| We investigated L-kynurenine distribution and metabolism in rats with experimental chronic renal failure of various severity, induced by unilateral nephrectomy and partial removal of contralateral kidney cortex. In animals with renal insufficiency the plasma concentration and the content of L-tryptophan in homogenates of kidney, liver, lung, intestine and spleen were significantly decreased. These changes were accompanied by increase activity of liver tryptophan 2,3-dioxygenase, the rate-limiting enzyme of kynurenine pathway in rats, while indoleamine 2,3-dioxygenase activity was unchanged. Conversely, the plasma concentration and tissue content of L-kynurenine, 3-hydroxykynurenine, and anthranilic, kynurenic, xanthurenic and quinolinic acids in the kidney, liver, lung, intestine, spleen and muscles were increased. The accumulation of L-kynurenine and the products of its degradation was proportional to the severity of renal failure and correlated with the concentration of renal insufficiency marker, creatinine. Kynurenine aminotransferase, kynureninase and 3-hydroxyanthranilate-3,4-dioxygenase activity was diminished or unchanged, while the activity of kynurenine 3-hydroxylase was significantly increased. We conclude that chronic renal failure is associated with the accumulation of L-kynurenine metabolites, which may be involved in the pathogenesis of certain uremic syndromes. |
| Key words: | L-kynurenine metabolites, experimental uremia, rats |
|
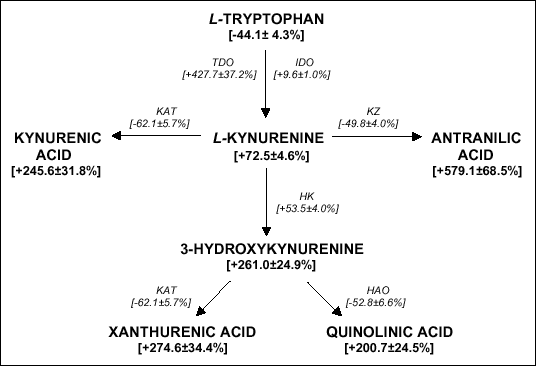
| Fig. 1. Scheme of kynurenine pathway. TDO - tryptophan 2,3-dioxygenase, IDO - indoleamine 2,3-dioxygenase, KAT - kynurenine aminotransferase, KZ - kynureninase, HK - kynurenine 3-hydroxylase, HAO - 3-hydroxyanthranilate-3,4-dioxygenase. The total conentration of TRP metabolites and activity of kynurenic pathway enzymes was prsented (bracketedes). Details are given in the text. |
| Table 1. The effect of experimental chronic renal failure of various severity (CRF 1-3) on biochemical parameters. |
 |
| Values are presented as means ± SEM, n = 8-10. Statistical significance vs control group: *p<0.05, **p<0.01, ***p<0.001. |
| Table 2. Plasma and tissues concentrations of L-kynurenine metabolites. |
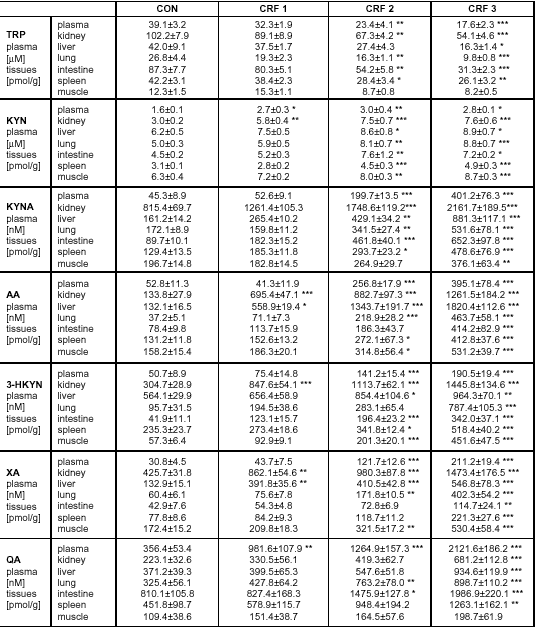 |
| Values are presented as means ± SEM, n = 8-10. Statistical significance vs control group: *p<0.05, **p<0.01, ***p<0.001. TRP - tryptophan, KYN - kynurenine, KAT - kynurenic acid, AA - anthranilic acid, 3-HKYN - 3-hydroxykynurenine, XA - xanthurenic acid, QA - quinolinic acid |
| Table 3. Activity of kynurenine pathway enzymes in chronic renal failures. |
 |
| Values are presented as means ± SEM, n = 8-10. Statistical significance vs control group: *p<0.05, **p<0.01, ***p<0.001. TDO - tryptophan 2,3-dioxygenase, IDO - indoleamine 2,3-dioxygenase, KAT - kynurenine aminotransferase, KZ - kynureninase, HK - kynurenine 3-hydroxylase, HAO - 3-hydroxyanthranilate-3,4-dioxygenase. |
| Table 4. The influence of products of L-kynurenine degradation on enzymes activity. |
 |
| Values are presented as mean ± SEM, n = 6-10. Statistical significance vs control group: **p<0.01, ***p<0.001. KAT - kynurenine aminotransferase, KZ - kynureninase, HAO - 3-hydroxyanthranilate-3,4-dioxygenase |
R e c e i v e d : February 1, 2003
A c c e p t e d : April 24, 2003
Dr Dariusz Pawlak, Department of Pharmacodynamics, Medical Academy, Mickiewicza Str. 2 C, 15-230 Bia³ystok, Poland. Tel/ fax: +48/85/7485601.
e-mail: dariuszpawlak@poczta.onet.pl