Plasma renin acitvity (PRA) has been found to either increase or decrease during acute hypoxia in humans as well as in animals (3-5). Among the reasons for these contradictory results are, e.g., lacking differentiation between resting states and exercise (4), uncontrolled salt- and water intake (5), and/or different experimental procedures such as CO2 inhalation to prevent the fall in PCO2 caused by hypoxia induced hyperventilation (see discussion section for further details).
Nifedipine is currently being used as an adjunct treatment of high-altitude pulmonary edema (HAPE) if supplemental oxygen is unavailable and descent is impossible (6), or if prophylaxis of HAPE is recommended (7), since nifedipine has been shown to reduce pulmonary arterial pressure and pulmonary vascular resistance during acute hypoxia. However, the possible inhibition of the hypoxia-induced decrease in PRA and Ang II concentration by nifedipine could be disadvantageous if it was to reduce urinary output.
Therefore, the objective of the present study was to find out whether nifidepine would inhibit the decrease in PRA, AngII, and PAC during acute hypoxia in conscious, resting dogs on a controlled salt and water intake. This was investigated together with the changes in hemodynamics and renal function that ensue from such a treatment.
Animals, maintenance, and diets
A total of 16 experiments was performed on eight purebred female Beagle dogs (body weight (wt) 12.2 ± 0.7 kg), two experiments on each dog. The intervals between the two experiments on the same dog were at least 14 days. The dogs were obtained from the Central Animal Facilities of the Humboldt-University in Berlin. The permission to perform the experiments was obtained from the Governmental Animal Protection Committee (AZ 0183/97).
A permanent tracheotomy was performed four to five weeks prior to the experiments (for details see Ref. 2). Thereafter, the dogs were trained to lie quietly on their right side on a padded animal table for at least 4 hours. The dogs were kept under standardized environmental conditions (for details see Ref.8). They were fed a low sodium diet, starting at least 7 days before the experiments. The diet consisted of minced beef (12 g), and boiled rice (58 g), and contained 0.5 mmol sodium, 91 ml water, and 3.5 mmol potassium (all values given per kg body weight per day). To ease the detection of small decreases in PRA, we stimulated the RAAS by a low sodium diet (0.5 mmol Na·kg bodywt-1·day-1), which was given for seven days before the experiments. If food intake is uncontrolled or rich in sodium, PRA and PAC values are often very low, and changes in PRA due to hypoxia and/or nifedipine infusion may be less apparent.
At least eight days prior to an experiment, 100 ml of the dog's own blood was collected via puncture of a foreleg vein and stored in a blood bag at 4°C (Biopack®, Biotrans, Dreieich, Germany). This blood served to replace the blood withdrawn for analysis during the experiments. No further fluid was administered during the experiment.
Experimental protocols
Preparation for the experiments started at 7:30 AM with the recording of the dog's body temperature and body weight. Thereafter, a foreleg vein was punctured and a creatinine infusion started for assessment of glomerular filtration rate (GFR) (exogenous creatinine clearance: priming dose 1.4 g for 30 min, maintenance infusion 4.7 mg/min). Urine was collected via a self retaining bladder catheter inserted through the urethra. An arterial line - for continuous blood pressure monitoring and blood sampling - and a pulmonary artery catheter were inserted using local anesthesia (for details see Ref. 8). After these procedures, the awake dog was placed on a padded animal table and positioned on its right side. The pressure transducers were adjusted to the level of the right atrium. The distance between transducer and table was recorded and also used for the next experiment in this individual dog. Finally, the tracheal tube was inserted, the cuff inflated, and the dog connected to the ventilator set to CPAP mode (CPAP level 4 mmHg). We used the widest tube that would fit the tracheostoma (mostly 8 mm I.D.) in order to decrease respiratory resistance. Thereafter, the conscious dogs were given 60 min to calm down and adjust to the experimental situation.
Each of the 8 dogs underwent two protocols in randomized order: control experiments (Control) and nifedipine experiments (Nifedipine). In Control experiments the dogs breathed room air (21 % O2, 79 % N2; normoxia) for one hour, followed by breathing a gas mixture containing 10 % O2 and 90 % N2 for two hours (hypoxia). In the Nifedipine experiments, the dogs also breathed room air for one hour (normoxia), after which time they were infused nifedipine (Adalat®, Bayer AG, Leverkusen, Germany; 1.5 µg·kg body wt-1·min-1) for 30 min before the two hours of hypoxia were started. During hypoxia the nifedipine infusion was continued at the same rate. The nifedipine solution was protected from light at all time. The additional sodium intake through the nifedipine solution was 0.02 mmol Na·kg body wt-1·h-1.
Heart rate (HR), mean arterial blood pressure (MAP), and central venous pressure (CVP) were measured continuously and the data stored on a personal computer. Cardiac output was measured using the thermodilution technique (5 ml injection volume at 5 - 10°C). Five consecutive measurements were performed and the highest and lowest values rejected. The mean cardiac output was calculated from the remaining three determinations and taken for calculation of systemic vascular resistance (SVR) by the standard formula.
Blood samples were taken at the end of each experimental hour to determine arterial blood gases, plasma electrolytes, creatinine, osmolarity, and hormone concentrations. The blood withdrawn was immediately replaced with an equal amount of the dog's own stored blood using a blood filter (TNSB-3, Biotest, Alzenau, Germany).
Urinary sodium, water, potassium, and creatinine excretions and osmolarity were measured hourly after complete evacuation of the urinary bladder (air washout). Exogenous creatinine clearance was calculated by the standard formula to assess glomerular filtration rate (GFR) (see above).
Assays
Blood samples for hormone analysis were collected into precooled Na-EDTA vials, and centrifuged at 4°C. The plasma was separated and stored at -22°C until analysis. Commercially available radioimmunoassay kits were used to measure plasma renin activity (PRA), concentration of angiotensin II (Ang II), aldosterone (PAC), atrial natriuretic peptide (ANP), antidiuretic hormone (ADH = vasopressin), and angiotensin converting enzyme activity (ACE) (for details see 2).
Plasma and urinary sodium and potassium concentrations were measured by flame photometry (Photometer Eppendorf, Hamburg, Germany), and creatinine with a creatinine analyzer (modified Jaffé reaction, Beckmann Instruments, Brea, USA). Plasma and urinary osmolarity was measured by cryoscopy (Osmometer, Roebling, Berlin, Germany). Blood gas analysis was performed at hourly intervals (ABL 505, Radiometer, Copenhagen, Denmark).
Statistical analysis
All values are given as means ± SE (n = 8). Intergroup comparison, i.e., Control vs. Nifedipine during the respective normoxia and hypoxia period, was performed using Student's t-test. For intra-group comparison (time course) a general linear model of analysis of variance (GLM ANOVA) for repeated measures was used (SPSS 9.0, Chicago, IL, USA). Post-hoc testing of means was performed with Student's t-test with Bonferroni correction for multiple comparisons. Statistical significance was considered at P < 0.05 (* = significant vs. normoxia, § = significant vs. Controls).
Minute ventilation, arterial blood gases, pH, plasma values
During hypoxia, arterial O2 tension (PaO2) decreased from ~95 Torr to 37 - 38 Torr and arterial carbon dioxide tension (PaCO2) from ~35 to 24 - 27 Torr in both protocols (P < 0.05) (Table 1). Minute ventilation increased about 1 - 1.9 l/min in both protocols during hypoxia (P < 0.05) (Table 1). Standard bicarbonate concentration (20 - 21.1 mmol/l), base excess (-4.5 -5.7 mmol/l), plasma sodium concentration (143 - 145 mmol/l) and plasma osmolarity (294 - 300 mosmol/l) were not different between both protocols and remained unchanged during hypoxia. Plasma potassium concentration decreased during hypoxia in both Control and Nifedipine experiments (P < 0.05) (Table 1).
| Table 1. Arterial blood gases, pH, plasma potassium concentration, and minute ventilation during Control and Nifedipine experiments. |
 |
| PaO2, arterial oxygen tension; PaCO2, arterial carbon dioxide tension; PK, plasma potassium concentration; VE, minute ventilation. Values measured during one hour of normoxia (21 % inspiratory O2 concentration) and two hours of hypoxia (10 % inspiratory O2 concentration). Means ± SE, n = 8; *P < 0.05 vs. normoxia. |
Hemodynamics
During hypoxia, MAP increased in Controls (P < 0.05) and decreased in Nifedipine experiments (P < 0.05) (Fig. 1). Heart rate did not change in Controls, but increased in Nifedipine experiments during hypoxia (P < 0.05) (Table 2). Cardiac output (CO) increased slightly during hypoxia in Controls (2.2 ± 0.2 to 2.6 ± 0.2 l/min, P < 0.05). The CO increase in Nifedipine experiments was more pronounced (2.2 ± 0.1 to 3.4 ± 0.3 l/min; P < 0.05) (Table 2). Systemic vascular resistance did not change during hypoxia in Controls, whereas it decreased in Nifedipine experiments (P < 0.05) (Fig. 1). Central venous pressure was similar in both protocols and remained unchanged throughout the experiments (Table 2).
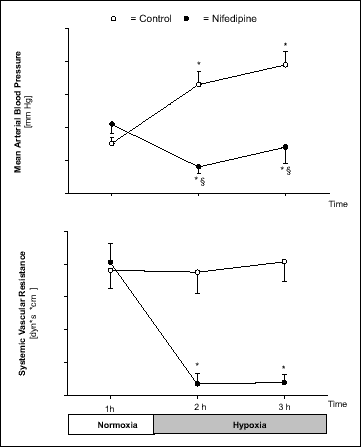 |
Fig. 1. Mean arterial pressure, and systemic vascular resistance during 1 h of normoxia (FiO2 = 0.21) and 2 h of hypoxia (FiO2 = 0.1) in Control and Nifedipine experiments. Values are means ± SE (n = 8). * P< 0.05 vs. normoxia, § P < 0.05 vs. Control. |
| Table 2. Hemodynamic parameters during Control and Nifedipine experiments. |
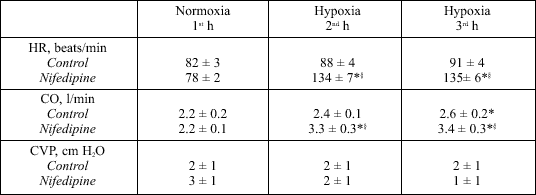 |
| HR, heart rate; CO, cardiac output; CVP, central venous pressure. Values measured during one hour of normoxia (21 % inspiratory O2 concentration) and two hours of hypoxia (10 % inspiratory O2 concentration). Means ± SE, n = 8; *P < 0.05 vs. normoxia, §P < 0.05 vs. Control. |
Plasma hormones
PRA and Ang II decreased during hypoxia in Controls (P < 0.05), but increased in Nifedipine experiments (P < 0.05) (Fig. 2). PRA correlated inversely with mean arterial pressure (MAP) in Controls (r = 0.71, P = 0.002), but not in Nifedipine experiments (r = 0.089, p = 0.742) (Fig. 3). In Controls PAC decreased swiftly with hypoxia, whereas in Nifedipine experiments the decrease was delayed (P < 0.05) (Fig. 2). Plasma concentrations of ANP (36 - 40 pg/ml), ACE (45 - 51 U/l), and ADH (0.2 - 0.6 pg/ml) were similar in both protocols and did not change during hypoxia in either protocol.
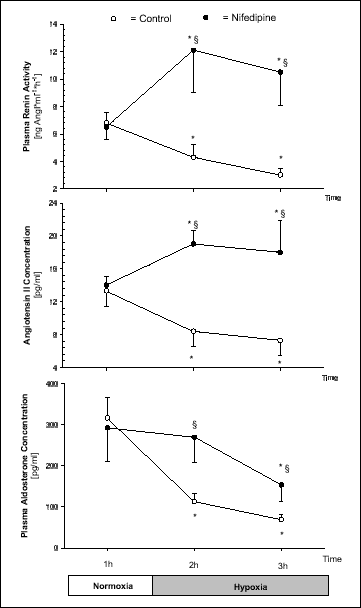 |
Fig. 2. Plasma renin activity, angiotensin II concentration, and plasma aldosterone concentration during 1 h of normoxia (FiO2 = 0.21) and 2 h of hypoxia (FiO2 = 0.1) in Control and Nifedipine experiments. Values are means ± SE (n = 8). * P< 0.05 vs. normoxia, § P < 0.05 vs. Control. |
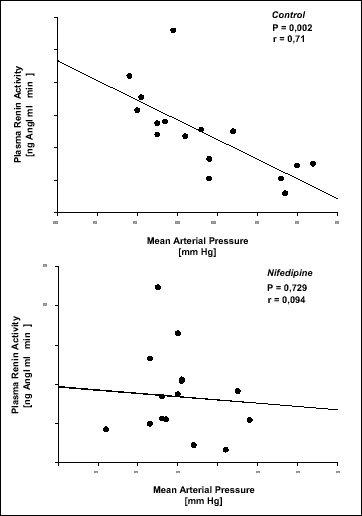 |
Fig. 3. Plasma renin activity of each dog in Control (n = 8) and Nifedipine (n = 8) experiments during normoxia and hypoxia plotted against mean arterial pressure at the same time. |
Renal function data
In Controls urine volume (P < 0.05) and urinary potassium excretion increased ~50 % during hypoxia (Table 3). In Nifedipine experiments urine volume decreased during hypoxia (P < 0.05), whereas urinary potassium excretion (P < 0.05) increased (Table 3). In Controls urinary sodium excretion, fractional excretion of sodium and glomerular filtration rate did not change during hypoxia (Table 3). In Nifedipine experiments glomerular filtration rate also did not change, whereas urinary sodium excretion and fractional excretion of sodium increased during hypoxia (P < 0.05) (Table 3). Urinary osmolarity decreased (P < 0.05) during hypoxia in Controls, but increased in Nifedipine experiments (P < 0.05) (Table 3).
| Table 3. Renal excretion parameters during Control and Nifedipine experiments. |
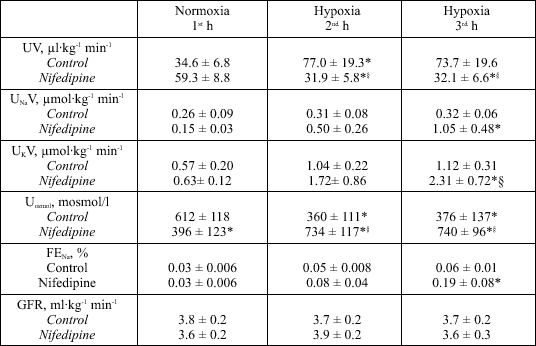 |
| UV, urine volume; UNaV, sodium excretion; UKV, potassium excretion; Uosmol, urinary osmolarity; FENa, fractional excretion of sodium; GFR, glomerular filtration rate, all values given per kilogram body weight (kg) and minute (min). Values were measured during one hour of normoxia (21 % inspiratory O2 concentration) and two hours of hypoxia (10 % inspiratory O2 concentration). Means ± SE, n = 8; *P < 0.05 vs. normoxia. §P < 0.05 vs. Control. |
The aim of the present study was to find out, whether the calcium channel antagonist nifedipine would inhibit the hypoxia-induced decrease in plasma renin activity. The experiments were performed on eight conscious, tracheotomized Beagle dogs. In a three hour protocol, the dogs were breathing room air for one hour, and a hypoxic gas mixture for two hours. The results demonstrated that nifedipine is able to inhibit the hypoxia-induced decrease in PRA.
The present study describes basic physiological and pathophysiological mechanisms during acute hypoxia in conscious, resting dogs with all their regulatory mechanisms intact. The significance of the results obtained is a matter of speculation. The decrease in PRA and AngII in Controls may result in less peripheral vasoconstriction and can be considered a physiological advantage to guarantee tissue oxygenation during hypoxia. The decrease of PAC at the same time may stimulate sodium and water excretion during hypoxia and improve oxygen transport by the resulting hemoconcentration.
Hormones
In Controls, plasma renin activity and angiotensin II concentration decreased during hypoxia (Fig. 1). This is in accordance with former studies from our laboratory (1, 2, 8), but there are also studies, in which PRA increased during acute hypoxia (4, 5). This can be explained by, e.g., uncontrolled sodium and water intake (5) and/or not differentiating between resting states and exercise (4). Furthermore, the experimental situation has to be taken into account, e.g., in some studies the arterial CO2 tension during hypoxia could not be reduced through hyperventilation by the subjects under investigation, because the studies were performed during general anesthesia with controlled mechanical ventilation, or CO2 had been mixed to the inspiratory gas (9), or the degree of hypoxia was very severe (FiO2 < 0.07). These conditions may all increase PRA and AngII levels, e.g., via a very high degree of stress and consecutively sympathetic stimulation (10). With our experimental setup (FiO2 = 0.1) and well trained conscious animals, who hyperventilated as a physiological response towards hypoxia, we minimized these non-physiological conditions.
Many adaptive responses to hypoxia involve the regulation of specific genes. These responses depend, e.g., on whether hypoxia is acute, chronic, or intermittent. In primary cultures of renal juxtaglomerular cells (JGC) of rats hypoxia per se had no influence on renin secretion and renin gene expression (11). Thus, other mediators have to play a role in regulating renin secretion from the juxtaglomerular cells during acute hypoxia. One potential mediator is adenosine, which has recently been suggested to mediate the renin decrease via inhibition of adenylate cyclase (1). Beside adenosine, mechanisms such as the stimulation of guanylate cyclase or phospholipase C can also mediate a decrease in PRA (12). Furthermore, ATP-sensitive potassium channel stimulators increase and inhibitors decrease plasma renin acitivity (13). A further intriguing feature of the control of renin secretion is called the "calcium paradox" and describes the observation that a decrease in cytoplasmatic calcium concentration increases the renin secretion from the juxtaglomerular cells and vice versa (14, 15). Our present Nifedipine experiments support this notion, because PRA and Ang II increased after application of the calcium channel antagonist instead of decreasing during hypoxia as in Controls (Fig. 1). The increase in PRA could be a direct effect of nifedipine on the juxtaglomerular cells (16), since it has been shown that L-type calcium channel blockade is able to stimulate renin release through a decrease in intracellular calcium (14).
In addition, an increase in renal sympathetic nerve activity (RSNA) may trigger the increase in PRA during hypoxia (17). In Controls, hypoxia decreased PRA, indicating that the RSNA was not significantly increased by hypoxia per se. In contrast, nifedipine is known to stimulate the cardiac sympathetic nervous system (e.g., tachycardia occurs) (18), and the renal sympathetic nervous system indirectly (19).
Furthermore, a decrease in mean arterial pressure, and thus renal perfusion pressure, may have increased PRA in Nifedipine experiments. In conscious dogs the threshold pressure for the pressure-dependent renin release has been determined to be about 89 mm Hg (20). Since the lowest MAP during Nifedipine experiments in our study was about 83 mm Hg (Fig. 1), it is unlikely that this decrease alone can explain the striking increase in PRA from 6.5 to 12.1 ng AngI·ml-1·h-1 during hypoxia. If PRA is plotted against mean arterial pressure (Fig. 3), the correlation is high only in Control experiments, whereas in Nifedipine experiments the mechanism of pressure-dependent renin release seems to be suspended, i.e., there is no correlation at all. Indicating, that the increase in PRA during hypoxia with simultaneous calcium antagonist treatment is mainly brought about by factors other than the slight decrease in MAP (Fig. 1).
Plasma aldosterone concentration decreased during hypoxia in both groups, but the effect was less pronounced in Nifedipine experiments (Fig. 2). The decrease of PAC in Control experiments is most likely due to a direct hypoxic inhibition of aldosterone secretion from the suprarenal glands (21). In Nifedipine experiments the increase in PRA and Ang II may counteract the hypoxia induced decrease in PAC.
Renal excretion
In Control experiments, urine volume and potassium excretion increased about fifty percent during hypoxia (Table 3). This is in accordance with previous data from our laboratory (8). Opposite to Controls, urine volume decreased during hypoxia in Nifedipine experiments (Table 3). The increase in angiotensin II concentration in Nifedipine experiments may partly account for this finding (22). An increase in ADH seems not to be involved since the ADH concentration did not differ between the normoxia and hypoxia period (see Results section).
The striking increase in potassium excretion during hypoxia in Nifedipine experiments (Table 3) may partly be explained by the greater aldosterone levels compared with Controls.
The unchanged urinary sodium excretion in Control experiments was accompanied by an increase in urine volume, resulting in a decrease in urinary osmolarity during hypoxia (Table 3). This is in accordance with former data from our laboratory obtained on a low sodium diet during hypoxia (1). Normally, a decrease in PAC during hypoxia - as observed in Controls - would be expected to increase sodium excretion ("high altitude natriuresis"; i.e., 23). On a low sodium diet, however, the organism has to defend against sodium losses and the hypoxia-induced decrease in Ang II and plasma aldosterone concentrations in Controls seems to be too small to bring about a measurable increase in renal sodium excretion under these circumstances. Interestingly, in Nifedipine experiments an increase in urinary sodium excretion of about 85% was observed during hypoxia (Table 3). This increase in sodium excretion occurred on the same low sodium diet as in Controls and is probably brought about by a direct tubular effect of nifedipine (24) since it occurred in the face of an increase in FENa without any change in the glomerular filtration rate (Table 3). This nifedipine induced natriuresis was not outweighed by the concomitant increase in Ang II and aldosterone concentrations observed during the hypoxia period in Nifedipine experiments.
Atrial natriuretic peptide (ANP) was suggested to initiate hypoxic natriuresis in conscious lambs (25). In our experiments, ANP concentrations did not change. Thus, ANP does not seem to play a pivotal role for acute hypoxic natriuresis in conscious dogs, which is in accordance with results in humans (26).
Hemodynamics
During the hypoxia period the Nifedipine dogs showed a decrease in systemic vascular resistance, a pronounced increase in cardiac output, a moderate decrease in MAP, and a striking increase in heart rate, while in Control experiments the systemic vascular resistance remained stable during hypoxia, because the slight increase in cardiac output paralleled the increase in mean arterial pressure (Fig. 1). It is speculated that the cardiovascular effects during hypoxia are mainly due to sympathetic stimulation of the cardiovascular system in Controls (27). The pharmacological properties of nifedipine, e.g., on the sympathetic nervous system (18), are able to modify and overrule the physiological cardiovascular effects of hypoxia (Fig. 1, Table 2).
We conclude that the hypoxia-induced decrease in PRA is among others a calcium dependent mechanism. The L-type voltage-dependent calcium channel antagonist nifedipine increases plasma renin activity during hypoxia possibly by decreasing intracellular calcium in the juxtaglomerular cells and/or by increasing renal sympathetic nerve activity.
Acknowledgment: The authors are indebted to Rainer Mohnhaupt for help with the statistics, to Birgit Brandt and Daniela Bayerl for expert technical assistance, and to April M. Kurzke for editorial help. This study was supported by a grant from the Deutsche Forschungsgemeinschaft to Gabriele Kaczmarczyk (Ka 526/5-2).
- Höhne C, Krebs MO, Arntz E, Boemke W, Kaczmarczyk G. Evidence that the renin decrease during hypoxia is adenosine-mediated in conscious dogs. J Appl Physiol 2001; 90: 1842-1848.
- Krebs MO, Boemke W, Simon S, Wenz M, Kaczmarczyk G. Acute hypoxic pulmonary vasoconstriction in conscious dogs decreases renin and is unaffected by losartan. J Appl Physiol 1999; 86: 1914-1919.
- Keynes RJ, Smith GW, Slater JD, Brown MM, Brown SE, Payne NN, Jowett TP, Monge CC. Renin and aldosterone at high altitude in man. J Endocrinol 1982; 92: 131-140.
- Neylon M, Marshall J, Johns EJ. The role of the renin-angiotensin system in the renal response to moderate hypoxia in the rat. J Physiol (Lond) 1996; 491: 479-488.
- Skwarski KM, Morrison D, Barratt A, Lee M, MacNee W. Effects of hypoxia on renal hormonal balance in normal subjects and in patients with COPD. Respir Med 1998; 92: 1331-1336.
- Hackett PH, Roach RC. High-altitude illness. N Engl J Med 2001; 345: 107-114.
- Bärtsch P, Maggiorini M, Ritter C, Noti P, Oelz O. Prevention of high-altitude pulmonary edema by nifedipine. N Engl J Med 1991; 325: 1284-1289.
- Höhne C, Boemke W, Schleyer N, Francis RCE, Krebs MO, Kaczmarczyk G. Low sodium intake does not impair short-term renal compensation of hypoxia-induced respiratory alkalosis in conscious dogs. J Appl Physiol 2002; 92: 2097-2104.
- Malpas SC, Shweta A, Anderson WP, Head GA. Functional responses to graded increases in renal nerve activity during hypoxia in conscious rabbits. Am J Physiol 1996; 271: R1489-R1499.
- Raff H, Roarty TP. Renin, ACTH, and aldosterone during acute hypercapnia and hypoxia in conscious rats. Am J Physiol 1988; 254: R431-R435.
- Ritthaler T, Schricker K, Kees F, Kramer B, Kurtz A. Acute hypoxia stimulates renin secretion and renin gene expression in vivo but not in vitro. Am J Physiol 1997; 272: R1105-1011.
- Jackson EK. ADENOSINE: A physiological brake on renin release. Annu Rev Pharmacol Physiol Toxicol 1991; 31: 1-35.
- Pratz J, Mondat S, Montier F, Cavero I. Effects of the K+ channel activators, RP52891, cromakalim and diazoxide, on the plasma insulin level, plasma renin activity an blood pressure in rats. J Pharmacol Exp Ther 1991; 258: 216-222.
- Hackenthal E, Paul M, Ganten D, Taugner R. Morphology, physiology, and molecular biology of renin secretion. Physiol Rev, 1990; 70:1067-1116.
- Wagner C, Krämer BK, Hinder M, Kieninger M, Kurtz A. T-type and L-type calcium channel blockers exert opposite effects on renin secretion and renin gene expression in conscious rats. Br J Pharmacol 1998; 124: 579-585.
- Schweda F, Riegger GAJ, Kurtz A, Krämer BK. Store-operated calcium influx inhibits renin secretion. Am J Physiol 2000; 279: F170-176.
- Denton KM, Shweta A, Anderson WP. Preglomerular and postglomerular resistance response to different levels of sympathetic activation by hypoxia. J Am Soc Nephrol 2002; 13: 27-34.
- Wenzel RR, Bruck H, Noll G, Schäfers RF, Daul AE, Philipp T. Antihypertensive drugs and the sympathetic nervous system. J Cardiovasc Pharmacol 2000; 35: 43-52.
- Huang BS, Leenen FH. Sympathoinhibitory effects of central nifedipine in spontaneously hypertensive rats on high versus regular sodium intake. Hypertension 1999; 33: 32-35.
- Finke R, Gross R, Hackenthal E, Huber J, Kirchheim HR. Threshold pressure for the pressure-dependent renin release in the autoregulating kidney of conscious dogs. Pflügers Arch 1983; 399: 102-110.
- Brickner RC, Jankowski B, Raff H. The conversion of corticosterone to aldosterone is the site of the oxygen sensitivity of the bovine adrenal zona glomerulosa. Endocrinology 1992; 130: 88-92.
- Keil J, Lehnfeld R, Reinhardt HW, Mohnhaupt R, Kaczmarczyk G. Acute effects of angiotensin II on renal haemodynamics and excretion in conscious dogs. Ren Physiol Biochem 1989; 12: 238-249.
- Swenson ER, Duncan TB, Goldberg SV, Ramirez G, Ahmad S, Schoene RB. Diuretic effect of acute hypoxia in humans: relationship to hypoxic ventilatory responsiveness and renal hormones. J Appl Physiol 1995; 78: 377-383.
- Cappuccio FP, Antonios TF, Markandu ND, Folkerd EJ, Sagnella GA, Sampson B, MacGregor GA. Acute natriuretic effect of nifedipine on different sodium intakes in essential hypertension: evidence for distal tubular effect? J Hum Hypertens 1994; 8: 627-630.
- Baertschi AJ, Teague WG. Alveolar hypoxia is a powerful stimulus for ANP release in conscious lambs. Am J Physiol 1989; 256: 990-998.
- Hildebrandt W, Otenbacher A, Schuster M, Swenson ER, Bärtsch P. Diuretic effect of hypoxia, hypocapnia, and hyperpnea in humans: relation to hormones and O2 chemosensitivity. J Appl Physiol 2000; 88: 599-610.
- Rowell L, Blackmon J. Human cardiovascular adjustments to acute hypoxemia. Clin Physiol 1987; 7: 349-376.