The vagus nerve has a wide peripheral distribution and is connected to the hypothalamus through a series of brain nuclei (7, 8). The vagus nerve contains sensory fibres that terminate in the nucleus tractus solitarius and area postrema of the brainstem (9). Pro-inflammatory cytokines are able to induce the release of CRH and arginine vasopressin (AVP) (10, 11). The mechanisms by which cytokines produced in the periphery can activate the hypothalamic CRH/AVP genes in neurons arising in the parvocellular paraventricular nucleus (pPVN) remain unclear (12). Interleukin-1alpha (IL-1alpha), interleukin-1ß (IL-1ß), interleukin-6 (IL-6), interleukin -2 (IL-2) and tumor necrosis factor-alpha (TNF-alpha) can stimulate ACTH release from the anterior pituitary in vitro and in vivo (13). These cytokines increase CRH mRNA and CRH immunoreactivity in the PVN (14) and release CRH and AVP from the hypothalamus. Interleukin (IL)-1ß is a proinflammatory cytokine produced not only in the immune system (e.g. lymphocytes and macrophage), but also in the brain neuronal and glial cells. Peripheral or central application of IL-1ß induces activation of the sympathetic and hypothalamic-pituitary-adrenal axis (15).
Increasing evidence suggest that vagal afferents transmit systemic cytokine information to the brain. Subdiaphragmatic vagotomy blocks or reduces increases in ACTH and corticosteroid secretion and brain norepinephrine changes that follow the intraperitoneal administration of LPS and IL-1ß. In addition, IL-1ß binding sites have been localized on paraganglia, which make afferent synaptic contact with vagal fibres, providing a potential mechanism for cytokine activation of vagal fibres (14, 16).
Vagotomy markedly inhibited stimulation of ACTH secretion by i.p. IL-1ß, indicating that the intra-abdominal stimulation of the vagus plays a critically important role in communicating with the brain and activating the HPA axis acutely (17).
Subdiaphragmatic vagotomy does not alter stimulation of ACTH secretion by not inflammatory stimuli e.g., insulin- or electrofootshock in comparison with sham-operated animals (10). Vagotomy may reduce the sensitivity of the hypothalamic paraventricular nucleus. This would result in either blunting or preventing the HPA response to any stimulus, including those independent of immune-to-brain pathways. Activation of the afferent vagus nerves itself can induce production of IL-1ß in the brain and activate the HPA axis. Therefore, the afferent vagus nerve may play an important role in transmitting peripheral signals to the brain (16). Subdiaphragmatic vagotomy moderately attenuated the IL-1ß-induced increase in plasma ACTH and corticosterone and slightly reduced the responses to catecholamine metabolism in mice. Therefore, the vagus nerve is not the main pathway by which abdominal IL-1ß affects HPA axis and brain catecholamines responses in that species (18).
The purpose of the present study was to evaluate the role of vagus nerve in the cholinergic stimulation, via muscarinic and nicotinic receptors, of the hypothalamic-pituitary-adrenal axis. The experiments were performed on conscious rats that underwent subdiaphragmatic vagotomy.
Adult male Wistar rats (200-250 g) were housed 6 per cage under a 12:12 h light dark cycle at a temperature of 20°C ± 2°C. The rats were allowed free access to rodent chow and drinking water. Animal care and handling throughout the experimental procedures were in accordance with Helsinki declaration. The experimental protocols were approved by the local Ethics Committee.
Vagotomy
Subdiaphragmatic vagotomy was performed on rats as previously described (19, 20). Briefly, after an overnight fast, rats were anesthetized using pentobarbital (25 mg/kg i.p.). After a midline laparotomy, the stomach and lower esophagus were gently exposed in the abdominal cavity. For the total vagotomy, the two vagal trunks were identified on the esophagus, separated from the surrounding tissues under a dissecting microscope and cut as high as possible on the esophagus below the diaphragm. All branches of the ventral subdiaphragmatic vagal nerves including the hepatic, intestinal and accessory celiac branches were cut in all animals. Sham animals were also prepared, by a similar procedure. The viscera were similarly handled, but no nerves were cut. The stomach was returned to its normal position, and the incision was closed in layers. Postoperatively, the animals were observed and weighted daily to monitor their health. Rats that failed to gain body mass were euthanized with pentobarbital. The effectiveness of vagotomy was verified at the end of the study by assessing the relative masses of the stomach (together with its content). Stomachs were carefully isolated, removed and weighted. Transection of the gastric branches causes food retention in the stomach. The mean relative weight of the stomach to the body weight in percent in vagotomized rats was 7.20 ± 3.85 versus 1.75 ± 0.42 in control sham-operated rats (p<0.001).
For drug administration into the lateral cerebral ventricle, the skulls of rats were prepared one day earlier under light ether anesthesia. Required doses of drugs were dissolved in 10 µl of saline and injected using Hamilton microsyringe.
Experimental design
The experiments were carried out in groups of rats in order to determine the selectivity of carbachol and nicotine to cholinergic muscarinic and nicotinic receptors and a site of their action on the HPA axis. In four groups of rats the effects of muscarinic receptor antagonist atropine sulphate, which easily penetrates the blood-brain barrier and atropine methyl bromide, which is not readily transported through this barrier, were given i.p. or i.c.v. 15 min earlier to antagonize the effect of muscarinic receptor agonist, carbachol administered by either route.
In subdiaphragmatically vagotomized rats the effect of carbachol given i.p. on ACTH and corticosterone secretion was determined and compared with the carbachol effect in sham operated rats. In last two groups of vagotomized and sham operated rats the effects of nicotine or saline injected i.p. on ACTH and corticosterone secretion were determined.
One hour after carbachol or nicotine injection the rats were rapidly decapitated without stress within 10 sec after removing the animal from a cage. Trunk blood was collected in plastic tubes on ice-cold bath, and plasma obtained after centrifugation was frozen at -80°C until later ACTH assay. Control animals injected with saline 1 h earlier were decapitated concurrently with the experimental groups to obtain basal plasma ACTH and serum corticosterone levels. For corticosterone determination the trunk blood was centrifuged at room temperature and serum aliquots were frozen until a further assay.
Plasma ACTH concentrations were measured using the double antibody 125I radioimmunoassay obtained from CIS Bio International and calculated as pg/ml of plasma. The concentration of serum corticosterone was determined fluorometrically and expressed as µg/100 ml.
Preparation of drugs
Drugs used in this study were: carbamylcholine hydrochloride (carbachol), (-)-Nicotine ([-]-methyl-2-3-pirydyl]pyrroline) hydrogen tartrate salt, atropine sulphate, atropine methylbromide and mecamylamine hydrochloride (Sigma). All drugs were dissolved in saline immediately before use. The required doses of drugs or saline were injected i.p. in a volume of 2 ml/kg or i.c.v. in a volume of 10 µl.
Analysis of data
The results were calculated as a groups mean ± standard error of the mean. Statistical evaluation was performed by an analysis of variance, followed by individual comparison with Duncan`s test. The results were considered significantly different when the probability value p was below 0.05.
Effect of atropine on carbachol-induced corticosterone secretion
Carbachol (0.5 mg/kg) given systemically considerably increased corticosterone secretion 1 h later (Fig. 1). Pretreatment with atropine bromide (AtrBr) (0.1 mg/kg i.p.) which does not easily penetrate the blood-brain barrier, 15 min earlier significantly reduced the carbachol-induced corticosterone response (Fig. 1A). On the other hand, corticosterone response induced by i.p. carbachol (0.5 mg/kg) was not markedly altered by atropine sulphate (AtrS) (0.1 µg per rat) given i.c.v. 15 min earlier (Fig. 1B). Corticosterone response to carbachol (2µg per rat) given i.c.v. was totally abolished by i.c.v. pretreatment with AtrS (0.1 µg). This result suggests that i.c.v. carbachol selectively activates central muscarinic neuronal receptors in structures involved in HPA axis stimulation (Fig. 2A). By contrast, i.p. pretreatment with Atr Br did not substantially alter the rise in corticosterone response to i.c.v. carbachol (2 µg)-given 15 min later. (Fig. 2B).
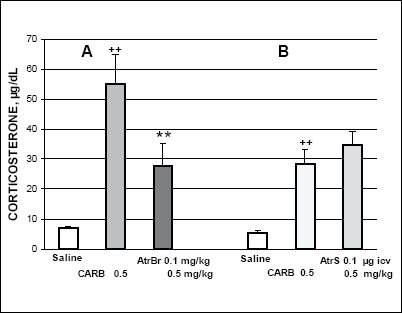 |
Fig. 1. Effects of atropine
hydrobromide (AtrBr) A, and atropine sulphate (AtrS) B on
the carbachol (CARB)-induced corticosterone secretion. AtrBr was given
i.p. and AtrS was injected i.c.v. 15 min before i.p. carbachol. One hour
after the last injection the rats were decapitated. ++p<0.01 vs. saline
control; **p<0.01 vs. carbachol treated group. In Fig. 1-4 values represent the mean ± SEM of 6 rats. |
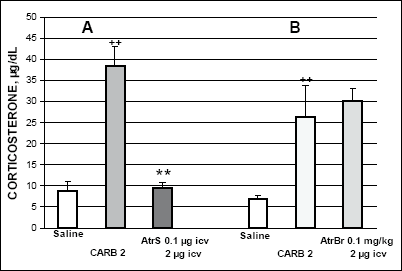 |
Fig. 2. Effects of atropine S A and atropine Br B on the carbachol-induced corticosterone secretion. AtrS was given i.c.v. and AtrBr was injected i.p. 15 min before i.c.v. carbachol. ++p<0.01 vs. saline control; **p<0.01 vs. carbachol treated group. |
These results indicate that significant increase in corticosterone secretion by systemically administered carbachol is induced in a major part by stimulation of peripheral cholinergic muscarinic receptors. On the other hand, carbachol given i.c.v. evokes considerable corticosterone response by stimulation of central cholinergic muscarinic receptors.
Effect of carbachol on ACTH and corticosterone secretion in vagotomized rats
These experiments were performed on rats with total subdiaphragmatic vagotomy performed 2 weeks earlier. Shame operated rats at the time of subdiaphragmatic vagotomy served as controls. In shame-operated rats treated with saline (2 ml/kg i.p.) the basal plasma ACTH levels (102.3 - 105.8 pg/ml) and serum corticosterone levels (2.0 - 8.2 µg/100 ml) were similar to the levels in intact rats. In shame-operated rats carbachol (0.2 and 0.5 mg/kg i.p.) dose dependently increased plasma ACTH levels (to 165.2 pg/ml and 224.3 pg/ml) and serum corticosterone levels (to 21.5 and 31.2 µg/dl, respectively). In vagotomized rats ACTH response to carbachol in a lower dose (0.2 mg/kg) was moderately diminished but was markedly increased after higher dose (0.5 mg/kg). (Fig. 3). In vagotomized rats carbachol in both doses induced substantially higher increase in corticosterone levels than in sham-operated rats (Fig. 3).
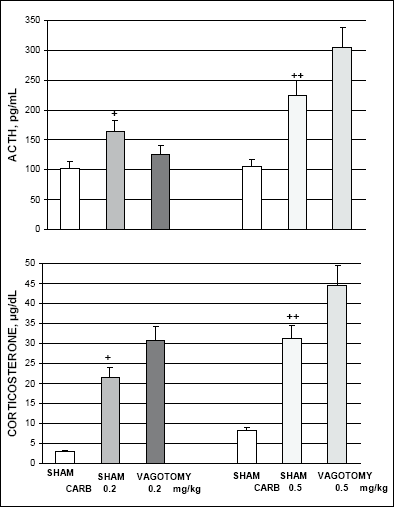 |
Fig. 3. Effect of subdiaphragmatic vagotomy on the carbachol (CARB)-induced ACTH and corticosterone secretion. White bars represent values in sham operated controls injected with saline. +p<0.05 and ++p<0.01 vs. sham control. |
Nicotine stimulates HPA axis response via selective cholinergic nicotinic receptors
We found that nicotine (5 mg/kg) given systemically stimulates ACTH and corticosterone secretion by a selective activation of cholinergic nicotinic but not muscarinic receptors. Pretreatment with i.c.v. mecamylamine (50 µg), a selective nicotinic receptor antagonist, abolished the stimulatory effect of nicotine on ACTH secretion. The nicotine-induced corticosterone response was significantly diminished, but not abolished by mecamylamine. Atropine, a muscarinic receptor antagonist, did not alter the nicotine-induced hormone secretion (5).
Nicotine-induced ACTH and corticosterone secretion in vagotomized rats
Nicotine (2.5 mg/kg i.p.) significantly increased plasma ACTH and serum corticosterone levels in sham operated rats 1 h after injection (Fig. 4). Subdiaphragmatic vagotomy considerably reduced the nicotine-induced ACTH response in comparison to the response in sham operated animals. On the other hand, vagotomy did not substantially alter the nicotine-induced corticosterone response in comparison with the response in sham operated controls. Vagotomy abolished the relatively weaker ACTH and corticosterone response to a lower dose of nicotine (1mg/kg i.p.) (Fig.4).
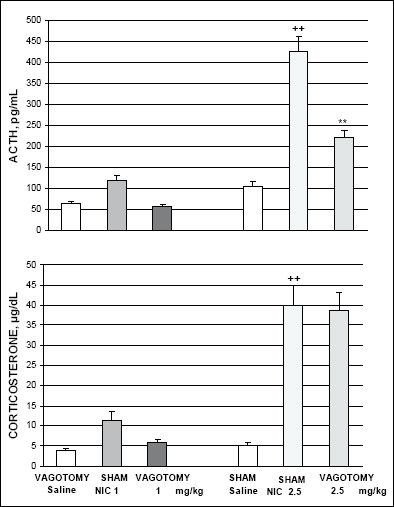 |
Fig. 4. Effect of subdiaphragmatic vagotomy on the nicotine (NIC)-induced ACTH and corticosterone secretion. ++p<0.01 vs. sham operated saline controls; **p<0.01 vs. nicotine-induced value in sham-operated rats. |
The present results indicate that carbachol given intraperitoneally or intracerebroventricularly considerably stimulates the HPA axis activity via cholinergic, predominantly muscarinc receptors. Atropine hydrobromide, which does not readily cross the blood-brain barrier from systemic circulation, significantly reduced, though did not abolish, the i.p. carbachol-induced corticosterone response, suggesting that a substantial part of this stimulation may be of central origin. Atropine sulphate, which after i.c.v. administration did not alter the i.p. carbachol-induced corticosterone response, corroborates this suggestion. This further suggests that cholinergic stimulation exerted peripherally may be in part transmitted via muscarinic pathways to central structures involved in activation of HPA axis. Stimulation of corticosterone secretion by i.c.v. carbachol was totally abolished by pretreatment with i.c.v. atropine. This indicates that centrally injected atropine totally prevented the muscarinic receptor-induced stimulation of CRH release from hypothalamic PVN with a subsequent activation of ACTH release from anterior pituitary corticotrophs and corticosterone secretion from adrenal cortex. On the other hand, atropine hydrobromide given i.p. was unable to substantially alter the i.c.v. carbachol-induced increase in corticosterone secretion, suggesting that this form of atropine does not act on central muscarinic system. Intracerebroventricularly injected carbachol stimulated neurons located along the lamina terminalis and the hypothalamus and this stimulation was reduced by pretreatment with i.c.v. muscarinic antagonist atropine (8). The present results suggest that part of the i.p. carbachol-induced stimulation may be transmitted via vagal afferents fibres to central vagal nuclei linked with cholinergic system-mediated activation of the HPA axis (9).
In the present experiment subdiaphragmatic vagotomy or sham vagotomy itself did not substantially alter the resting plasma ACTH and serum corticosterone levels, measured 2 weeks after surgery, in agreement with other report (20). Subdiaphragmatic vagotomy moderately diminished the increase in ACTH secretion induced by a lower dose of carbachol (0.2 mg/kg) but markedly augmented this increase evoked by a higher dose (0.5 mg/kg). This finding suggests that ACTH response to a lower dose of carbachol is mediated by vagal afferents. The response to a higher dose of carbachol may involve additional neuronal or humoral pathways e.g. catecholaminergic-mediated signaling. The inherent mediation of nitric oxide and prostaglandins in the cholinergic-induced stimulation of the HPA axis at different levels is also well documented (10, 11, 21, 22).
The present result may suggest that, depending on the intensity of cholinergic system activation, intact vagus nerve transmits either stimulatory or inhibitory signal evoked by stimulation of the HPA axis by lower or higher dose of carbachol, respectively. In either dose carbachol substantially augmented, in a dose-dependent manner, corticosterone secretion in vagotomized rats in comparison with the carbachol-induced increase in sham operated rats.
Adrenal cortex response is regulated not only by plasma ACTH level, but also by cholinergic and adrenergic innervations. In the human adrenal cortex cholinergic and noradrenergic fibres had a near-identical distribution, with varicose noradrenergic fibres located immediately alongside non-varicose cholinergic nerve bundles in all cortical zones. Noradrenergic innervation has a role in the modulation of cortical endocrine secretion, while cholinergic nerves are mainly implicated in the control of medullar secretion (23). Inner zone cells, isolated from bovine adrenal cortex, secrete cortisol in response to both adrenergic and cholinergic agonists. Cells show a dose-dependent cAMP response but no increased membrane phosphoinositide turnover. Cortisol secretion stimulated by catecholamines is blocked by adrenergic ß-receptor, but not by a-receptor. Muscarinic, but not nicotinic receptor antagonists selectively blocked acetylcholine-induced cortisol secretion in both freshly isolated and cultured cells (24).
The vagus nerve is involved in transmitting cytokine signals to the brain and the induction of brain signals (25 - 27). Peripheral vagal afferents activated by interleukin 1ß (IL-1ß) induce stimulation of HPA axis (28). On the other hand the efferent vagus nerve fibres inhibit pro-inflammatory cytokine release and protect against systemic inflammation which was termed "the cholinergic anti-inflammatory pathway" (29 - 31). A stronger HPA response to carbachol in vagotomized rats in comparison with the response in shame operated controls in the present experiment may result from the cholinergic modulation of cytokine release and a subsequent increase of HPA axis response to i.p. carbachol. Higher dose of carbachol may increase the noradrenergic activity in the hypothalamic PVN and subsequently the HPA activity (28).
There are several lines of evidence to indicate that the increase in hypothalamic noradrenaline induces the stimulation of the HPA axis. Noradrenaline is crucial for the stimulation of CRH in the PVN, which plays a major role in the release of ACTH from the anterior pituitary, which in turn increases secretion of corticosterone (15, 32). In addition, our earlier in vivo data indicate that central alpha1- and ß-adrenoceptors are considerably involved in the carbachol-induced ACTH and corticosterone secretion in rats (33).
In sham-operated rats nicotine in relatively low dose (1 mg/kg i.p.) induced rather weak increase in ACTH and corticosterone secretion and subdiaphragmatic vagotomy totally prevented this increase. Nicotine in a larger dose (2.5 mg/kg i.p.) considerably increased both ACTH and corticosterone secretion in sham operated rats. Subdiaphragmatic vagotomy considerably reduced the nicotine-induced increase in ACTH secretion, whereas the increase in corticosterone secretion remained unchanged. Nicotine given systemically easily penetrates the blood-brain barrier and reaches hypothalamic PVN to stimulate, by multiple pathways, the release of CRH, which induces ACTH release from pituitary corticotrophs. In the stimulatory effect of nicotine on HPA axis adrenergic system and alpha1-adrenergic receptors are significantly involved (34, 35).
The mechanism of a significant reduction in ACTH response to nicotine in vagotomized rats is not yet clear. It seems to depend on changes in the central parts of the HPA axis. Since vagotomy does not decrease plasma levels of cytokines, the blood-borne communication is not sufficient to produce cytokine-induced effects (29). The CRH and ACTH responses to nicotine may, in part, be mediated by vagal afferents. The CRH cells in the hypothalamic PVN are innervated by neurons localized in the nucleus of the solitary tract, the principal central recipient of viscerosensory input. It remains to be verified if i.p. administered nicotine transmits inhibitory signals to hypothalamic PVN secretory neurons via vagal afferents and central vagal nuclei. Interleukin 2 (IL-2) and IL-2 receptors are present in different brain regions under normal and pathophysiological conditions. IL-2 stimulates hypothalamic CRH and AVP release and that of pituitary ACTH. IL-2 releases AVP from both the hypothalamus and the amygdala and involves NO-mediated signaling (36). In the cholinergic stimulation of HPA axis in vivo both NO and prostaglandins are significantly involved in both basal and stress conditions (22, 37). Acetylcholine is known to stimulate CRH release in a dose-related fashion. Nicotinic as well as muscarinic receptors play an important role in CRH release, and they both act via stimulation of IL-2 receptors (38).
Systemically administered nicotine acts directly on adrenal medulla and postganglionic sympathetic neurons may stimulate corticosterone response. Therefore, subdiaphragmatic vagotomy did not markedly influence the corticosterone response induced by a direct adrenergic stimulation. Observed alterations in serum corticosterone levels in the present experiment did not parallel those of ACTH. There are now anatomical and functional evidence for paracrine mode of regulation of the adrenal cortex. The adrenal gland possesses islets of medullary chromaffin tissue within the three zones of the cortex, which suggests a cross talks between these cell types (39). Thus, frequently, ACTH levels do not parallel the elevated corticosterone (40).
- Rhodes ME, Billings TE, Czambel RK, Rubin RT. Pituitary-adrenal responses to cholinergic stimulation and acute mild stress are differentially elevated in male and female M2 muscarinic receptor knockout mice. J Neuroendocrinol 2005; 17: 817-826.
- Pisarska M, Mulchahey JJ, Sheriff S, Geracioti TD,Jr, Kasckow JW. Regulation of corticotropin-releasing hormone in vitro. Peptides 2001; 22: 705-712.
- Kasckow JW, Aguilera G, Mulchahey JJ, Sheriff S, Herman JP. In vitro regulation of corticotropin-releasing hormone. Life Sci 2003; 73: 769-781.
- Bugajski J, Gadek-Michalska A, Bugajski AJ. Effect of constitutive- and inducible-cyclooxygenase in the carbachol-induced pituitary-adrenocortical response during social stress. J Physiol Pharmacol 2002; 53: 453-462.
- Bugajski J, Gadek-Michalska A, Bugajski AJ. Involvement of prostaglandins in the nicotine-induced pituitary-adrenocortical response during social stress. J Physiol Pharmacol 2002; 53: 847-857.
- Cooper E. Nicotinic acetylcholine receptors on vagal afferent neurons. Ann N Y Acad Sci 2001; 940: 110-118.
- Berthoud HR, Neuhuber WL. Functional and chemical anatomy of the afferent vagal system. Auton Neurosci: Basic Clin 2000; 85; 1-17.
- Xu Z, Ross MG, Johnson AK. Intracerebroventricular carbachol induces FOS immunoreactivity in lamina terminalis neurons projecting to the supraoptic nucleus. Brain Res 2001; 895: 104-110.
- Zhu W, Umegaki H, Suzuki Y, Miura H, Iguchi A. Involvement of the bed nucleus of the stria terminalis in hippocampal cholinergic system-mediated activation of the hypothalamo-pituitary-adrenocortical axis in rats. Brain Res 2001; 916: 101-106.
- Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev 1999; 79: 1-71.
- John CD, Buckingham JC. Cytokines: regulation of the hypothalamo-pituitary- adrenocortical axis. Curr Opin Pharmacol 2003; 3: 78-84.
- Itoi K, Jiang YQ, Iwasaki Y, Watson SJ. Regulatory mechanism of corticotropin-releasing hormone and vasopressin gene expression in the hypothalamus. J Neuroendocrinol 2004; 16: 348-355.
- McCann SM, Kimura M, Yu WH, Mastronardi CA, Rettori V, Karanth S. Cytokines and pituitary hormone secretion. Vitam Horm 2001; 63: 29-62.
- Fleshner M, Goehler LE, Schwartz BA, et al. Thermogenic and corticosterone response to intravenous cytokines (IL-1ß and TNF-a) are attenuated by subdiaphragmatic vagotomy. J Neuroimmunol 1998; 86: 134-141.
- Ueta Y, Kannan H, Higuchi T, Negoro H, Yamaguchi K, Yamashita H. Activation of gastric afferents increases noradrenaline release in the paraventricular nucleus and plasma oxytocin levels. J Auton Nrerv Syst 2000; 78: 69-76.
- Bucinskaite V, Kurosawa M, Lundeberg T. Effect of interleukin-1ß on subdiaphragmatic vagal efferents in the rat. Auton Neurosci: Basic & Clinical 2000; 85: 93-97.
- Kapcala LP, He JR, Gao Y, Pieper JO, DeTolla LJ. Subdiaphragmatic vagotomy inhibits intra-abdominal interleukin-1ß stimulation of adrenocorticotropin secretion. Brain Res 1996; 728: 247-254.
- Wieczorek M, Swiergiel AH, Pournajafi-Nazarloo H, Dunn AJ. Physiological and behavioral responses to interleukin-1ß and LPS in vagotomized mice. Physiol Behav 2005; 85: 500-511.
- Hansen MK, Yaishi P, Chen Z, Krueger JM. Vagotomy blocks the induction of interleukin-1ß (IL-1ß) mRNA in the brain of rats in response to systemic IL-1ß. J Neurosci 1998; 18: 2247-2253.
- Hansen MK, Nguyen KT, Fleshner M, et al. Effects of vagotomy on serum endotoxin, cytokines, and corticosterone after intraperitoneal lipopolysaccharide. Am J Physiol Regul Integ Comp Physiol 2000; 278: R331-R336.
- Gadek-Michalska A, Spyrka J, Bugajski J. Psychosocial stress affects the involvement of prostaglandins and nitric oxide in the lipopolysaccharide-induced hypothalamic-pituitary-adrenal response. J Physiol Pharmacol 2005; 56: 287-298.
- Bugajski AJ, Gadek-Michalska A, Bugajski J. The involvement of nitric oxide and prostaglandins in the cholinergic stimulation of hypothalamic-pituitary-adrenal response during crowding stress. J Physiol Pharmacol 2006; 57: 463-477.
- Gilchrist AB, Leake A, Charlton BG. Innervation of the human adrenal cortex: stimultaneous visualization using acetylcholinesterase histochemistry and dopamine beta-hydroxylase immunochemistry. Acta Anat (Basel) 1993; 146: 31-35.
- Walker SW, Lightly ER, Clyne C, Williams BC, Bird IM. Adrenergic and cholinergic regulation of cortisol secretion from the zona fasciculata/reticularis of bovine adrenal cortex. Endocr Res 1991; 17: 237-265.
- Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain communication: a review and analysis of alternative mechanisms. Life Sci 1995; 57: 1011-1026.
- Goehler LE, Gaykema RPA, Nguyen KT, Lee JE, Tidlers FJH. Interleukin-1ß in immune cells of the abdominal vagus nerve: a link between the immune and neurons systems? J Neurosci 1999; 19: 2799-2806.
- Gaykema RPA, Dijkstra I, Tilders FJH. Subdiaphragmatic vagotomy suppresses endotoxin-induced activation of hypothalamic corticotropin-releasing hormone neurons and ACTH secretion. Endocrinology 1995; 136: 4717-4720.
- Fleshner M, Goehler LE, Hermann J, Relton JK, Maier SF, Watkins LR. Interleukin-1ß induced corticosterone elevation and hypothalamic NE depletion is vagally mediated. Brain Res Bull 1995; 37: 605-610.
- Borovikova LV, Ivanova S, Zhang M, et al. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000; 405: 458-462.
- Pavlov VA, Tracey KJ. The cholinergic anti-inflammatory pathway. Brain Behav Immun 2005; 19: 493-499.
- Ulloa L. The vagus nerve and the nicotinic anti-inflammatory pathway. Nature Rev: Drug Discov 2005; 4: 673-684.
- MohanKumar SMJ, MohanKumar PS, Quadri SK. Effects of bacterial lipopolysaccharide on central monoamines and fever in the rat: involvement of the vagus. Neurosci Lett 2000; 284: 159-162.
- Bugajski J, Borycz J, Gadek-Michalska A. Involvement of the central noradrenergic system in cholinergic stimulation of the pituitary-adrenal response. J Physiol Pharmacol 1998; 49: 285-292.
- Gadek-Michalska A, Bugajski J, Bugajski AJ, Glod R. Effect of adrenergic antagonists and cyclooxygenase inhibitors on the nicotine-induced hypothalamic-pituitary-adrenocortical activity. J Physiol Pharmacol 2002; 53: 275-287.
- Okada S, Shimizu T, Yokotani K. Extrahypothalamic corticotropin-releasing hormone mediates (-)-nicotine-induced elevation of plasma corticosterone in rats. Eur J Pharmacol 2003; 473: 217-223.
- Raber J, Koob GF, Bloom FE. Interleukin-2 (IL-2) induces corticotropin-releasing factor (CRF) release from the amygdala and involves a nitric oxide-mediated signaling: comparison with the hypothalamic response. J Pharmacol Exp Ther 1995; 272: 815-824.
- Gadek-Michalska A, Bugajski J. Nitric oxide mediates the interleukin-1ß- and nicotine induced hypothalamic-pituitary-adrenocortical response during social stress. J Physiol Pharmacol 2005; 56: 491-503.
- Karanth S, Lyson K, McCann SM. Effects of cholinergic agonists and antagonists on interleukin-2-induced corticotropin-releasing hormone release from the mediobasal hypothalamus. Neuroimmunomodulation 1999; 6: 168-174.
- Delarue C, Contesse V, Lenget S, et al. Role of neurotransmitters and neuropeptides in the regulation of the adrenal cortex. Rev Endocr Metab Disord 2001; 2: 253-267.
- Bornstein SR, Chrousos GP. Adrenocorticotropin (ACTH)- and non-ACTH-mediated regulation of the adrenal cortex: neural and immune inputs. J Clin Endocrinol Metab 1999; 84: 1729-1736.