Inflammatory bowel disease (IBD) is a chronic relapsing inflammation of the lower part of the gastrointestinal tract. The main forms of IBD are ulcerative colitis and Crohns disease. In the recent decades an unquestionable worldwide increase in the incidence of IBD has been observed. The pathogenesis of IBD remains still not fully understood. It has been postulated that different environmental and genetic factors in combination with the microbial intestinal flora trigger an event which activates intestinal immune (T cells, monocytes, eosinophils, B-cells, neutrophils,
etc.) and non-immune systems (epithelial, mesenchymal cells
etc.). Through secretion of mediators and expression of adhesion molecules, immune and non-immune cells exchange signals resulting in further cell activation and amplification of the production of cytokines, growth factors, nitric oxide, reactive oxygen species, eicosanoids, antibodies
etc. culminating in inflammation and tissue damage (1, 2).
Ghrelin is a 28-amino acid peptide mainly produced in the gastric mucosa. It shares several properties of growth hormone-releasing hormone (GHRH) and acts
via growth hormone secretagogue receptor (GHS). Interestingly, the presence of an octanoyl group at Ser 3 is essential for its activity (3). Apart from a potent GH-releasing action, ghrelin shows a number of physiological activities such as stimulation of appetite and gastrointestinal motility, cardiovascular effects,
etc. (4). Our own studies demonstrated a strong gastroprotective effect of this peptide against acute gastric lesions induced by stress, ethanol or ischemia-reperfusion (5-7).
The fact that the ghrelin receptors are expressed by monocytes/macrophages,
B cells and dendritic cells indicates a possible participation of ghrelin in
the modulation of colonic inflammatory response in IBD (8). However, the potential
role of ghrelin in colitis has not been studied extensively. Recently, Gonzalez-Rey
and Delgado (9) demonstrated for the first time the anti-inflammatory action
of ghrelin in two different experimental models of colitis (TNBS and DDS-induced
colitis) in mice by showing an inhibitory effect of ghrelin on proinflammatory
cytokines and Th1-mediated inflammatory response. The strong anti-inflammatory
activity of ghrelin was accompanied by a significant reduction in the systemic
inflammatory response. Importantly, it was demonstrated that growth hormone
(GH) and IGF-1 do not mediate the effects of ghrelin (9). As a possible important
mechanism responsible for the anti-inflammatory activity of ghrelin was postulated
the downregulation of nuclear factor
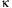
B
(NF
B) (10). However,
the role of ghrelin in colitis remains still controversial since some groups
even postulated the proinflammatory actions of ghrelin
via increase in
NF
B activity (11).
The aim of the present study was: 1) to examine the expression the mRNAs expression
of ghrelin and TNF-
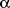
in the inflamed colonic mucosa of patients with UC, 2) to determine the effect
of exogenous ghrelin on the time course of healing of TNBS-induced colitis in
rats, 3) to assess the effects of ghrelin treatment on mRNA expression for inducible
NO synthase (iNOS) and protein expression for cyclooxygenase-2 (COX-2) and peroxisome
proliferators-activated receptor
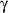
(PPAR
)
and; 4) to analyze the involvement of sensory nerves in the anti-inflammatory
actions of ghrelin on colonic mucosa in rats with TNBS-induced colitis.
MATERIAL AND METHODS
Expression of ghrelin mRNA in colonic mucosa of patients with UC
The biopsy samples were obtained during colonoscopic investigation from sigma and colon ascendens in 15 patients with active and histologically proved UC and in 15 sex- and age matched healthy controls. The expression of ghrelin was assessed by RT-PCR. Human study protocol was approved by Ethics Committee of the University of Erlangen-Nuremberg, Erlangen, Germany and informed consent was obtained from all subjects before they entered the study.
TNBS-induced colitis and treatment groups of rats
All animal experiments were approved by the Local Institutional Animal Care
and Use Committee at the Jagiellonian University College of Medicine, Cracow,
Poland and were carried out in accordance with European Union guidelines for
the handling and use of laboratory animals. Acute colitis was induced according
to published methods (12). Briefly, rats were anesthetized with ketamine and
colitis was induced by intra-rectal administration of 15 mg of 2,4.6-trinitrobenzene
sulphonic acid (TNBS) in 15% ethanol. Following treatment groups (each including
6 animals) received: 1) vehicle (control); 2) TNBS; 3) ghrelin applied in a
dose of 20 µg/kg i.p. starting at 3
rd day after
the induction of the TNBS colitis); 4) L-NNA at a dose 20 mg/kg i.p. to inhibit
NO synthase activity starting at 3
rd day after
induction of TNBS colitis; 5) L-NNA in a dose of 20 mg/kg i.p. co-adminstered
with ghrelin in a dose 20 µg/kg i.p. starting at 3
rd
day after induction of TNBS colitis.
In the separate experiments, the involvement of sensory neurons in the healing by ghrelin of TNBS-colitis was tested. For this purpose the animals were treated with capsaicin (Sigma) injected s.c. for 3 consecutive days at a respective dose of 25, 50 and 50 mg/kg about 2 weeks before induction of TNBS colitis. All injections of capsaicin were performed under ether anesthesia to counteract the respiratory impairment associated with the injection of this agent. To check the effectiveness of the capsaicin denervation, a drop of 0.1 mg/ml solution of capsaicin was instilled into the eye of each rat and the protective wiping movements were counted as previously described. Control rats received vehicle injections. All animals pretreated with capsaicin showed negative wiping movement test, confirming functional denervation of the capsaicin sensitive nerves. Administration of ghrelin or vehicle saline was applied in rats without or with colitis induced by 3 days earlier by TNBS.
Reverse transcription polymerase chain reaction (RT-PCR)
The human biopsy samples from colon were collected for determination of ghrelin mRNA expression. The colonic samples were excised from intact rats and from those treated with vehicle (control), ghrelin and/or L-NNA with or without induction of TNBS-colitis for the determination of mRNA and protein expression for iNOS using RT-PCR with specific primers. Immediately after the measurement of the area of colonic mucosal damage the mucosal specimens were scraped off on ice using a slide glass and immediately snap frozen in liquid nitrogen and stored at -80°C until analysis. Total RNA was extracted from mucosal samples using a guanidium isothiocyanate/phenol chloroform single step extraction kit from Stratagene (Heidelberg, Germany). Following precipitation, RNA was resuspended in RNAse-free TE buffer and its concentration was estimated by absorbance at 260 nm wavelength. Furthermore, the quality of each RNA sample was determined by running the agarose-formaldehyde electrophoresis. RNA samples were stored at -80°C until analysis.
Single stranded cDNA was generated from 5 µg of total cellular RNA using Moloney
murine leukemia virus reverse transcriptase (MMLV-RT) (Stratagene, Heidelberg,
Germany) and oligo-(dT)-primers (Stratagene, Heidelberg, Germany). Briefly,
5 µg of total RNA was uncoiled by heating (65°C for 5 min) and then reverse
transcribed into complementary DNA (cDNA) in a 50 µl reaction mixture that contained
50 U MMLV-RT, 0.3 µg oligo-(dT)-primer, 1 µl RNase Block Ribonuclease Inhibitor
(40U/µl), 2 µl of a 100 mM/l mixture of deoxyadenosine triphosphate (dATP),
deoxythymidine triphosphate (dTTP), deoxyguanosine triphosphate (dGTP) and deoxycytidine
triphosphate (dCTP), 10 mM/l Tris-HCl (pH=8.3), 50 mM KCl, 5 mM MgCl
2).
The resultant cDNA (2 µl) was amplified in a 50 µl reaction volume containing
2 U Taq polymerase, dNTP (200 µM each) (Pharmacia, Germany), 1.5 mM MgCl
2,
5 µl 10x polymerase chain reaction buffer (50 mM KCl, 10 mM Tris-HCl, pH=8.3)
and specific primers for ß-actin, ghrelin, TNF
and iNOS used at final concentration of 1 mM (all reagents from Takara, Shiga,
Japan). The mixture was overlaid with 25 µl of mineral oil to prevent evaporation.
The polymerase chain reaction mixture was amplified in a DNA thermal cycler
(Perkin-Elmer-Cetus, Norwalk, CT) and the incubation and thermal cycling conditions
were as followed; denaturation at 94°C for 1 min, annealing at 60°C for 45 sec
and extension 72°C for 2 min. The number of cycles was 30 for ß-actin,
TNF-
and 33 cycles
for ghrelin, respectively. The nucleotide sequence of the primers were as follows;
human preproghrelin sense: GAG GAT GAA CTG GAA GTC CG; antisense CAT TTA TTC
GCC TCC TGA GC; human TNF-
sense; GAG TGA CAA GCC TGT AGC CCA TGT TGT AGC A; antisense GCA ATG ATC CCA
AAG TAG ACC TGC CCA GAC T; rat ß-actin: sense TTG TAA CCA ACT GGG ACG
ATA TGG; antisense GAT CTT GAT CTT CAT GGT GCT AGG; rat iNOS: sense: CAG TGG
CAA CAT CAG GTC.; antisense: GGT CTC GGA CTC CAA TCT . The primer sequences
were based on the sequences of the published cDNAs.
Polymerase chain reaction products were detected by electrophoresis on a 1.5%
agarose gel containing ethidium bromide. Location of predicted products was
confirmed by using 100-bp ladder (Takara, Shiga, Japan) as a standard size marker.
The gel was then photographed under UV transillumination. The intensity of PCR
products was measured using video image analysis system (Kodak Digital Science)
as described earlier. The signal for investigated mRNA was standardized against
that of the ß-actin mRNA from each sample and the results were expressed
as analyzed ghrelin and iNOS mRNA/ß-actin mRNA ratio as described earlier
(6).
Western blot
For Western blot analysis, proteins were extracted from the same colonic mucosa
samples as mentioned above. Approximately 10 µg of total protein extracts was
loaded on SDS-polyacrylamide gels and run at 40 mA, followed by transfer onto
nitrocellulose membrane (Protran, Schleicher and Schuell, Germany) by electroblotting.
solution of 3% BSA (Sigma Aldrich, Germany) in TBS/Tween-20 buffer (137 mmol
NaCl, 20 mmol Tris-HCl, pH 7.4, 0.1 % Tween-20) was used to block filters for
at least 1 h at room temperature. Specific primary antibody against PPARg and
COX-2 (rabbit polyclonal, dilution 1:200; Santa Cruz, USA) or ß-actin
(mouse monoclonal, dilution 1:3000; Sigma Aldrich, Germany) was added to the
membrane, followed by an anti-rabbit-IgG or anti-mouse-IgG HRP-horseradish peroxidase
conjugated secondary antibody (dilution 1:40 000 or 1:20 000) dissolved in 1%
non-fat milk in TBS-Tween-20 buffer. Incubation of primary antibody was followed
by 3 washes with TBS-Tween-20 buffer for 5 min. incubation of the secondary
antibody was followed by 6 washes for 5 min. Immunocomplexes were detected by
the SuperSignal West Pico Chemiluminescent Kit (Pierce, USA). Thereafter, the
developed membrane was exposed to an X-ray film (Kodak, Wiesbaden, Germany).
Statistical analyses
Results are expressed as means ± S.E.M. from 6 rats per group. Statistical significance
of difference was determined using analysis of variance (one-way ANOVA). Further
statistical analysis for
post hoc comparisons was carried out using Bonferroni/Duncan
test or Students
t-test when appropriate. Differences were considered
statistically at p<0.05 and this is indicated with an asterisk, cross or slash
in the figures.
RESULTS
In control healthy subjects weak but detectable mRNA expressions for ghrelin
and TNF-
in colonic
biopsies were observed. In contrast, patients with UC showed significantly higher
mRNA expression of ghrelin and TNF-
in colonic mucosa. The expression of ghrelin and TNF-
was significantly higher in the sigma than in the colon ascendens (
Fig. 1AB).
 |
Fig. 1. Densitometric analysis
of mRNA expression of ghrelin in colonic mucosa (Colon ascendens and sigma/rectum)
of patients with UC and healthy controls, presented as a ratio of ghrelin
mRNA over GAPDH mRNA (upper panel-A). Densitometric analysis of mRNA expression
of TNF- in
colonic mucosa (Colon ascendens and sigma/rectum) of patients with UC
and controls, presented as a ratio of ghrelin mRNA over GAPDH mRNA (lower
panel-B). Asterisk indicates significant increase of this ratio as compared
to that observed in colonic mucosa in healthy controls. Cross indicates
significant increase above the value observed in colon ascendens. |
As shown in the
Fig. 2, patients with left sided colitis showed an increased
mRNA expression of ghrelin and TNF
only in the sigma, but not in the colon ascendens. In contrast, patients with
pancolitis showed an increased mRNA expression for ghrelin and TNF
both in sigma and colon ascendens.
 |
Fig. 2. Representative RT-PCR
showing the expression of mRNAs for ghrelin, TNF-
and GAPDH (house-keeping gene) in the mucosa of colon ascendens (1) and
in sigma (2) of healthy controls and in patients with left-sided ulcerative
colitis (UC 1) and patients with pancolitis (UC 2). |
The intrarectal administration of TNBS in rats induced severe mucosal injury
characterized by necrosis of the epithelium and focal ulcerations of the mucosa.
The colonic mucosal blood flow was reduced by about 42%. The treatment with
ghrelin was associated with a significant reduction in the mean lesion area
in the colon (by 50%) and significant increase in colonic blood flow as compared
to that observed in vehicle-treated animals with TNBS colitis. As shown in the
Fig. 3, the co-administration of NO synthase blocker, L-NNA to rats with
TNBS colitis, caused a significant aggravation in the mean lesion area and a
profound reduction in the colonic blood flow as compared to those in vehicle-treated
TNBS colitis. The administration of L-NNA before application of ghrelin in TNBS-induced
colitis rats completely abolished the healing effects of ghrelin on colonic
mucosa and also eliminated the vasodilatatory effect of this peptide (
Fig.
3).
 |
Fig. 3. Mean area (mm2)
of colonic lesions induced by TNBS and accompanying changes in the colonic
blood flow measured in intact rats treated with vehicle (saline) or ghrelin
at a dose of 20 µg/kg i.p. with or without pre-treatment with L-NNA (20
mg/kg i.p.). Mean ± SEM of 6 rats per group. Asterisk (*) indicates significant
changes compared to vehicle treated rats with TNBS colitis; cross (+)
indicates significant changes compared to the vehicle-treated TNBS rats
and the slash (#) indicates significant change compared to that observed
in ghrelin-treated TNSB rats. |
To study the role of sensory nerves in the healing effects of ghrelin on the
colonic mucosa, in separate experiments TNBS was administered to rats with capsaicin-ablated
sensory nerves. The inactivation of sensory nerves caused a significant increase
in mean lesion area and a significant decrease in colonic mucosal blood flow
compared to those in rats without capsaicin deactivation (
Fig. 4). In
rats with inactivated sensory nerves by capsaicin the healing and vasodilatatory
effects of ghrelin in colon mucosa were significantly smaller when compared
to those observed with ghrelin in rats without capsaicin deactivation (
Fig.
4).
 |
Fig. 4. Mean area (mm2)
of colonic lesions induced by TNBS and accompanying changes in the colonic
blood flow measured in rats with or without denervation of sensory nerves
with capsaicin and treated with vehicle (control) or ghrelin at a dose
of 20 µg/kg i.p. Mean ±SEM of 6 rats per group. Asterisk (*) indicates
significant change observed in ghrelin-treated rats compared to that in
vehicle-treated controls; cross (+) indicates significant change compared
to vehicle (saline)-treated TNBS colitis rats; asterisk with cross (*
+) indicate significant change as compared to ghrelin-treated rats without
capsaicin deneravation. |
At the mRNA level, the intrarectal administration of TNBS resulted in the increase
in ghrelin mRNA expression starting from day 3 and reaching its maximum at day
7
th after induction of colitis (
Fig. 5).
At days 10 and 14 after induction of colitis with TNBS, the ghrelin expression
decreased to the level observed in the control rats without TNBS colitis but
instead administered with saline (
Fig. 5).
 |
Fig. 5. The mRNA expression for ghrelin in colonic mucosa of intact rats and in colonic mucosa of rats at days 3, 7, 10 and 14 after induction of TNBS colitis. Means ± SEM of 6 rats per group. Asterisk (*) indicates a significant change compared to intact colonic mucosa. |
The iNOS mRNA expression measured by RT-PCR was almost negligible in vehicle-treated
control (intact colon) rats. The induction of TNBS colitis was associated with
a small but significant increase in the mRNA expression for iNOS observed at
14
th day upon the induction of colitis by TNBS.
The treatment with ghrelin of rats with TNBS-induced colitis resulted in a significant
and strong upregulation of mRNA for iNOS (
Fig. 6).
 |
Fig. 6. The mRNA expression for iNOS in the colonic mucosa of intact rats treated with vehicle and in colonic mucosa of rats with TNBS colitis after 14 days upon induction of the colitis without or with ghrelin treatment at 14 days after induction of TNBS colitis. Mean ± SEM of 6 rats per group. Asterisk (*) indicates a significant change compared to intact colonic mucosa and asterisk with cross (*+) indicate significant change compared to that observed in rats with TNBS colitis without ghrelin treatment. |
At protein level, the expression of COX-2 in intact colon mucosa was detected
as a very weak signal. In contrast, the PPAR
expression was detected as a strong signal in the intact colonic mucosa. The
induction of colitis by TNBS was associated with a significant increase in COX-2
expression observed at day 14 upon this induction and this was accompanied by
a significant decrease in PPAR
expression (
Fig. 7). Treatment with ghrelin the expression significantly
increased expression of COX-2 protein above that observed in rats with TNBS
colitis alone without ghrelin treatment, while the PPAR
expression remained at the same low level as in rats with TNBS colitis without
ghrelin treatment (
Fig. 7).
 |
| Fig.
7. Protein expression of COX-2 and PPAR
in intact colonic mucosa and in mucosa of rats with TNBS induced colitis
(14 days after induction of colitis) without or with pretreatment with
ghrelin (20 µg/kg i.p.). Mean ± SEM of 5-6 rats per group. Asterisk (*)
indicates a significant change compared to intact colonic mucosa. Asterisk
with cross (*+) indicate significant change compared to that observed
in rats with TNBS colitis without administration of ghrelin. |
DISCUSSION
The results of this study demonstrate that ghrelin may be implicated in the colonic inflammation as an anti-inflammatory and vasodilatatory substance, which could be possibly triggered by the damage to colonic mucosa. This notion is supported by the fact that the mRNA expression of ghrelin in the colonic mucosa of patients with UC was significantly upregulated in the area of inflamed colonic mucosa. The exact role of ghrelin in the colonic inflammation in humans remains unclear but results of our study are in keeping with the observation of Karmiris
et al. (13) showing that the serum levels of ghrelin are significantly increased in patients with IBD.
To elucidate the role of ghrelin in the modulation of colitis, the experimental colits with TNBS was induced in rats. In this model that mimics many features of human colitis, 2,4,6-trinitrobenzene sulfonic acid (TNBS) is delivered intrarectally and the inflammation results from a covalent binding of the haptenizing agent to autologous host proteins with subsequent stimulation of a delayed-type hypersensitivity to TNBS-modified self antigens (14). Using such rats with colitis we demonstrated a strong anti-inflammatory and vasodilatatory effect of exogenous ghrelin, which reduced the mean lesion area in colon by about 50% and increased significantly the colonic mucosal blood flow. The amelioration of colonic mucosal injury by ghrelin was accompanied by a significant upregulation of inducible NO synthase indicating an involvement of NO in the healing effect of ghrelin on experimental colitis. In addition, the concurrent treatment with of NO-synthase inhibitor (L-NNA) completely abolished the anti-inflammatory effects of ghrelin against TNBS-induced mucosal injury. These results are in keeping with the previous reports showing the contribution of endogenous NO in the protective effect of ghrelin against acute gastric mucosal injury (6, 7, 15). However, the exact role of iNOS in colonic inflammation remains still controversial and is the subject of intense investigation. A substantial number of the experimental studies demonstrated an improvement of colitis with iNOS inhibition (16). There are also several reports showing an exacerbation of colitis with application of iNOS inhibitors (17). It is known that iNOS expression is regulated by several pro-inflammatory cytokines and it functions to generate high levels of NO
via oxidative metabolism of L-arginine. NO seems to have a dual role in colitis leading to opposite results in IBD models. On one hand, NO leads to vasodilatation, host defense and inhibition of neutrophil activation and platelet aggregation. On the other hand, NO is a highly reactive free radical and may rapidly react with active oxygen to generate peroxynitrate, which causes severe detrimental effects on colonic mucosa (18).
The present study demonstrated a significant downregulation of peroxisome proliferators-activated
receptor
(PPAR
),
a member of the nuclear receptor superfamily of transcriptor factors. Previous
studies demonstrated that PPAR
plays a crucial role in the control of intestinal inflammation and its resolution
(19). Numerous studies demonstrated that the activation of PPAR
by specific ligands causes an amelioration of colitis (20, 21). The anti-inflammatory
effects of PPAR
agonists were partly mediated by inhibition of NF
B
and p38 MAPK signaling pathways (22). In the present study we demonstrated that
the healing effects of ghrelin treatment have no significant influence on PPAR
expression that remained at decreased level as compared to rats with TNBS colitis
without ghrelin treatment.
An important finding of the present study was the increase in COX-2 protein
expression in colonic mucosa in rats with TNBS-induced colitis treated with
ghrelin. This fact supports the role of COX-2 derived prostaglandins in ghrelin-mediated
healing of TNBS-induced colitis. In agreement with our present observation,
the exacerbation of colitis by COX-2 specific inhibitors was currently documented
(23). In addition, the recent study by Tsubouchi
et al (24) revealed
a significant increase in mucosal PGE
2 generation
in the experimental colitis that was attenuated by specific COX-2 inhibitor
such as rofecoxib but not COX-1 specific inhibitor such as SC-560. All these
data together indicate an involvement of COX-2 derived prostaglandins in healing
of colonic lesions by ghrelin and these results are corraborative with our previous
findings that ghrelin attenuated gastric damage induced in acute model of ischemia-reperfusion
in rats (5-7).
Of physiologic relevance is the observation that deactivation of sensory nerves by capsaicin was associated with aggravation of TNBS-induced colitis and the partial attenuation of protective effect of ghrelin. This observation suggests an importance of integrity of sensory nerves in the healing effects of ghrelin as shown in our previous studies in the different models of acute gastric injury (6, 7). Our results are in agreement with the earlier studies showing an aggravation of colonic inflammation by sensory denervation with capsaicin and an improvement in healing of colonic injury by neuropeptide CGRP released from these nerves (25, 26).
Taken together, our study demonstrates that ghrelin expression is upregulated
in colonic mucosa of patients with UC and that treatment with this peptide exerts
a beneficial anti-inflammatory effect on experimental colitis, resulting in
acceleration of colonic lesions healing. The amelioration of experimental colitis
by ghrelin is, at least partly, due to the increased release of NO, stimulation
of COX-2-derived PGE
2 and depends on the integrity
of sensory nerves. Ghrelin does not influence the PPAR
expression considered as protective factor in IBD (17) that is according to
our present study significantly decreased in TNBS-induced colitis.
Conflict of interest statement: None declared.
REFERENCES
- Sands BE. Inflammatory bowel disease: past, present, and future. J Gastroenterol
2007; 42(1); 16-25.
- Aoi Y, Terashima S, Ogura M, Nishio N, Kato S, Takeuchi K. Roles of nitric
oxide (NO) synthesis in healing of dextran sulfate sodium-induced rat colitis.
J Physiol Pharmacol 2008; 59: 315-336.
- Kojima M, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing
acylated peptide from stomach. Nature 1999; 402: 656-660.
- Kojima M Kangawa K. Ghrelin: Structure and Function. Physiol Rev 2005;
85: 495-522
- Konurek PC, Pajdo R, Nikiforuk A et al. Ghrelin-a new gastroprotective
factor in gastric mucosa. J Physiol Pharmacol 2004; 55(2): 325-336.
- Konturek PC, Brzozowski T, Walter B, Burnat G, Hahn EG, Konturek SJ. Ghrelin-induced
gastroprotection against ischemia-reperfusion injury involves an activation
of sensory afferent nerves and hyperemia mediated by nitric oxide. Eur J
Pharmacol 2006; 235: 171-181.
- Brzozowski T, Konturek PC, Sliwowski Z et al. Neural aspects of
ghrelin-induced gastroprotection against mucosal injury induced by noxious
agents. J Physiol Pharmacol 2006; 57 (Suppl 6): 63-76.
- Dixit VD, Pyle R.S, Collins GD et al. Ghrelin inhibits leptin-
and activation-induced proinflammatory cytokine expression by human monocytes
and T-cells. J Clin Invest 2004; 14: 57-66.
- Gonzalez-Rey ECA, Delgado M. Therapeutic action of ghrelin in a mouse
model of colitis. Gastroenterology 2006; 130: 1707-1720.
- Li WG, Liu X, Wang L et al. Ghrelin inhibits proinflammatory responses
and nuclear factor kB activation in human endothelial cells. Circulation
2004; 109: 2221-2226.
- Zhao DZY, Zeng H, Moyer MP, Mantzoros CS, Pothoulakis C. Ghrelin stimulates
interleukin-8 gene expression through protein kinase C-mediated NF-kB pathway
in human colonic epithelial cells. J Cell Biochem 2006; 97: 1317-1327.
- Inokuchi Y, Kawana Y, Nagashima Y, Kihara M, Umemura S. Amelioration of
2,4,6-trinitrobenzene sulphonic acid induced colitis in angiotensinogen
gene knockout mice. Gut 2005; 54: 349-356.
- Karmiris KI, Xidakis C, Polychronaki M, Voudouri T, Kouroumalis EA. Circulating
levels of leptin, adiponectin, resistin and ghrelin in inflammatory bowel
disease. Inflamm Bowel Dis 2006; 12(2): 100-105.
- Hoffmann JC, Kuhl AA, Hohne W, Zeitz M. Animal models of inflammatory
bowel disease: an overview. Pathobiology 2003; 70: 121-130.
- Sibilia V, Pagani F, Rapetti D et al. Ghrelin protects against
ethanol-induced gastric ulcers in rats: studies on the mechanisms of action.
Endocrinology 2003; 144: 353-359.
- Dudhgaonkar SP, Tandan SK, Kumar D, Raviprakash V, Kataria M. Influence
of simultaneous inhibition of cyclooxygenase-2 and inducible nitic oxide
symthase in experimental colitis in rats. Inflammopharmacology 2007; 15:
188-195.
- Dikopoulus N, Nussler AK, Liptay S et al. Inhibition of nitric
oxide synthesis by aminoguaniidine increases intestinal damage in the acute
phase of rat TNB-colitis. Eur J Clin Invest 2001; 31: 234-239.
- Cross RK. Nitric oxide in inflammatory bowel disease. Inflamm Bowel Dis
2003; 9: 179-189.
- Dubuquoy L, Thuru X, Peyrin-Biroulet L at al. PPAR
as a new therapeutic target in inflammatory bowel disease. Gut 2006; 55(9):
1341-1349.
- Adachi M, Morimura K, Shah Y et al. Peroxisome proliferator activated
receptor gamma in colonic epithelial cells protects against experimental
inflammatory bowel disease. Gut 2006; 55(8): 1067-1069.
- Takaki K, Tsuruta O, Toyonaga A, Sata M. Attenuation of experimental colonic
injury by thiazolidinedione agents. Inflamm Res 2006; 55(1): 10-15.
- Sanchez-Hidalgo MMA, Villegas I, Alarcon de la Lastra C. Rosiglitazone,
a PPAR
ligand, modulates signal transduction pathways during the development of
acute TNBS-induced colitis in rats. Eur J Pharmacol 2007; 562(3): 247-258.
- Zhang L, Dong XY. Effects and mechanism of the selective COX-2 inhibitor,
celecoxib, on rat colitis induced by trinitrobenzene sulfonic acid. Chin
J Dig Dis 2004; 5(3): 110-114.
- Tsobouchi R, Aoi Y, Nishio H, Terashima S, Kato S, Takeuchi K. Healing
impairment effect of cyclooxygenase inhibitors on dextran sulfate sodium-induced
colitis in rats. Digestion 2006; 74(2): 91-100.
- Reinshagen M, Sottili M, French S, Sternini C, Eysselein VE. Action of
sensory neurons in an experimental colitis model of injury and repair. Am
J Physiol 1996; 270: G79-G86.
- Reinshagen M, Ernst S, Geerling I et al. Calcitonin gene-related
peptide mediates the protective effect of sensory nerves in a model of colonic
injury. J Pharmacol Exp Ther 1998; 286(2): 657-661.