A very important effect of PTX is inhibition of inflammatory response, especially the downregulation of TNF. PTX suppresses TNF gene transcription, expression of TNF mRNA (7, 8), and secretion of TNF protein in macrophages and monocytes (9, 7, 10). Accordingly, the attenuation of morbidity and mortality in septic shock has been attributed to capability of PTX of suppressing the induction of TNF (11, 12).
PTX is a non-specific inhibitor of phosphodiesterases (PDEs), which decreases hydrolysis of both cAMP and cGMP, and thereby augments cyclic nucleotide-dependent signal transduction (13, 14). The exact mechanisms underlying the therapeutic effects of PTX are not fully recognized. It was postulated that decreased synthesis of TNF results from cAMP elevation, protein kinase-A (PKA) activation and cytosolic calcium decline (15). On the other hand, PTX may attenuate oxidative stress by induction of manganese superoxide dismutase (MnSOD) (16), and direct scavenging of free-radicals (17). In consequence, it inhibits activity of NF
Interestingly, some studies suggested one more mechanism contributing to the activity of PTX. Experiments performed in fibrosarcoma L292 cells showed that PTX strongly induces the expression of heme oxygenase-1 (HO-1), a cytoprotective and anti-inflammatory enzyme, degrading heme to biliverdin, iron ions and carbon monoxide (CO) (20). Cytoprotective effects of PTX were imitated by incubation of cells with hemin (an HO-1 activator) or with CORM (a compound releasing CO), while reduced in the presence of ZnPPIX (zinc protoporphyrin-IX, an HO-1 inhibitor) (20). These observations strongly suggested that effects of PTX may at least partly depend on the induction of HO-1.
The similarities between cellular effects of HO-1 upregulation and PTX treatment are striking. Thus, both PTX and HO-1 have anti-inflammatory properties, as exemplified by inhibition of IL-1ß, IL-6, IL-8, TNF or GM-CSF and induction of IL-10 (21-23). Furthermore, they both were shown to reduce proliferation, migration and adhesion of leukocytes to endothelial cells through inhibition of CD25 and ICAM-1 (24, 25). Potential involvement of HO-1 in regulation of PTX signaling appears to be particularly important, because of the human HO-1 promoter polymorphism. The presence of less active alleles, resulting in lower HO-1 activation, was demonstrated as a factor increasing risk of cardiovascular complications, at least in some populations of patients (26-29). Thus, one can expect that also efficacy of PTX may be influenced by this polymorphism and may be low in patients with less active HO-1 variants.
The aim of our paper was to verify this supposition. Therefore we investigated the involvement of HO-1 pathway in anti-inflammatory activities of PTX in vitro (in murine and human macrophages and endothelial cells), and in vivo, in murine model of endotoxemia. The obtained results clearly show that PTX acts independently of HO-1.
Reagents
PTX, triton X-100, phenylmethyl sulfonyl fluoride, leupeptin, aprotinin, bicinchonic acid protein assay kit, BCIP/NBT alkaline phosphate substrate solution, HEPES, MTT, LPS, hemin and G418 antibiotic were purchased from Sigma (Poznan, Poland). Tin protoporphyrin-IX (SnPPIX) was from Porphyrin Products (Logan, UT, USA). The total RNA extraction kit, reverse transcription system, PCR system, agarose, and Cytotox-96 NonRadioactive Cytotoxicity Assay were obtained from Promega (Madison, WI, USA). SuperFect transfection reagents were purchased from Qiagen (Hilden, Germany). ELISA kits for human and mouse TNF were obtained from R&D Systems (Minneapolis, MN, USA). BrdU incorporation ELISA was obtained from Roche Diagnostics (Mannheim, Germany). ELISA kit for human HO-1 and Rabbit anti-HO-1 polyclonal IgG were obtained from Stressgen Biotechnologies (Victoria, BC, Canada). DyNAmoTM HS SYBR® Green qPCR kit was purchased from FinnZymes (Espoo, Finland). Nitrocellulose membrane HybondECL was procured from Amersham Pharmacia Biotech (Boston, MA). Mouse anti-tubulin polyclonal IgG was purchased from Calbiochem. Anti-mouse monoclonal HRP-linked IgG and anti-rabbit monoclonal HRP-linked IgG were obtained from Cell Signaling Technology (Danvers, MA, USA). Expression plasmid pcDNA3.1 was obtained from Invitrogen (San Diego, CA, USA), while heparin and endothelial cell growth supplement from Upstate Biotechnology (Charlottesville, VA, USA). Adeno-X adenoviral expression system, adeno-X rapid titer ELISA kit and anti-VCAM-1 antibodies were purchased from BD Bioscience (Erembodegem, Belgium), while TransAM ELISA for measurements of NF
Cell culture
The human monocyte U937 cell line was kindly provided by Dr. Guenter Weigel, Vienna, Austria. The cells were cultured in RPMI-1640 medium supplemented with 10% FCS, penicillin (100 U/ml), streptomycin (10 µg/ml) and L-glutamine (2 mM). Both murine macrophage J774 cell line (ATCC, Manassas, VA, USA) and murine brain microvascular endothelial cell-1 line MBEC-1 (kindly provided by Dr. Joanna Bereta, Krakow, Poland) were cultured in DMEM-HG medium supplemented with 10% FCS, L-glutamate (2 mM), penicillin (100 U/ml), and streptomycin (100 µg/ml). HUVEC were freshly isolated from human umbilical veins of newborn babies by collagenase digestion. HUVEC were grown in M199 medium supplemented with 20% FCS, HEPES (20 mM), L-glutamine (2 mM), heparin/endothelial cell growth supplement (30 ng/ml), penicillin (100 U/ml), and streptomycin (100 µg/ml). Cells of the second to fourth passages were used in experiments. Human embryonic kidney cells (HEK 293) obtained by courtesy of Dr. Stefan Kochanek (Ulm, Germany), and used for production of adenoviral vectors, were maintained in
Stable transfection
Cells were transfected with pcDNA3.1+ plasmid vector harboring human HO-1 (pcDNA-HO-1) or with empty plasmid (pcDNA) using SuperFect® Transfection Reagent. Procedure was carried out according to vendors instruction, with 0.5 mg of DNA and 2.5 ml of SuperFect per one well in a 24-well plate. Transfected cells were selected in the presence of G418 antibiotic (1 mg/ml) for 3 weeks. Presence of stably introduced plasmids and overexpression of transgene were confirmed with PCR, RT-PCR and western-blotting, respectively. Cells were then cultured with G418 at the dose of 200 mg/ml.
Transduction with adenoviral vectors
Endothelial cells were infected with Ad-HO-1 (harboring human HO-1 cDNA) or Ad-GFP (containing control GFP gene) at the dose of 10 multiplicity of infection (MOI). Vectors were poured into the cells in 30% volume of normal amount of media. The remaining medium was added after 2 h. After 24 hours, medium was changed and cells were stimulated with PTX (100 µM and 1 mM) for 24 h.
Cell viability assay
Cell viability was assessed by colorimetric measurement of lactate dehydrogenase (LDH) release, and MTT reduction assays according to the manufacturers protocols.
Real-time RT-PCR
Total RNA was isolated from cells by acid guanidinium thiocyanate-phenol-chloroform extraction as described earlier (30). Reverse transcription with oligo-dT primers was performed on 2 µg of total RNA, for 1 hr at 42°C, using MMLV reverse transcriptase, according to vendors instruction. Real-time polymerase chain reactions using qRT-PCR DyNAmo HS SYBR Green qPCR Kit, were performed in samples containing 50 ng cDNA, 0.5 µl of primers, 7.5 µl of DyNAmo SYBR Green master-MIX, and nuclease-free water added to 15 µl of total volume. Reaction was carried out using the following protocol: 95°C for 10 min, followed by 40 three-step cycles 95°C for 30 s, 60°C for 60 s, 72°C for 45 s, and by 72°C for 5 minutes as a final elongation. The primers specific for HO-1 (5 GTG GAA MCG CTT YAC RTA GYG C 3 and 5 CTT TCA GAA GGG YCA GGT GWC C 3), and for housekeeping EF2 (5 GCG GTC AGC ACA ATG GCA TA 3 and 5 GAC ATC ACC AAG GGT GTG CAG 3). Relative quantification of gene expression was calculated based on
ELISA assays
ELISAs for human and murine TNF (measured in conditioned media or in plasma) and for human HO-1 (measured in cell lysates) were performed according to the vendors protocols.
Western blotting
Expression of HO-1 protein was checked in cells cultured in six-well plates or in murine organs. Cells or tissues were lysed in ice-cold lysis buffer (1% Triton X-100, 1 µg/ml phenylmethyl sulfonyl fluoride, 1 µg/ml leupeptin, and 1 µg/ml aprotinin) and centrifuged for 20 minutes, 8000g, at 4°C. Clear supernatants were collected, and protein samples (15-20 µg/well) were subjected to electrophoresis in 12% sodium dodecyl sulfate-polyacrylamide electrophoresis gel followed by transfer to nitrocellulose membrane HybondECL. Then, membranes were probed with polyclonal antibodies against HO-1, treated with secondary antibodies linked with horseradish peroxidase (diluted 1:10,000 in Tris-buffered saline with 3% albumin), and visualized using BCIP/NBT blue liquid substrate.
Animal experiments
All animal protocols were approved by the Institutional Animal Care and Use Committee at the Jagiellonian University. All mice were maintained on a standard diet and water ad libitum.
Eight week old C57BL/6 were used to assess the expression of HO-1 after treatment with PTX. To this aim, animals were injected intraperitoneally (i.p) with PTX (10 mg/kg body weight, total volume of 100 ml) for 7 consecutive days. Control mice received the same volume of vehicle (PBS). Then, mice were sacrificed and HO-1 protein in the liver, kidney, spleen, heart and skin was analyzed using western-blot.
Second experiment was designed to determine the role of HO-1 in PTX activities. Here, eight week old HO-1-/- and HO-1+/+ mice of C57BL/6×FVB background (obtained thanks to courtesy of Dr. Anupam Agarwal, Birmingham, USA) were treated with PTX as described above, and then subjected to endotoxemia induced by i.p. injection of LPS (5 µg/kg body mass). After 1.5 hours, animals were sacrificed, and the peritoneal cavities were washed with 1 ml of Ca/Mg-free PBS. Aliquots of the lavage fluids were stained in Turks solution (0.01% crystal violet in 3% acetic acid), and total infiltrating leukocytes were counted using a Burker chamber under a light microscope and then seeded in a 24 well plate (100,000/well) in 1 ml of RPMI medium supplemented with 10% FCS. Concentrations of cytokines released to the culture media were measured 24 h later, using ELISA. Proportion of different leukocyte populations was assessed using flow cytometry and FSC/SSC analysis. Additionally, mice were bled for measurement of plasma TNF, and their aortas were isolated for immunohistochemical staining.
Immunohistochemistry
Frozen aorta sections (8 µm) were air-dried and fixed for 10 minutes in acetone at 4°C. After blocking of nonspecific binding sites with 10% goat serum (in 0.1% Triton X-100 and 0.05% tween in PBS solution for 90 minutes), slides were incubated with primary antibodies (rat anti-mouse CD106 (VCAM-1) diluted 1:50) overnight at 4°C. After a PBS rinse, the slides were incubated with anti-rat IgG-PE conjugated antibodies diluted 1:250 in PBS for 45 min at room temperature. After a PBS rinse, the slides were air-dried and mounted.
Statistical analysis
In vitro experiments were performed in duplicates or triplicates and repeated at least three times. In vivo experiments were carried out in 5 animals per group. Results are presented as mean ± SD. Statistical significance was determined using student t-test (comparison of two groups) or ANOVA followed by Tukey test (comparison of more than two groups).
Expression of HO-1 in cells incubated with PTX
Murine monocytes (J774), human monocytes (U937), murine endothelial cells (MBEC-1), and human primary endothelial cells (HUVEC) were incubated with PTX at the wide range of concentrations from 0.1 µM to 1 mM for 24 h. Even the highest doses applied did not influence cell viability, as checked using LDH activity and MTT reduction assays (data not shown).
Effect of PTX on expression of HO-1 was measured at mRNA level by qRT-PCR and at protein level using ELISA (for human cells) or western-blotting (for murine cells). Surprisingly, we did not observe an induction of HO-1 in any cell line studied, except some experiments where the very high and potentially toxic doses of PTX (3 mM or 10 mM) were used. In such extreme conditions HO-1 mRNA expression could be induced up to 3-fold in endothelial cells and up to 40-fold in monocytes, but the results were very variable. Instead, within the range of non toxic concentrations (0.1 µM - 1 mM), PTX dose-dependently decreased expression of HO-1, to about 40-50% of control values in all cell lines, as demonstrated both at mRNA and protein levels (Fig. 1). The same modest downregulation of HO-1 expression by PTX was observed after 6 h incubation period, as well as in cells treated concomitantly with typical HO-1 inducers, namely with 10 µM hemin, 10 µM cobalt protoporphyrin (CoPPIX), or 10 µM 15-deoxy-
 |
Fig. 1. Effect of PTX (0.1 µM 1 mM, 24 h) on expression of HO-1 mRNA (qRT-PCR) and protein (ELISA or western-blotting) in cultured cells. A J774 cells, HO-1 mRNA; B J774 cells, HO-1 protein; C U937 cells, HO-1 mRNA; D U937 cells, HO-1 protein; E MBEC-1 cells, HO-1 mRNA; F MBEC-1, HO-1 protein; G HUVEC, HO-1 mRNA, H HUVEC, HO-1 protein. Each bar represents mean ± SD of at least three experiments. * - P < 0.05 in comparison with control. Pictures show representative western-blots. |
Anti-inflammatory effects of PTX in cells of different HO-1 activity
To determine whether modulation of HO-1 activity may influence the anti-inflammatory action of PTX, we checked the effect of PTX on synthesis of TNF in cells preincubated with 10 µM hemin (HO-1 activator) or 10 µM tin protoporphyrin-IX (SnPPIX, HO-1 inhibitor).
MBEC-1 did not produce measurable amounts of TNF protein, therefore the experiments were performed in U937, J774, and HUVEC cells. Mean concentrations of TNF released to the culture media by control cells after a 24 h incubation were 19.4 ± 2.3 pg/ml, 26.2 ± 3.4 pg/ml, 20.1 ± 3.7 pg/ml, respectively. In all cell lines studied, PTX at the concentrations of 100 µM and 1 mM significantly decreased synthesis of TNF, as measured by ELISA. The extent of reduction was similar, regardless of stimulation or inhibition of HO-1 activity, indicating that HO-1 does not play a role in PTX signaling (Fig. 2). Instead, PTX at the concentrations of 100 µM and 1 mM downregulated DNA binding activity of NF
 |
Fig. 2. Effect of PTX (100 µM and 1 mM, 24 h) on synthesis of TNF in cells preincubated with hemin (10 µM, HO-1 activator) or SnPPIX (10 µM, HO-1 inhibitor), measured by ELISA. A U937 cells; B J774 cells; C HUVEC cells. Each bar represents mean ± SD of at least three experiments, presented as a percentage of control value, obtained for intact cells. * - P < 0.05 in comparison with cells incubated without PTX; # - P<0.05 in comparison to intact cells. |
Next, to avoid possible non-specific effects of hemin or SnPP, we checked the influence of PTX on TNF synthesis in cells genetically modified to overexpress HO-1. To this aim we established U937 and J774 cell lines overexpressing HO-1 after a stable transfection with pcDNA-HO-1 plasmid. Control cell lines were stably transfected with the empty pcDNA construct. To overexpress HO-1 in HUVEC cells we transduced them using adenoviral vectors harboring HO-1 cDNA or control GFP cDNA (transfection efficacy was estimated as ~80%). In all cases strong overexpression of HO-1 has been confirmed by western blot or ELISA (data not shown). Likewise pharmacological modulation of HO-1 activity, also genetic overexpression of HO-1 did not influence inhibitory effect of PTX on synthesis of TNF (Fig. 3). Noteworthy, cell lines stably overexpressing HO-1 released less TNF than their control counterparts. Similar tendency was observed in HUVEC transduced with AdHO-1 vectors, but here this effect did not reach statistical significance.
 |
Fig. 3. Effect of PTX (100 µM and 1 mM, 24 h) on synthesis of TNF in cells overexpressing HO-1, measured by ELISA. A U937 cells stably transfected with pcDNA-HO-1 or with control, empty pcDNA plasmid; B J774 cells stably transfected with pcDNA-HO-1 or with control, empty pcDNA plasmid; C HUVEC cells transduced with adenoviral vectors harboring HO-1 (Ad-HO-1) or control GFP cDNA (Ad-GFP). Each bar represents mean ± SD of at least three experiments, presented as a percentage of control values. * - P < 0.05 in comparison with cells incubated without PTX; # - P<0.05 in comparison to control cell (transfected with empty plasmid or transduced with Ad-GFP vectors), incubated without PTX. |
We also decided to investigate the effect of HO-1 on the postulated cytoprotective potential of PTX (20). To this aim we treated control (GFP expressing) or HO-1 overexpressing endothelial cells with PTX (100 µM) and then with high dose of H2O2 (100 µM) for 24 h. Both in the presence or absence of PTX, HO-1 showed similar tendency to protect HUVEC cells, whereas in MBEC-1 this effect was negligible. PTX (100 µM) did not show any cytoprotection regardless of the HO-1 expression level (Fig. 4).
 |
Fig. 4. Effect of PTX (100 µM) on viability of endothelial cells transduced with adenoviral vectors harboring HO-1 (Ad-HO-1) or control GFP cDNA (Ad-GFP), treated with H2O2 (100 µM, 24 h). A HUVEC; B MBEC-1. MTT reduction assay. Each bar represents mean ± SD of at least three experiments, presented as a percentage of control values. * - P < 0.05 in comparison with control. |
Finally, we checked whether HO-1 promoter polymorphism, characteristic for human populations, may modulate the efficacy of PTX-mediated inhibition of TNF production. Here we employed HUVEC cells of different HO-1 promoter genotype: i) cells carrying at least one allele with short sequence of GT repeats (N
 |
Fig. 5. Effect of PTX (100 µM) on synthesis of TNF in control or LPS-stimulated (100 ng/ml) HUVEC cells of different genotype of HO-1 promoter: carrying at least one allele with short sequence of GT repeats (S) or without such alleles (no S). Each bar represents mean ± SD of at least 30 cell batches, presented as a percentage of control values. * - P < 0.05 in comparison with control. |
Expression of HO-1 in mice treated with PTX
To determine the effect of PTX on HO-1 expression not only in cell cultures but also in vivo, we decided to inject C57Bl mice with PTX (10 mg/kg, i.p., 100 µl/mouse), for seven consecutive days. Control animals were injected with the same volume of PBS. Then, mice were sacrificed and expression of HO-1 protein in their livers, spleens, kidneys, hearts, and skins was analyzed using western-blotting. As shown in Fig. 6, injections of mice with PTX did not influence the expression of HO-1 in any organ studied.
 |
Fig. 6. Expression of HO-1 protein in the liver, spleen, kidney, heart and skin of mice injected i.p. with PTX (10 mg/kg) for seven consecutive days. Control mice injected with PBS. Western blot; 5 animals per group. |
Modulation of inflammatory response by PTX in mice of different HO-1 genotype
To conclusively establish whether HO-1 plays any role in anti-inflammatory activities of PTX in vivo, we performed experiments in C57Bl/FVB mice devoid of HO-1 gene (HO-1 KO) and in mice of the same genetic background, but with normal level of HO-1 (HO-1 WT). KO and WT mice were injected i.p. with PTX (10 mg/kg, 100 µl/mouse) or with PBS (100 µl) for seven consecutive days. Then they were injected i.p. with LPS (5 µg/kg) for 1.5 h. As an additional control served the intact, untreated WT and KO animals.
After sacrificing the mice, we first checked number of leukocytes in their peritoneal cavities. Calculating the cells in the Burker chamber showed that intact mice had relatively small amounts of peritoneal leukocytes, regardless of the HO-1 status (672,000 ± 312,000 cells and 599,000 ± 369,000 cells in WT and KO, respectively). Intraperitoneal injection with LPS led to the rapid increase in leukocyte number already in 1.5 h. This effect was significantly less pronounced in the HO-1 deficient individuals (3,140,000 ± 1,177,709 cells and 1,256,000 ± 95,433 cells in WT and KO, respectively). As shown in Fig. 7A, also PTX considerably influenced leukocyte motility infiltration of peritoneal cavity in response to LPS was much stronger in animals treated with PTX than in those injected with PBS. Flow cytometric analysis showed that these differences occurred in lymphocytes and granulocytes but not in a monocyte population (Fig. 7B). Expression of HO-1 did not influence effects of PTX, which were very similar in the WT and KO animals (Fig. 7).
 |
Fig. 7. Effect of PTX on leukocytes infiltration in peritoneal cavity of the wild type and HO-1 deficient mice injected with LPS (5 µg/kg, 1.5 h), pretreated or not with PTX (10 mg/kg, i.p. for seven consecutive days). A Total leukocyte number, calculated using Burker chamber and presented as a percentage of reference value (number of cells after treatment with LPS only); control intact animals, non-injected with LPS. B Number of lymphocytes, monocytes and granulocytes in peritoneal cavity. Cells were calculated using Burker chamber, while proportion of leukocyte populations was determined using flow cytometer and FSC/SSC analysis. Each bar represents mean ± SD of measurements for 5 animals.* - P<0.05 in comparison with mice injected with LPS alone. |
We measured also concentration of TNF in plasma of mice and production of TNF by peritoneal leukocytes, cultured ex vivo for 24 h. In intact animals of both HO-1 genotypes, TNF in the blood was undetectable using ELISA method. However, at 1.5 h after i.p. injection with LPS, mice had significantly increased level of TNF. Concentration of TNF was higher in HO-1 deficient mice (1,819.45 ± 642.59 pg/ml), than in their wild-type counterparts (636.08 ± 444.09 pg/ml). One-week exposure to PTX resulted in a tendency to decrease the production of TNF in animals responding to LPS, as demonstrated by lower concentration of the cytokine in the blood (Fig. 8A). The level of this inhibition did not depend on expression of HO-1 and was comparable in WT and KO animals.
 |
Fig. 8. Effect of PTX on production of TNF by the wild type and HO-1 deficient mice injected with LPS (5 µg/kg, 1.5 h), pretreated or not with PTX (10 mg/kg, i.p. for seven consecutive days). A Concentration of TNF in the blood plasma. B Concentration of TNF released to culture medium by peritoneal leukocytes incubated ex vivo (100,000/ml) for 24 h. TNF was measured by ELISA. Each bar represents mean ± SD of measurements for 5 animals. * - P<0.05 in comparison with mice injected with LPS alone. |
Accordingly, the leukocytes harvested from the peritoneal cavity of LPS-injected animals produced measurable amounts of TNF, and production was stronger in the cells obtained from HO-1 deficient mice. Importantly, generation of TNF was significantly decreased in leukocytes collected from PTX-treated animals. Again, inhibitory effect of PTX was independent of HO-1 genotype and was similar in HO-1 KO and HO-1 WT cells.
Finally, we checked the effect of PTX on the inflammatory reaction in the blood vessels of LPS-injected mice. To this aim we collected the aortas of intact or LPS-injected animals of both genotypes to perform an immunohistochemical staining for VCAM-1 expression. As shown in Fig. 9, lack of HO-1 gene was associated with stronger expression of VCAM-1 in intact animals. At 1.5 h after i.p. injection with LPS, VCAM-1 in aortal intima was upregulated, to similar values in HO-1 deficient and wild-type mice. PTX very potently reduced expression of VCAM-1. Like in all previous experiments, its activity was independent of HO-1 and the extent of reduction was comparable in mice of both HO-1 genotypes (Fig. 9).
 |
Fig. 9. Effect of PTX on expression of VCAM-1 protein in aorta of wild type and HO-1 deficient mice injected with LPS (5 µg/kg, 1.5 h), pretreated or not with PTX (10 mg/kg, i.p. for seven consecutive days). Immunohistochemical staining. A Percent of fields with positive staining per one specimen. Each bar represents mean ± SD of measurements for 5 animals. * - P<0.05 in comparison with mice injected with LPS alone. B representative pictures. |
Starting point to our study was the hypothesis based on experiments done in L292 fibroblasts, that PTX in the range of 0.1 mM to 1.0 mM strongly induces expression of HO-1 and that cytoprotective action of PTX is mediated via HO-1 pathway, possibly through elevated synthesis of CO (20). Our aim was to investigate the role of HO-1 in anti-inflammatory and cytoprotective effects of PTX in monocytes and endothelial cells.
HO-1, a cytoprotective, anti-inflammatory and proangiogenic enzyme, can be induced by numerous factors, including important drugs, such as rapamycin, statins, dopamine, probucol, or even aspirin (31-33). It has been suggested in many papers that activation of HO-1 pathway, leading to removal of prooxidative heme and increased production of CO and biliverdin, may contribute to therapeutic effects of these medicines (34). In most cases, however, these presumptions derived from the cell culture experiments only, where relatively high doses of the drugs were used. Thus, studies in animal models, especially employing HO-1 deficient mice are desired to validate the actual role of HO-1.
Therapeutic plasma levels of PTX and its active metabolites range from 10-6 to 10-4 M (5). Therefore, in vitro we used the concentrations of 10-7 -10-3 M, while in vivo we applied PTX at the dose of 10 mg/kg, relevant to clinical trials (35). First, we decided to check the effect of PTX on expression of HO-1 in cultured murine and human monocytes and endothelial cells, and in several organs of PTX-injected mice.
The obtained results show that PTX slightly decreases expression of HO-1 in all cell lines studied, both at mRNA and protein levels. Inhibitory effects of PTX was observed also if HO-1 was induced by hemin, CoPPIX or 15d-PGJ2. We detected an upregulation of HO-1 only in some experiments, when very high, cytotoxic doses of PTX were used (data not shown). The reason for such discrepancy with results published earlier (20) is not clear and may be associated with the different cell types used. It seems, however, that PTX-mediated inhibition of HO-1 expression is not surprising, as the promoter of HO-1 contains binding sites for NFkB and AP-1 (36), the transcription factors attenuated by PTX (18, 19), as demonstrated also in our experimental settings. In accordance, PTX significantly decreased LPS-induced upregulation of placental HO-1 in mice (37). Similar inhibition of intestinal and lung HO-1 generation was also reported in rats treated with hypertonic saline/PTX infusion (38, 39). In our experiments PTX injected i.p. for seven days to the mice, although effective in reduction of TNF generation, did not influence HO-1 expression in kidney, liver, skin, spleen and heart.
Then, to investigate whether different levels of HO-1 activity may influence anti-inflammatory efficacy of PTX, we employed three experimental models: i) monocytes and endothelial cells incubated in the presence of pharmacological inducer or inhibitor of HO-1, ii) cells stably or transiently overexpressing HO-1, iii) mice with the knocked-out HO-1 gene. Results obtained in all models univocally evidence that HO-1 pathway does not play a role in PTX signaling.
Thus, in contrast to the results reported by Oh and colleagues (20), we found that preincubation of cells with the HO-1 activator hemin or with HO-1 inhibitor SnPP neither mock nor modulate activity of PTX. We also did not observe any influence of HO-1 overexpression on effects of PTX in cells stably transfected with pcDNA-HO-1 plasmid or transiently transduced with adenoviral vectors harboring HO-1 cDNA. In all experimental settings we demonstrated exactly the same inhibition of TNF production. We also checked the cytoprotective effects of PTX in endothelial cells treated with H2O2 to investigate the potential role of HO-1 in this activity, but we did not obtain any protection by PTX, regardless of the HO-1 expression. Thus, we conclude that involvement of HO-1 in PTX action is rather limited to some cell lines only, and HO-1 cannot be regarded as a mediator of PTX signaling.
Importantly, this conclusion is confirmed by experiments carried out in the animal model of endotoxemia. Seven-day pretreatment with PTX strongly inhibited LPS-induced production of TNF, reduced endothelial expression of VCAM-1, and augmented peritoneal infiltration of leukocytes. The observed effects were very similar in HO-1 knock-out and wild type mice, giving strong evidence that HO-1 is not involved in PTX activities. Worth mentioning that apart from analysis of TNF we performed similar in vitro and in vivo measurements of IL-1ß, IL-6, IL-8/KC, ICAM-1, and E-selectin. Again, we found that HO-1 did not modulate effects of PTX on expression of any cytokine (data not shown).
TNF, the major target in anti-inflammatory effects of PTX, is crucial in triggering the lethal effects of septic shock (40). PTX not only reduces TNF generation in vitro and in vivo (10, 18, 41), but also potently inhibits endotoxin-induced neutrophil degranulation (42), and decreases the binding of activated T cells to endothelial cells (43). As costimulation of neutrophils and cytokines may play an important role in organ injury in sepsis (42), PTX was shown to be of great benefit in different models of animal septic shock (11, 12, 18, 44). Pretreatment of mice or rats with PTX resulted also in the reduction of liver, kidney and lung injury during endotoxemia (12, 45, 46), and in prevention of hepatitis (47). Finally, experiments performed in rats suggested also that PTX may exert gastroprotective effects, both through inhibition of TNF synthesis and attenuation of oxidative stress, and through enhancement of gastric microcirculation (48, 49). Our study confirms anti-inflammatory effect of PTX in acute endotoxemia, as illustrated by decreased generation of TNF and VCAM-1 in mice.
Moreover, we found that pretreatment with PTX strongly increased number of cells recruited in the peritoneal cavity in response to LPS, either in the wild-type or in HO-1 deficient mice. Similar effects were observed earlier in mice after cecal ligation and puncture, where PTX increased cell influx to the peritoneum (50). It is in agreement with opinion that PTX acts on the activated neutrophils, increasing their chemotaxis (51). In fact, PTX was shown to restore impaired chemotactic efficiency of neutrophils incubated with serum of patients subjected to IL-2 therapy (52). Our data suggest that PTX increases peritoneal infiltration of both granulocytes and lymphocytes 1.5 h after LPS stimulation. Very early tissue sequestration of neutrophils in response to LPS (even 10 minutes after intravenous injection) were observed earlier (53). Interestingly, number of infiltrating cells is lower in HO-1 knock-outs, what might be associated with weaken migratory capabilities of HO-1 deficient cells (54).
Today, PTX is widely used for treatment of occlusive arterial diseases and cerebrovascular disorders (1, 55, 56). It has been also suggested as a potential drug in treatment of sepsis. Actually, inhibition of TNF and better cardiopulmonary function was demonstrated in several clinical trials (57-61), and some reports showed even improved survival of septic patients (62). PTX was also reported to be helpful in preventing the systemic inflammation in patients undergoing cardiopulmonary bypass (63-65), and in reduction of TNF synthesis in patients with severe Plasmodium falciparum malaria (66). Clinical results are, however, ambiguous and some trials failed to show any beneficial effects of PTX in sepsis (67) and malaria (68) or even in decrease of TNF production (35).
According to earlier postulate (20) that PTX activity is mediated by HO-1 pathway, one could suppose that efficacy of PTX as an anti-inflammatory agent depends on polymorphism of HO-1 promoter resulting in different HO-1 activity in patients enrolled. This could be a possible explanation for the discrepant results of clinical trials. Our studies, performed in endothelial cells of different genotypes excluded this possibility, showing that PTX is an efficient anti-inflammatory compound, regardless of variants of HO-1 promoter.
In summary, we demonstrated that PTX dose-dependently decreases the expression of HO-1 in cultured human and murine monocytes and endothelial cells, whereas does not modulate significantly in vivo HO-1 expression in murine organs. Anti-inflammatory efficacy of PTX is independent of HO-1 pathway, as demonstrated in cells of different HO-1 levels and in HO-1 deficient mice.
Acknowledgements: We thank Dr. Anupam Agarwal, University of Alabama at Birmingham, for providing the HO-1-/- mice. The technical support by Aneta Gierbuszewska MSc, Tina Kubitzki MSc, Malgorzata Ostachowicz MSc, and Aleksandra Sierpniowska MVM, is gratefully appreciated. This work was supported by grant 105/P05/2004 from Polish Ministry of Science and Higher Education. A.J. is a recipient of the Wellcome Trust Senior Research Fellowship in Biomedical Science. H.W. is a recipient START fellowship from Foundation for Polish Science.
Conflict of interests: None declared.
- Solerte SB, Fioravanti M, Cerutti N et al. Retrospective analysis of long-term hemorheologic effects of pentoxifylline in diabetic patients with angiopathic complications. Acta Diabetol 1997; 34: 67-74.
- Sliwa K, Woodiwiss A, Kone VE et al. Therapy of ischemic cardiomyopathy with the immunomodulating agent pentoxifylline: results of a randomized study. Circulation 2004; 109: 750-755.
- Aviado DM, Porter JM. Pentoxifylline: a new drug for the treatment of intermittent claudication. Mechanism of action, pharmacokinetics, clinical efficacy and adverse effects. Pharmacotherapy 1984; 4: 297-307.
- Jull A, Waters J, Arroll B. Pentoxifylline for treatment of venous leg ulcers. Lancet 2002; 359: 1550-1554.
- Ward A, Clissold SP. Pentoxifylline. A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy. Drugs 1987; 34: 50-97.
- Minhas BS, Ripps A. Methods for enhancement of sperm function. Front Biosci 1996; 1: 65-71.
- Rieckmann P, Weber F, Gunther A et al. Pentoxifylline, a phosphodiesterase inhibitor, induces immune deviation in patients with multiple sclerosis. J Neuroimmunol 1996; 64: 193-200.
- Martins AMC, Nobre ACL, Almeida AC et al. Thalidomide and pentoxifylline block the renal effects of supernatants of macrophages activated with Crotalus durissus cascavella venom. Braz J Med Biol Res 2004; 37: 1525-1530.
- Lin HI, Chu SJ, Wang D, Feng NH. Pharmacological modulation of TNF production in macrophages. J Microbiol Immunol Infect 2004; 37: 8-15.
- Tong Z, Chen B, Dai H, Bauer PC, Guzman J, Costable U. Extrinsic allergic alveolitis: inhibitory effects of pentoxifylline on cytokine production by alveolar macrophages. Ann Allergy Asthma Immunol 2004; 92: 234-239.
- Lundblad R, Ekstrom P, Giercksky KE. Pentoxifylline improves survival and reduces tumor necrosis factor, interleukin-6, and endothelin-1 in fulminant intra-abdominal sepsis in rats. Shock 1995; 3: 210-215.
- Tukov FF, Luyendyk JP, Ganey PE, Roth RA. The role of tumor necrosis factor alpha in lipopolysaccharide/ranitidine-induced inflammatory liver injury. Toxicol Sci 2007; 100: 267-280.
- Bielekova B, Lincoln A, McFarland H, Martin R. Therapeutic potential of phosphodiesterase-4 and -3 inhibitors in Th1-mediated autoimmune diseases. J Immunol 2000; 164: 1117-1124.
- Beshay E, Croze F, Prudhomme GJ. The phosphodiesterase inhibitors pentoxifylline and rolipram suppress macrophage activation and nitric oxide production in vitro and in vivo. Clin Immunol 2001; 98: 272-279.
- Blaine TA, Pollice PF, Rosier RN, Reynolds PR, Puzas JE, OKeefe RJ. Modulation of the production of cytokines in titanium-stimulated human peripheral blood monocytes by pharmacological agents. The role of cAMP-mediated signaling mechanisms. J Bone Joint Surg Am 1997; 79: 1519-1528.
- Bhandari V. Developmental differences in the role of interleukins in hyperoxic lung injury in animal models. Front Biosci 2002; 7: 1624-1633.
- Bhat VB, Madyastha KM. Antioxidant and radical scavenging properties of 8-oxo derivatives of xanthine drugs pentoxifylline and lisofylline. Biochem Biophys Res Commun 2001; 288: 1212-1217.
- Coimbra R, Melbostad H, Loomis W, Tobar M, Hoyt DB. Phosphodiesterase inhibition decreases NFkB activation and shifts the cytokine response toward anti-inflammatory activity in acute endotoxemia. J Trauma 2005; 59: 575-582.
- Ji Q, Zhang L, Jia H, Xu J. Pentoxifylline inhibits endotoxin-induced NFkB activation and associated production of proinflammatory cytokines. Ann Clin Lab Sci 2004; 34: 427-436.
- Oh GS, Pae HO, Moon MK et al. Pentoxifylline protects L929 fibroblasts from TNFa toxicity via the induction of heme oxygenase-1. Biochem Biophys Res Commun 2003; 302: 109-113.
- Sarady JK, Otterbein SL, Liu F, Otterbein LE, Choi AM. Carbon monoxide modulates endotoxin-induced production of granulocyte macrophage colony-stimulating factor in macrophages. Am J Respir Cell Mol Biol 2002; 27: 739-745.
- Song R, Ning W, Liu F et al. Regulation of IL-1b-induced GM-CSF production in human airway smooth muscle cells by carbon monoxide. Am J Physiol Lung Cell Mol Physiol 2003; 284: 50-56.
- Bulger EM, Garcia I, Maier RV. Induction of heme-oxygenase 1 inhibits endothelial cell activation by endotoxin and oxidant stress. Surgery 2003; 134: 146-152.
- Wagener, da Silva JL, Farley T. Differential effects of heme oxygenase isoforms on heme mediation of endothelial intracellular adhesion molecule 1 expression. J Pharmacol Exp Ther 1999; 291: 416-423.
- Ishikawa K, Navab M, Leitinger N, Fogelman AM, Lusis AJ. Induction of heme oxygenase-1 inhibits the monocyte transmigration induced by mildly oxidized LDL. J Clin Invest 1997; 100: 1209-1216.
- Chen YH, Chau LY, Lin MW et al. Heme oxygenase-1 gene promoter microsatellite polymorphism is associated with angiographic restenosis after coronary stenting. Eur Heart J 2004; 25: 39-47.
- Schillinger M, Exner M, Minar E et al. Heme oxygenase-1 genotype and restenosis after balloon angioplasty: a novel vascular protective factor. J Am Coll Cardiol 2004; 43: 950-957.
- Schillinger M, Exner M, Mlekusch W et al. Heme oxygenase-1 gene promoter polymorphism is associated with abdominal aortic aneurysm. Thromb Res 2002; 106: 131-136.
- Chen YH, Lin SJ, Lin MW et al. Microsatellite polymorphism in promoter of heme oxygenase-1 gene is associated with susceptibility to coronary artery disease in type 2 diabetic patients. Hum Genet 2002; 111: 1-8.
- Jozkowicz A, Huk I, Nigisch A, Cisowski J, Weigel G, Dulak J. Prostaglandin-J2
upregulates expression of matrix metalloproteinase-1 independently of activation
of peroxisome proliferator-activated receptor-
 .
Acta Biochim Pol 2003; 50: 677-687.
.
Acta Biochim Pol 2003; 50: 677-687. - Visner GA, Lu F, Zhou H, Liu J, Kazemfar K, Agarwal A. Rapamycin induces heme oxygenase-1 in human pulmonary vascular cells: implications in the antiproliferative response to rapamycin. Circulation 2003; 107: 911-916.
- Rieder CRM, Williams AC, Ramsden DB. Selegiline increases heme oxygenase-1 expression and the cytotoxicity produced by dopamine treatment of neuroblastoma SK-N-SH cells. Braz J Med Biol Res 2004; 37: 1055-1062.
- Deng YM, Ben JW, Witting PK, Stocker R. Probucol protects against smooth muscle cell proliferation by upregulating heme oxygenase-1. Circulation 2004; 110: 1855-1860.
- Dulak J, Deshane J, Jozkowicz A, Agarwal A. Heme oxygenase-1 and carbon monoxide in vascular pathology: focus on angiogenesis. Circulation 2008; 117: 231-241.
- Szczepanik AM, Wordliczek J, Serednicki W, Siedlar M, Czupryna A. Pentoxifylline does not affect nociception if administered postoperatively. Pol J Pharmacol 2004; 56: 611-616.
- Hill-Kapturczak N, Sikorski E, Voakes C, Garcia J, Nick HS, Agarwal A. An internal enhancer regulates heme- and cadmium-mediated induction of human heme oxygenase-1. Am J Physiol Renal Physiol 2004; 285: 515-523.
- Zhang C, Li XY, Zhao L, Wang H, Xu DX. Lipopolysaccharide (LPS) up-regulates the expression of haem oxygenase-1 in mouse placenta. Placenta 2007; 28: 951-957.
- Deree J, Martins J, de Campos T et al. Pentoxifylline attenuates lung injury and modulates transcription factor activity in hemorrhagic shock. J Surg Res 2007; 143: 99-108.
- Deree J, de Campos T, Shenvi E, Loomis WH, Hoyt DB, Coimbra R. Hypertonic saline and pentoxifylline attenuates gut injury after hemorrhagic shock: the kinder, gentler resuscitation. J Trauma 2007; 62: 818-827.
- Cerami A, Tracey KJ. Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu Rev Med 1994; 45: 491-503.
- Strieter RM, Remick DG, Ward PA et al. Cellular and molecular regulation of tumor necrosis factor-a production by pentoxifylline. Biochem Biophys Res Commun 1998; 155: 1230-1236.
- van Leenen D, van der Poll T, Levi M et al. Pentoxifylline attenuates neutrophil activation in experimental endotoxemia in chimpanzees. J Immunol 1993; 151: 2318-2325.
- Bruynzeel I, van der Raaij LM, Willemze R, Stoof TJ. Pentoxifylline inhibits human T-cell adhesion to dermal endothelial cells. Arch Dermatol Res 1997; 289: 189-193.
- Schonharting MM, Schade UF. The effect of pentoxifylline in septic shock - new pharmacologic aspects of an established drug. J Med 1989; 20: 97-105.
- Wang W, Zolty E, Falk S, Basava V, Reznikov L, Schrier RW. Pentoxifylline protects against endotoxin-induced acute renal failure in mice. Am J Physiol Renal Physiol 2006; 291: 1090-1095.
- Michetti C, Coimbra R, Hoyt DB, Loomis W, Junger W, Wolf P. Pentoxifylline reduces acute lung injury in chronic endotoxemia. J Surg Res 2003; 115: 92-99.
- Shirin H, Bruck R, Aeed H et al. Pentoxifylline prevents concanavalin A-induced hepatitis by reducing tumor necrosis factor-a levels and inhibiting adhesion of T lymphocytes to extracellular matrix. J Hepatol 1998; 29: 60-67.
- Kwiecien S, Brzozowski T, Konturek PC, Pawlik MW, Kwiecien N, Konturek SJ. Gastroprotection by pentoxyfilline against stress-induced gastric damage. Role of lipid peroxidation, antioxidizing enzymes and proinflammatory cytokines. J Physiol Pharmacol 2004; 55: 337-55.
- Kwiecien S, Pawlik MW, Sliwowski Z et al. Involvement of sensory afferent fibers and lipid peroxidation in the pathogenesis of stress-induced gastric mucosa damage. J Physiol Pharmacol 2007; 58: 149-162.
- Hadjiminas DJ, McMasters KM, Robertson SE, Cheadle WG. Enhanced survival from cecal ligation and puncture with pentoxifylline is associated with altered neutrophil trafficking and reduced interleukin-1ß expression but not inhibition of tumor necrosis factor synthesis. Surgery 1994; 116: 348-355.
- Lenoble GM. New aspects of the pharmacology of pentoxifylline. J Mal Vasc 1989; 14: 35-41.
- Fossat C, Sainty D, Stoppa AM, Maraninchi D, Vague J. Pentoxifylline in vitro reverses neutrophil chemotactic deficiency induced by interleukin-2 treatment. Nouv Rev Fr Hematol 1992; 34: 61-64.
- Gebska A, Olszanecki R, Korbut R. Endotoxaemia in rats: role of leukocyte sequestration in rapid pulmonary nitric oxide synthase-2 expression. J Physiol Pharmacol 2005; 56: 299-311.
- Deshane J, Chen S, Caballero S et al. Stromal cell-derived factor 1 promotes angiogenesis via a heme oxygenase-1 dependent mechanism. J Exp Med 2007; 204: 605-618.
- Porter JM, Cutler BS, Lee BY et al. Pentoxifylline efficacy in the treatment of intermittent claudication: Multicenter controlled doubled-blind trial with objective assessment of chronic occlusive arterial disease patients. Am Heart J 1982; 104: 66-72.
- Meininger V, Asselain B, Guillet P et al. Pentoxifylline in ALS. A doubled-blind, randomized, multicenter, placebo-controlled trial. Neurology 2006; 66: 88-92.
- Zeni F, Pain P, Vindimian M et al. Effects of pentoxifylline on circulating cytokine concentrations and hemodynamics in patients with septic shock: results from a double-blind, randomized, placebo-controlled study. Crit Care Med 1996; 24: 207-214.
- Staudinger T, Presterl E, Graninger W et al. Influence of pentoxifylline on cytokine levels and inflammatory parameters in septic shock. Intensive Care Med 1996; 22: 888-893.
- Farkas G, Nagy Z, Marton J, Mandi Y. Relevance of cytokine production to infected pancreatic necrosis. Acta Chir Hung 1997; 36: 86-88.
- Staubach KH, Schroder J, Stuber F, Gehrke K, Traumann E, Zabel P. Effect of pentoxifylline in severe sepsis: results of a randomized, double-blind, placebo-controlled study. Arch Surg 1998; 133: 94-100.
- Lauterbach R, Zembala M. Pentoxifylline reduces plasma tumor necrosis
factor-
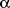 concentration
in premature infants with sepsis. Eur J Pediatr 1996; 155: 404-409.
concentration
in premature infants with sepsis. Eur J Pediatr 1996; 155: 404-409. - Lauterbach R, Pawlik D, Kowalczyk D, Ksycinski W, Helwich E, Zembala M. Effect of the immunomodulating agent, pentoxifylline, in the treatment of sepsis in prematurely delivered infants: a placebo-controlled, double-blind trial. Crit Care Med 1999; 27: 807-814.
- Tsang GM, Allen S, Pagano D, Wong C, Graham TR, Bonser RS. Pentoxifylline preloading reduces endothelial injury and permeability in cardiopulmonary bypass. ASAIO J 1996; 42: 429-434.
- Boldt J, Brosch C, Lehmann A, Haisch G, Lang J, Isgro F. Prophylactic use of pentoxifylline on inflammation in elderly cardiac surgery patients. Ann Thorac Surg 2001; 71: 1524-1529.
- Cagli K, Ulas MM, Ozisik K et al. The intraoperative effect of pentoxifylline on the inflammatory process and leukocytes in cardiac surgery patients undergoing cardiopulmonary bypass. Perfusion 2005; 20: 45-51.
- Wenisch C, Looareesuwan S, Wilairatana P et al. Effect of pentoxifylline on cytokine patterns in the therapy of complicated Plasmodium falciparum malaria. Am J Trop Med Hyg 1998; 58: 343-347.
- Selim K, Huseyin C, Ibrahim KH, Hasan BU, Kazim U, Huseyin K. Effect of pentoxifylline on tumor necrosis factor-a and interleukin-6 levels in neonatal sepsis. Med J Malaysia 2004; 59: 391-394.
- Hemmer CJ, Hort G, Chiwakata CB et al. Supportive pentoxifylline in falciparum malaria: no effect on tumor necrosis factor-a levels or clinical outcome: a prospective, randomized, placebo-controlled study. Am J Trop Med Hyg 1997; 56: 397-403.