res 2006; 69: 164-177.
- Wei K, Serpooshan V, Hurtado C, et al. Epicardial FSTL1 reconstitution regenerates the adult mammalian heart. Nature 2015; 525: 479-485.
- Bell SP, Sack MN, Patel A, Opie LH, Yellon DM. Delta opioid receptor stimulation mimics ischemic preconditioning in human heart muscle. J Am Coll Cardiol 2000; 36: 2296-2302.
- Schultz JJ, Hsu AK, Nagase H, Gross GJ. TAN-67, a delta 1-opioid receptor agonist, reduces infarct size via activation of Gi/o proteins and KATP channels. Am J Physiol 1998; 274: H909-H914.
- Michelsen MM, Stottrup NB, Schmidt MR, et al. Exercise-induced cardioprotection is mediated by a bloodborne, transferable factor. Basic Res Cardiol 2012; 107: 260-269.
- Dickson EW, Hogrefe CP, Ludwig PS, Ackermann LW, Stoll LL, Denning GM. Exercise enhances myocardial ischemic tolerance via an opioid receptor-dependent mechanism. Am J Physiol Heart Circ Physiol 2008; 294: H402-H408.
- Borges JP, Verdoorn KS, Daliry A, et al. Delta opioid receptors: the link between exercise and cardioprotection. PLoS One 2014; 9: e113541. doi: 10.1371/journal.pone.0113541
- Galvao TF, Matos KC, Brum PC, Negrao CE, Luz PL, Chagas AC. Cardioprotection conferred by exercise training is blunted by blockade of the opioid system. Clinics (Sao Paulo) 2011; 66: 151-157.
- Miller LE, McGinnis GR, Peters BA, et al. Involvement of the -opioid receptor in exercise-induced cardioprotection. Exp Physiol 2015; 100: 410-421.
- Kavazis AN, McClung JM, Hood DA, Powers SK. Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am J Physiol Heart Circ Physiol 2008; 294: H928-H935.
- Alleman RJ, Stewart LM, Tsang AM, Brown DA. Why does exercise ‘trigger’ adaptive protective responses in the heart? Dose Response 2015; May 4. doi: 10.2203/dose-response.14-023.Alleman.
- Lee Y, Min K, Talbert EE, et al. Exercise protects cardiac mitochondria against ischemia-reperfusion injury. Med Sci Sports Exerc 2012; 44: 397-405.
- Frasier CR, Moore RL, Brown DA. Exercise-induced cardiac preconditioning: how exercise protects your achy-breaky heart. J Appl Physiol 2011; 111: 905-915.
- Kavazis AN, Alvarez S, Talbert E, Lee Y, Powers SK. Exercise training induces a cardioprotective phenotype and alterations in cardiac subsarcolemmal and intermyofibrillar mitochondrial proteins. Am J Physiol Heart Circ Physiol 2009; 297: H144-H152.
- Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab 2015; 21: 206-214.
- Ong SB, Dongworth RK, Cabrera-Fuentes HA, Hausenloy DJ. Role of the MPTP in conditioning the heart - translatability and mechanism. Br J Pharmacol 2015; 172: 2074-2084.
- Magalhaes J, Goncalves IO, Lumini-Oliveira J, et al. Modulation of cardiac mitochondrial permeability transition and apoptotic signaling by endurance training and intermittent hypobaric hypoxia. Int J Cardiol 2014; 173: 40-45.
- Marcil M, Bourduas K, Ascah A, Burelle Y. Exercise training induces respiratory substrate-specific decrease in Ca2+-induced permeability transition pore opening in heart mitochondria. Am J Physiol Heart Circ Physiol 2006; 290: H1549-H1557.
- Pons S, Martin V, Portal L, et al. Regular treadmill exercise restores cardioprotective signaling pathways in obese mice independently from improvement in associated co-morbidities. J Mol Cell Cardiol 2013; 54: 82-89.
- Ciminelli M, Ascah A, Bourduas K, Burelle Y. Short term training attenuates opening of the mitochondrial permeability transition pore without affecting myocardial function following ischemia-reperfusion. Mol Cell Biochem 2006; 291: 39-47.
- Muntean DM, Sturza A, Danila MD, Borza C, Duicu OM, Mornos C. The role of mitochondrial reactive oxygen species in cardiovascular injury and protective strategies. Oxid Med Cell Longev 2016; 2016: 8254942. doi: 10.1155/2016/8254942
- Golbidi S, Laher I. Molecular mechanism in exercise-induced cardioprotection. Cardiol Res Pract 2011; 2011: 972807. doi: 10.4061/2011/972807.
- Hamilton KL, Quindry JC, French JP, et al. MnSOD antisense treatment and exercise-induced protection against arrhythmias. Free Radic Biol Med 2004; 37: 1360-1368.
- Lennon SL, Quindry JC, Hamilton KL, et al. Elevated MnSOD is not required for exercise-induced cardioprotection against myocardial stunning. Am J Physiol Heart Circ Physiol 2004; 287: H975-H980.
- Taylor RP, Starnes JW. Reactive oxygen species are not a required trigger for exercise-induced late preconditioning in the rat heart. Am J Physiol Regul Integr Comp Physiol 2012; 303: R968-R974.
- Inoue I, Nagase H, Kishi K, Higuti T. ATP-sensitive K+ channel in the mitochondrial inner membrane. Nature 1991; 352: 244-247.
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature 1983; 305: 147-148.
- Ardehali H. Role of the mitochondrial ATP-sensitive K+ channels in cardioprotection. Acta Biochim Pol 2004; 51: 379-390.
- Gross GJ, Peart JN. KATP channels and myocardial preconditioning: an update. Am J Physiol Heart Circ Physiol 2003; 285: H921-H930.
- Parra VM, Macho P, Domenech RJ. Late cardiac preconditioning by exercise in dogs is mediated by mitochondrial potassium channels. J Cardiovasc Pharmacol 2010; 56: 268-274.
- Jang YH, Kim JH, Lee YC. Mitochondrial ATP-sensitive potassium channels play a role in reducing both myocardial infarction and reperfusion arrhythmia in remote ischemic preconditioned hearts. Anesth Pain Med 2017; 7: e42505. doi: 10.5812/aapm.42505
- Hutter JJ, Mestril R, Tam EK, Sievers RE, Dillmann WH, Wolfe CL. Overexpression of heat shock protein 72 in transgenic mice decreases infarct size in vivo. Circulation 1996; 94: 1408-1411.
- Demirel HA, Hamilton KL, Shanely RA, Tumer N, Koroly MJ, Powers SK. Age and attenuation of exercise-induced myocardial HSP72 accumulation. Am J Physiol Heart Circ Physiol 2003; 285: H1609-H1615.
- Quindry JC, Hamilton KL, French JP, et al. Exercise-induced HSP-72 elevation and cardioprotection against infarct and apoptosis. J Appl Physiol 2007; 103: 1056-1062.
- Totzeck M, Hendgen-Cotta UB, Rassaf T. Nitrite-nitric oxide signaling and cardioprotection. Adv Exp Med Biol 2017; 982: 335-346.
- Lefer DJ. Nitrite therapy for protection against ischemia-reperfusion injury. Am J Physiol Renal Physiol 2006; 290: F777-F778.
- Zweier JL, Wang P, Samouilov A, Kuppusamy P. Enzyme-independent formation of nitric oxide in biological tissues. Nat Med 1995; 1: 804-899.
- Babai L, Szigeti Z, Parratt JR, Vegh A. Delayed cardioprotective effects of exercise in dogs are aminoguanidine sensitive: possible involvement of nitric oxide. Clin Sci (Lond) 2002; 102: 435-445.
- Hajnal A, Nagy O, Litvai A, Papp J, Parratt JR, Vegh A. Nitric oxide involvement in the delayed antiarrhythmic effect of treadmill exercise in dogs. Life Sci 2005; 77: 1960-1971.
- Taylor RP, Olsen ME, Starnes JW. Improved postischemic function following acute exercise is not mediated by nitric oxide synthase in the rat heart. Am J Physiol Heart Circ Physiol 2007; 292: H601-H607.
- Farah C, Nascimento A, Bolea G, et al. Key role of endothelium in the eNOS-dependent cardioprotection with exercise training. J Mol Cell Cardiol 2017; 102: 26-30.
- Gangoiti P, Camacho L, Arana L, et al. Control of metabolism and signaling of simple bioactive sphingolipids: Implications in disease. Prog Lipid Res 2010; 49: 316-334.
- Huwiler A, Kolter T, Pfeilschifter J, Sandhoff K. Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochim Biophys Acta 2000; 1485: 63-99.
- Beresewicz A, Dobrzyn A, Gorski J. Accumulation of specific ceramides in ischemic/reperfused rat heart; effect of ischemic preconditioning. J Physiol Pharmacol 2002; 53: 371-382.
- Dobrzyn A, Knapp M, Gorski J. Effect of acute exercise and training on metabolism of ceramide in the heart muscle of the rat. Acta Physiol Scand 2004; 181: 313-319.
- Bielawska AE, Shapiro JP, Jiang L, et al. Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am J Pathol 1997; 151: 1257-1263.
- Borodzicz S, Czarzasta K, Kuch M, Cudnoch-Jedrzejewska A. Sphingolipids in cardiovascular diseases and metabolic disorders. Lipids Health Dis 2015; 14: 55. doi: 10.1186/s12944-015-0053-y
- Knapp M. Cardioprotective role of sphingosine-1-phosphate. J Physiol Pharmacol 2011; 62: 601-607.
- Li N, Zhang F. Implication of sphingosine-1-phosphate in cardiovascular regulation. Front Biosci (Landmark Ed) 2016; 21: 1296-1313.
- Alvarez SE, Milstien S, Spiegel S. Autocrine and paracrine roles of sphingosine-1-phosphate. Trends Endocr Metab 2007; 18: 300-307.
- Takabe K, Paugh SW, Milstien S, Spiegel S. ‘Inside-out’ signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev 2008; 60: 181-195.
- Baranowski M, Charmas M, Dlugolecka B, Gorski J. Exercise increases plasma levels of sphingoid base-1 phosphates in humans. Acta Physiol (Oxf) 2011; 203: 373-380.
- Knapp M, Baranowski M, Czarnowski D, et al. Plasma sphingosine-1-phosphate concentration is reduced in patients with myocardial infraction. Med Sci Monit 2009; 15: CR490-CR493.
- Knapp M, Lisowska A, Zabielski P, Musial W, Baranowski M. Sustained decrease in plasma sphingosine-1-phosphate concentration and its accumulation in blood cells in acute myocardial infarction. Prostaglandins Other Lipid Mediat 2013; 106: 53-61.
- Knapp M, Zendzian-Piotrowska M, Blachnio-Zabielska A, Zabielski P, Kurek K, Gorski J. Myocardial infarction differentially alters sphingolipid levels in plasma, erythrocytes and platelets of the rat. Basic Res Cardiol 2012; 107: 294-310.
- Sattler K, Levkau B. Sphingosine-1-phosphate as a mediator of high-density lipoprotein effects in cardiovascular protection. Cardiovasc Res 2009; 82: 201-211.
- Egom EE, Mohamed TM, Mamas MA, et al. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am J Physiol Heart Circ Physiol 2011; 301: H1487-H95.
- Bilinska M, Rudnicki S, Beresewicz A. Delayed attenuation of myocardial ischemia with repeated exercise in subjects with stable angina: a possible model for the second window of protection? Basic Res Cardiol 2000; 95: 418-23.
- Lambiase PD, Edwards RJ, Cusack MR, Bucknall CA, Redwood SR, Marber MS. Exercise-Induced Ischemia initiates the second window of protection in humans independent of collateral recruitment. J Am Coll Cardiol 2003; 41: 1174-1182.
- Crisafulli A, Melis F, Tocco F, et al. Exercise-induced and nitroglycerin-induced myocardial preconditioning improves hemodynamics in patients with angina. Am J Physiol Heart Circ Physiol 2004; 287: H235-H242.
- Koning MM, Gho BC, van Klaarwater E, Opstal RL, Duncker DJ, Verdouw PD. Rapid ventricular pacing produces myocardial protection by nonischemic activation of KATP+ channels. Circulation 1996; 93: 178-186.
- Domenech RJ, Macho P, Velez D, Sanchez G, Liu X, Dhalla N. Tachycardia preconditions infarct size in dogs: role of adenosine and protein kinase C. Circulation1998; 97: 786-794.
- Bergman BC, Tsvetkova T, Lowes B, Wolfel EE. Myocardial glucose and lactate metabolism during rest and atrial pacing in humans. J Physiol 2009; 587: 2087-2099.
- Camici P, Marraccini P, Marzilli M, et al. Coronary hemodynamics and myocardial metabolism during and after pacing stress in normal humans. Am J Physiol 1989; 257: E309-E317.
- Bergman BC, Tsvetkova T, Lowes B, Wolfel EE. Myocardial FFA metabolism during rest and atrial pacing in humans. Am J Physiol Endocrinol Metab 2009; 296: E358-E366.
- Wojcik B, Baranowski M, Chabowski A, Gorski J. Effect of atrial pacing on the level of bioactive sphingolipids in the heart ventricles of the rat. J Physiol Pharmacol 2015; 66: 385-389.
- Wojcik B, Harasim E, Zabielski P, Chabowski A, Gorski J. Effect of tachycardia on lipid metabolism and expression of fatty acid transporters in the heart ventricles of the rat. J Physiol Pharmacol 2015; 66: 691-699.
- Wojcik B, Miklosz A, Zabielski P, Chabowski A, Gorski J. Effect of tachycardia on mRNA and protein expression of the principal components of the lipolytic system in the rat’s heart ventricles. J Physiol Pharmacol 2017; 68: 731-736.
- Burelle Y, Wambolt RB, Grist M, et al. Regular exercise is associated with a protective metabolic phenotype in the rat heart. Am J Physiol Heart Circ Physiol 2004; 287: H1055-H1063.
- Dai Z, Wu F, Yeung EW, Li Y. IGF-1Ec expression, regulation and biological function in different tissues. Growth Horm IGF Res 2010; 20: 275-281.
- Goldspink G, Booth F. General remarks - mechanical signals and gene expression in the muscle. Am J Physiol 1992; 262: R327-R328.
A c c e p t e d : April 24, 2018