The oral mucosal responses to ethanol cytotoxicity are manifested by the elevation in proinflammatory cytokine production, enhancement in epithelial cell apoptosis, disturbances in epidermal growth factor and nitric oxide signaling pathways, and the impairment in prostaglandin generation, (13-15). Moreover, salivary glands of alcoholics and animals exposed to ethanol display the evidence of fatty infiltration, acinar cell swelling, and change in cytoplasm vacuolation accompanied by cellular degeneration and even atrophy (16, 17). Interestingly, recent evidence indicates that the critical event responsible for rapid changes in prostaglandin production is the selective channeling of arachidonic acid substrate, released from membrane glycerophospholipids by the action of cPLA2 enzyme, to the site of cyclooxygenase-2 (COX-2) action for prostaglandin synthesis (18, 19). The activity of cPLA2 is tightly regulated by post-translational mechanism involving MAPK/ERK-dependent enzyme protein phosphorylation and calcium influx that facilitate the enzyme translocation from cytosol to phospholipid-rich membrane (20, 21). There are also reports suggesting that cPLA2 is a downstream effector of ERK in leptin signaling through Src, and that leptin-induced responses mediated by ERK involve epidermal growth factor receptor (EGFR) transactivation (22-24).
Review of the pertinent literature indicates that the signaling cross-talk involving EGFR transactivation is implicated in the regulation of a wide variety of cell functions of significance to oral mucosal repair and integrity maintenance, including cellular proliferation, differentiation, survival, and migration to the site of injury (24, 25). In general, the signals triggered by EGFR transactivation with the involvement of Src are short of duration and result in transient activation of ERK, which does not undergo nuclear translocation (26), and hence could be utilized for a direct activation through phosphorylation a number of cytosolic proteins of significance to cellular survival, including cPLA2. Moreover, there are indications that Src-dependent transactivation pathway is involved in the induction of matrix metalloproteinase-9 (MMP-9), which promotes selective release of membrane-anchored EGFR ligands that subsequently activate EGFR to initiate ERK phosphorylation (27, 28).
Recently, using rat sublingual salivary gland acinar cells, we demonstrated that leptin protection of the acinar cells against ethanol cytotoxicity involves Src kinase-mediated cPLA2 activation (11). In this study, we provide evidence that transactivation of EGFR is involved in the signaling cascade that leads to cPLA2 activation and up-regulation in PGE2 generation, and thus mediate leptin protection of salivary gland acinar cells against ethanol cytotoxicity.
Sublingual gland cell preparation
The acinar cells of sublingual gland were collected from freshly dissected rat salivary glands by passage of the trimmed glandular tissue through a 50 mesh metal grid (29). The minced tissue was then suspended in five volumes of ice-cold Dulbeccos modified (Gibco) Eagles minimal essential medium (DMEM), supplemented with fungizone (50 µg/ml), penicillin (50 U/ml), streptomycin (50 µg/ml), and 10% fetal calf serum, and dispersed into single cells and cell clusters by trituration with a glass homogenizer, and settled by centrifugation. After three consecutive rinses with DMEM, the cells were resuspended in the medium to a concentration of 2 x 107 cell/ml. The viability of cell preparations before and during the experimentation, assessed by Trypan blue dye exclusion assay, was greater than 98%.
Ethanol-induced cytotoxicity
Aliquots of the acinar cells suspension (1 ml) were transferred to DMEM in culture dishes and incubated for 2 h at 37ºC under 95% O2/5% CO2 atmosphere in the absence and the presence of 3% of ethanol (11). In the experiments evaluating the effect of leptin (mouse recombinant, Sigma), janus kinase (JAK) inhibitor AG490 (Calbiochem), Src kinase inhibitor PP2 (Calbiochem), EGF and EGFR kinase inhibitor AG1478 (Sigma), ERK1/2 inhibitor PD98059 (Calbiochem), PKC inhibitor Ro318220, and a broad spectrum matrix metalloproteinase inhibitor (MMP), GM6001 Calbiochem), the cells were first treated for 30 min with the indicated dose of the agent or vehicle followed by 2 h incubation with ethanol (11). At the conclusion of incubation, the aliquots of cell suspension from the control and various experimental conditions were centrifuged at 250 x g for 5 min and the supernatants used for the measurement of cytotoxicity using TOX-7 lactate dehydrogenase assay kit in accordance with the manufacturers (Sigma) instructions.
PGE2 and leptin quantification
The aliquots of the acinar cells suspension from the control and various experimental conditions were centrifuged at 1500 x g for 5 min and the conditioned medium supernatant collected. PGE2 assays were carried out using a PGE2 EIA kit (Cayman) and 100µl aliquots of the spent medium supernatant, according to the manufacturers instruction. The amount of PGE2 released by the acinar cell incubates into the medium was expressed in pg/ml (9). Leptin content in the acinar sublingual gland cells was measured with a mouse leptin enzyme-linked immunometric assay according to manufacturers (Calbiochem) instruction.
Acinar cell cPLA2 activity assay
The measurement of cPLA2 activity in the acinar salivary gland cells following various experimental conditions was carried out using cPLA2 assay kit (Cayman). Following experimental treatments, the cells were homogenized in 1 ml of 50 mM Hepes buffer, pH 7.4, containing 1 mM EDTA, centrifuged at 10,000 x g for 15 min at 4 °C and the supernatants filtered through an Amicon YM30 filter concentrators (m.w. cut-off 30,000) to remove any contamination with secretory PLA2, followed by 15 min incubation with 5 µM of calcium-independent PLA2 inhibitor, bromoenol lactone. The aliquots (10µl) of such prepared cell lysates were the subjected to cPLA2 assay according to manufacturers instruction.
Phospholipid labelling and [3H]arachidonic acid release assay
Arachidonic acid release from the acinar cells of salivary gland into the incubation medium was used to assess the activity of cPLA2 (23). Aliquots of the cell suspension (1 ml) were labeled with 20 µCi of [5,6,8,9,11,12,14,15-3H]arachidonic acid for 4 h, washed with DMEM containing 5% albumin to remove free radiolabel, and resuspended in fresh DMEM free of albumin. The cells were then treated with the indicated dose of the agent of interest, incubated for 2 h in the presence of 3% ethanol (11), and following centrifugation the supernatant was analyzed for the released [3H]arachidonic acid by scintillation spectrometry.
MMP-9 assay
The acinar cell suspensions from the control and experimental treatments were centrifuged at 300g for 5 min and the conditioned medium supernatant collected. The assays of matrix metalloproteinase-9 (MMP-9) were carried out using a MMP-9 ELISA kit (Calbiochem) and 100µl aliquots of spent culture medium, according to manufacturers instruction.
Measurement of EGFR tyrosine kinase transactivation
The measurements of EGFR transactivation was conducted with PhosphoDetect Elisa kit (Calbiochem). The acinar cells from the experimental treatments were washed with phosphate-buffered saline, treated with the receptor extraction buffer and centrifuged at 1500 x g for 10 min at 4 °C. The supernatant was incubated at room temperature for 2 h with anti-EGFR antibody followed by 30 min incubation with protein A agarose and centrifuged (30). The pellet was suspended in kinase reaction buffer and 100 µl aliquots were used for EGFR phosphotyrosine assays following the manufacturers instructions.
Western blotting procedures
The acinar cells from the control and experimental treatments were collected by centrifugation, washed with phosphate-buffered saline, and resuspended in ice-cold lysis buffer (11). Following brief sonication, the cell lysates were centrifuged at 12,000g for 10 min, and the supernatants were subjected to protein determination using BCA protein assay kit (Pierce). The samples were then resuspended in loading buffer, boiled for 5 min, and subjected to SDS-PAGE using 50 µg protein/lane (11). The separated proteins were transferred onto nitrocellulose membranes, blocked with 5% skim milk, and incubated with the antibodies against the phosphorylated proteins at 4 °C for 16 h (11). After 1 h incubation with the horseradish peroxidase-conjugated secondary antibody, the phosphorylated proteins were revealed using an enhanced chemiluminescence detection kit (Pierce). Membranes were stripped by incubation in 1M Tris-HCl (pH 6.8), 10% SDS, and 10 mM dithiotreitol for 30 min at 55 °C, and reprobed with antibodies against the proteins of interest. Immunoblotting was performed using specific antibodies directed against EGFR and phospho-EGFR (Tyr1173).
Data analysis
All experiments were carried out using duplicate sampling and the results are expressed as means ±SD. Analysis of variance (ANOVA) was used to determine significance and the significance level was set at P < 0.05.
Building on our recent finding that leptin protection of salivary gland acinar cells against ethanol cytotoxicity stems from its ability to impact the events of cPLA2 activation for the increase in arachidonic acid release for prostaglandin synthesis (11), we investigated further the factors affecting signaling pathways that modulate cPLA2 activity and arachidonic acid release. Using rat sublingual gland acinar cells exposed to incubation with ethanol in conjunction with lactate dehydrogenase cytotoxicity, we demonstrated that preincubation with leptin at the previously determined optimal concentration of 1 µg/ml (11), resulted in a nearly complete protection against ethanol-induced cytotoxicity (Fig. 1). At the same time, the acinar cell assays of the content of endogenous leptin gave the mean value of 32.1 pg/mg protein.
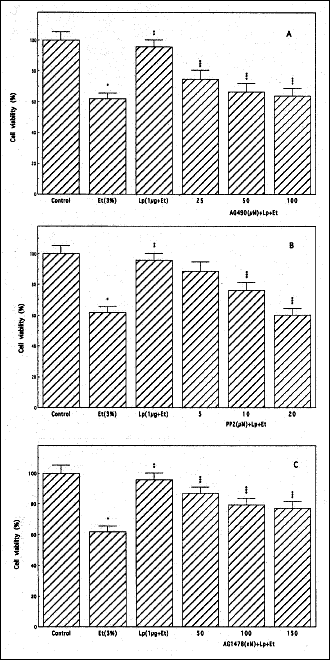 |
Fig. 1. Effect of Janus kinase (JAK) inhibitor, AG490 (A), Src kinase inhibitor, PP2 (B), and EGFR kinase inhibitor, AG1478 (C) on the leptin (Lp)-induced protection of rat sublingual salivary gland acinar cells against ethanol (Et) cytotoxicity. The cells, preincubated with the indicated concentrations of AG490, PP2, and AG1478, were treated with Lp at 1 µg/ml and incubated for 2 h in the presence of 3% Et. The cell-free aliquots of the medium were assayed for lactate dehydrogenase release. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of Et alone. ***P < 0.05 compared with that of Lp+Et. |
Thus using pharmacological concentration of leptin at 1 µg/ml, we found that the protective effect of this pleiotropic hormone against the ethanol-induced salivary gland acinar cell toxicity was subject to suppression by AG490, a specific inhibitor of JAK (Fig.1A), PP2, a selective inhibitor of tyrosine kinase Src (Fig. 1B), as well as an inhibitor EGFR kinase, AG1478 (Fig. 1C). Further results reveled that all three agents evoked also the inhibition in leptin-induced up-regulation in arachidonic acid release and the acinar cell capacity for PGE2 generation (Fig. 2). These findings point to the involvement of EGFR transactivation in the processes of leptin-induced PGE2 generation as well as to the role of cPLA2 activation in the protective mechanism of leptin action against ethanol cytotoxicity in salivary glands.
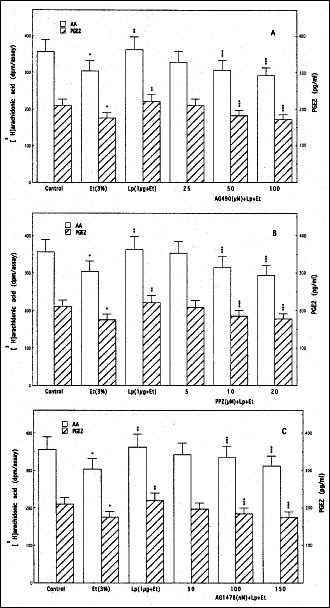 |
Fig. 2. Effect of JAK inhibitor, AG490 (A), Src kinase inhibitor, PP2 (B), and EGFR kinase inhibitor, AG1478 (C), on leptin (Lp)-induced changes in cPLA2 activation and PGE2 generation by sublingual salivary gland acinar cells in the presence of ethanol (Et). The [3H]arachidonic acid-labeled cells, preincubated with the indicated concentrations of AG490, PP2, and AG1478, were treated with Lp at 1µg/ml and incubated for 2 h in the presence of 3% Et. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of Et alone. ***P < 0.05 compared with that of Lp+Et. |
Since cPLA2 activation for the rapid release of arachidonic acid involves MAPK/ERK-dependent enzyme protein phosphorylation on Ser505 that plays a crucial role in Ca2+-dependent translocation of the enzyme from cytosol to phospholipid-rich membrane (20, 21), we next assessed the acinar cell cPLA2 activation by leptin by measuring cPLA2 enzymatic activity following various treatments. As summarized in Fig. 3, the cytotoxic effect of ethanol was reflected in a 30% drop in the acinar cell cPLA2 activity, while preincubation with leptin countered the ethanol effect and evoked a 1.6-fold increase in the cPLA2 activity. Moreover, the leptin-induced up-regulation in the acinar cell cPLA2 activity was subject to suppression by ERK1/2 inhibitor, PD98059 as well as the inhibitors of JAK (AG490), Src (PP2), and EGFR (AG1478) kinases.
 |
Fig. 3. Effect of JAK inhibitor, AG490 (AG4), Src kinase inhibitor, PP2, EGFR inhibitor, AG1478 (AG1), and ERK1/2 inhibitor, PD98059 (PD), on leptin (Lp)-induced cPLA2 activity. The acinar cells, preincubated with 100µM AG4 or 20 µM PP2 or 150 nM AG1 or 30 µM PD, were treated with 1µg/ml Lp and incubated for 2 h in the presence of 3% ethanol (Et). The cell homogenates were centrifuged and the supernatants subjected to measurements of cPLA2 activity. Values represent the means ± SD of five experiments *P < 0.05 compared with that of control. **P < 0.05 compared with that of Et. ***P < 0.05 compared with that of Lp+Et. |
To characterize further the involvement of EGFR transactivation in leptin-induced signaling leading to up-regulation in salivary gland acinar cell cPLA2 activation we monitored the requirements for EGFR protein tyrosine kinase (PTK) activation. As shown in Fig. 4, we observed that leptin at its optimal concentration (1µg/ml) for the suppression of the cytotoxic effect of ethanol elicited a 1.9-fold stimulation in the acinar cell EGFR PTK activity, and the effect was not only subject to the suppression by EGFR kinase inhibitor AG1478, but also JAK kinase inhibitor, AG490 and Src kinase inhibitor, PP2. However, the leptin-induced up-regulation in EGFR PTK activity was not affected by the preincubation with MAPK/ERK inhibitor, PD98059. These data together with the results on cPLA2 activation are indicative of the involvement of JAK and Src in EGFR PTK activation as well as the requirement for EGFR transactivation in leptin-induced up-regulation of cPLA2 activity through ERK activation.
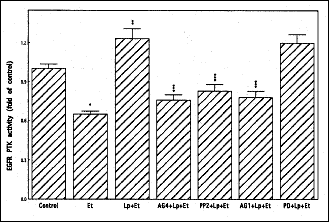 |
Fig. 4. Effect of leptin (Lp) on epidermal growth factor receptor (EGFR) protein tyrosine kinase (PTK) activity in sublingual salivary gland acinar cells exposed to ethanol (Et). The cells, preincubated with 100 µM AG490 (AG4), 150 nM AG1478 (AG1), 20µM PP2 or 30 µM PD98059 (PD), were treated with 1µg/ml Lp and incubated for 2 h in the presence of 3% Et. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of ethanol alone. ***P < 0.05 compared with that of Lp+Et. |
As there are reports that membrane-anchored EGFR ligands in response to external stimuli are cleaved and processed by matrix metalloproteinases to promote receptor engagement (24-26, 28), we next assessed the role of metalloproteinases in leptin-induced EGFR transactivation in the acinar cells. We found that preincubation of the acinar cells with a broad-spectrum metalloproteinase inhibitor, GM6001 led to a concentration-dependent suppression in the protective effect of leptin against ethanol cytotoxicity (Fig. 5). Moreover, the GM6001 caused also the suppression (up to 29.3%) in leptin-induced EGFR PTK activity and blocked the leptin-induced EGFR phosphorylation, but had no effect on EGFR phosphorylation induced by EGF ligand (Fig. 6). These findings thus point to the involvement of matrix metalloproteinases in the event of leptin-induced EGFR transactivation that results in a signaling cascade leading to cPLA2 activation and up-regulation in PGE2 generation.
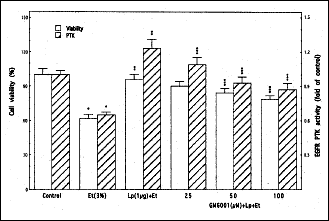 |
Fig. 5. Effect of matrix metalloprotease inhibitor, GM6001 on leptin (Lp)- induced EGFR PTK activity and the protection of sublingual salivary gland acinar cells against ethanol (Et) cytotoxicity. The cells, preincubated with the indicated concentrations of GM6001, were treated with 1 µg/ml Lp and incubated for 2 h in the presence of 3% Et. Values represent the means ± SD of five experiments. *P < 0.05 compared with that of control. **P < 0.05 compared with that of Et alone. ***P < 0.05 compared with that of Lp+Et. |
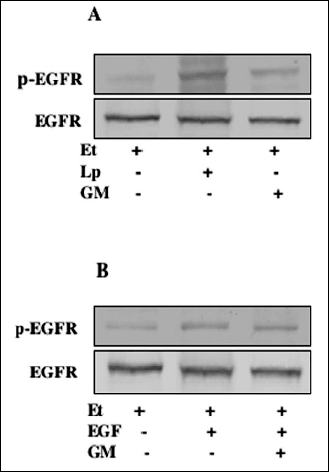 |
Fig. 6. Effect of leptin (Lp) and epidermal growth factor (EGF) on EGFR tyrosine phosphorylation in the acinar cells exposed to ethanol (Et). The cells, preincubated with 100 µM GM6001 (GM), were treated with 1 µg/ml Lp (A) or 10 ng/ml EGF (B) and incubated for 2 h in the presence of Et. Cell lysates were resolved on SDS-PAGE, transferred to nitrocellulose and probed with anti-phosphotyrosine antibody (p-EGFR), and after stripping reprobed with anti-EGFR antibody. The immunoblots shown are representative of three experiments. |
Recent literature data indicate that Src kinase-dependent EGFR transactivation occurs with the involvement of matrix metalloproteinase, MMP-9 (27, 28). Therefore, we further evaluated the effect of leptin on the acinar cell MMP-9 production. As shown in Fig. 7, leptin at its optimal concentration for the suppression of the cytotoxic effect of ethanol evoked a 2.1-fold increase in the acinar cell MMP-9 secretion, and the observed effect was subject to suppression by pretreatment with Src kinase inhibitor, PP2 as well as JAK kinase inhibitor, AG490 and metalloprotease inhibitor, GM6001. We have also observed that preincubation with EGFR kinase inhibitor, AG1478 and PKC inhibitor, Ro318220 did not produce any discernible impact on the leptin-induced MMP-9 secretion (Fig. 7). Thus, the induced up-regulation in the acinar cell MMP-9 secretion associated with the countering effect of leptin against ethanol cytotoxicity occurs upstream of EGFR, with the involvement of JAK and Src, and does not appear to show the requirement for PKC participation.
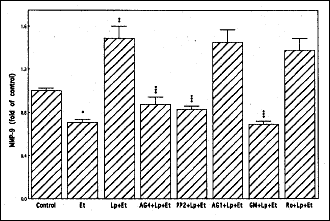 |
Fig. 7. Effect of JAK inhibitor, AG490 (AG4), Src inhibitor PP2, EGFR inhibitorAG1478 (Ag1), matrix metalloprotease inhibitor, GM6001 (GM), and PKC inhibitor Ro318220 (Ro) on leptin(Lp)-induced changes in MMP-9 generation by the acinar cells exposed to ethanol (Et). The cells, preincubated with 100 µM AG4, 20µM PP2, 150nM AG1, 100 µM GM or 10µM Ro, were treated with 1 µg/ml Lp, incubated for 2 h in the presence of Et, and the medium analyzed for MMP-9. *P < 0.05 compared with that of control. **P < 0.05 compared with that of Et alone. ***P < 0.05 compared with that of Lp+Et. |
Investigations into the functional role of leptin released locally within peripheral tissues and secreted into saliva by the acinar cells of salivary glands, have brought to the forefront the importance of this peptide hormone to the processes of mucosal defense and repair along alimentary tract, including that of oral cavity (4-8). The cytokine released locally within the oral cavity has emerged as an important integrator of intracellular signaling pathways that are of significance to the maintenance of soft oral tissue homeostasis. Indeed, the increase in leptin level characterizes oral mucosal responses to injury, and the exogenous leptin is known to accelerate wound repair and protects the acinar cells of salivary gland against cytotoxic effect of ethanol (7, 11, 12, 31). As the diminished secretion of saliva and oral mucosal inflammatory changes are well-recognized consequences of alcohol abuse on the health of oral cavity (14-17, 32), we focused our attention on the understanding the way leptin protects the acinar cells of salivary glands against ethanol cytotoxicity by examining functional and signaling cross-talk between responses induced by leptin and those involving EGFR transactivation.
The available data indicate that the prominent manifestation of oral mucosal and the acinar cell ethanol cytotoxicity is the impairment in prostaglandin generation, and that the critical event responsible for rapid changes in prostaglandin production is the release of arachidonic acid from membrane phospholipids by the action of cPLA2 enzyme (9, 11, 13). The literature evidence, furthermore, implies that cPLA2 is a downstream effector of ERK in leptin signaling, and we have shown recently that leptin-induced cPLA2 activation for the increase in arachidonic acid release for prostaglandin generation requires MAPK/ERK participation (11, 22-24). Hence, using rat sublingual salivary gland acinar cells exposed to ethanol at the concentration range that impairs the cell capacity for mucin synthesis and prostaglandin generation (13, 33), we examined the mechanism of leptin-induced cPLA2 activation for the increase in PGE2 generation and the role of EGFR transactivation in this process.
The results of our findings revealed that the protective effect of leptin against ethanol cytotoxicity was associated with the increased EGFR protein tyrosine kinase and cPLA2 activation, and characterized by a marked increase in arachidonic acid release, and PGE2 generation. Furthermore, a significant loss in the induced up-regulation in PGE2 generation and the countering capacity of leptin on the ethanol-induced toxicity was attained with JAK kinase inhibitor AG490, Src kinase inhibitor PP2 and EGFR inhibitor AG1478, as well as MAPK/ERK inhibitor PD98059. These findings, together with the demonstrated dependence of cPLA2 activity on the enzyme protein phosphorylation (20, 21), attest to the involvement of EGFR transactivation in the processes of leptin-induced PGE2 generation as well as to the role of cPLA2 activation in the acinar cell protection against ethanol cytotoxicity. Indeed, as revealed earlier, the post-translational cPLA2 activation through phosphorylation provides for rapid cellular responses that require selective release and channeling of arachidonic acid to the site of COX-2 action for prostaglandin synthesis (18-20).
In concordance with the prevailing evidence for the involvement of Src kinase in the signaling pathways associated with EGFR transactivation (26-28), the results of our findings suggest that leptin-induced EGFR transactivation is dependent on JAK and Src kinase, and that ERK activation is dependent on EGFR transactivation. We found that leptin-induced increase in the acinar cell EGFR PTK activity was not only subject to suppression by EGFR kinase inhibitor, AG1478, but also to JAK and Src kinase inhibitors, AG490 and PP2. However, the induced up-regulation in EGFR PTK activity was not affected by MAPK/ERK inhibitor, PD98059. Hence, ERK activation by leptin for the increase in cPLA2 activity and PGE2 generation is clearly dependent on the event of EGFR tyrosine kinase activation. This is also in keeping with the reported association of Src kinase with EGFR and its activation by phosphorylation at the tyrosine residues (26, 34, 35). Moreover, our results lend further support to a prevalent concept that Src is an upstream effector of EGFR transactivation (26, 27, 34-36).
Recent studies indicate that Src kinase-dependent events involving cross talk between EGFR and Src that result in transient ERK activation occur with the involvement of matrix metalloproteinases (24-28). Indeed, the Src kinase-dependent EGFR transactivation has been demonstrated as an essential requirement for MMP-9 (type IV collagenase) activation in human tracheal smooth muscle cells, MMP-1 activation was observed in association with EGFR-induced esophageal keratinocytes migration, and the requirement for matrix metalloproteinase was shown to be an essential element of EGFR-mediated signaling induced in neuronal cell by gonadotropin-releasing hormone receptor activation (26, 27, 37). In our study, reported herein, we employed a broad-spectrum metalloprotease inhibitor, GM6001, capable of membrane-anchored HB-EGF cleavage inhibition (24, 27, 28). We found that GM6001 not only evoked suppression in the protective effect of leptin against ethanol cytotoxicity and the inhibition in leptin-induced EGFR PTK activity, but also blocked the leptin-induced EGFR phosphorylation. However, the GM6001 had no effect on the acinar cell EGFR phosphorylation induced by the exogenous EGF ligand. These findings thus point to the involvement of matrix metalloproteinases in the event of leptin-induced EGFR transactivation that results in a signaling cascade leading to cPLA2 activation and up-regulation in PGE2 generation.
Moreover, since MMP-9 as well as proteases of ADAM class ( a disintegrin and metalloprotease family) are known to respond to external stimuli by cleavage of membrane-anchored growth factors, including ligands of EGFR to promote receptor engagement (26, 27), we also evaluated the effect of leptin on the acinar cell MMP-9 production. Interestingly, we observed that protection by leptin against cytotoxic effects of ethanol was associated with the increase in the acinar cell MMP-9 production, and that the induced MMP-9 secretion was subject to the suppression by Src inhibitor PP2, as well as JAK inhibitor AG490 and metalloproteinase inhibitor GM6001. However, the leptin-induced production of MMP-9 was not affected by EGFR inhibitor AG1478 or PKC inhibitor Ro318220. These data suggest that the induced up-regulation by leptin in the acinar cell MMP-9 secretion occurs upstream of EGFR, with the involvement of JAK and Src, and does not require PKC participation.
Taken together, our findings point to the involvement of MMP-9 in the event of leptin-induced EGFR transactivation that results in the signaling cascade leading to cPLA2 activation and up-regulation in the acinar cell PGE2 generation. Hence, leptin protection of salivary gland acinar cells against ethanol cytotoxicity stems from the ability of this pluripotent cytokine to affect rapid and selective release of arachidonic acid at the site of COX-2 action to counter the detrimental effect of ethanol on prostaglandin generation.
Conflict of interests: None declared.
This article is dedicated to Prof. Stanislaw J. Konturek, M.D., on the occasion of his 77th birthday.
- Friedman JM, Halaas JL. Leptin and regulation of body weight in mammals. Nature 1998; 395: 763-770.
- Hill RA, Margetic S, Pegg GC, Gazzola C. Leptin: its pharmacokinetics and tissue distribution. Int J Obes 1998; 22: 765-770.
- Faggioni R, Feingold KR, Grunfeld C. Leptin regulation of the immune responses and immunodeficiency of malnutrition. FASEB J 2001; 15: 2565-2571.
- Bado A, Lavasseur S, Attoub S et al. The stomach is a source of leptin. Nature 1998; 394: 790-793.
- Konturek PC, Brzozowski T, Sulekova Z et al. Role of leptin in ulcer healing. Eur J Pharmacol 2001; 414: 87-97.
- Slomiany BL, Slomiany A. Leptin suppresses Porphyromonas gingivalis lipopolysaccharide interference in with salivary mucin synthesis. Biochem Biophys Res Commun 2003; 312: 1099-1103.
- Slomiany BL, Slomiany A. Role of endothelin-1-dependent up-regulation of leptin in oral mucosal repair. J Physiol Pharmacol 2005; 56: 531-541.
- Groschl M, Rauh M, Wagner R et al. Identification of leptin in human saliva. J Clin Endocrinol Metab 2001; 86: 5234-5239.
- Slomiany BL, Slomiany A. Alterations by indomethacin in proinflammatory consequences of salivary gland cytosolic phospholipase A2 activation by Porphyromonas gingivalis: role of leptin. J Appl Res 2007; 7: 127-134.
- Takahashi N, Waelput W, Guisez Y. Leptin is an endogenous protective protein against the toxicity exerted by tumor necrosis factor. J Exp Med 1999; 189: 207-212.
- Slomiany BL, Slomiany A. Leptin protection of salivary gland acinar cells against ethanol cytotoxicity involves Src kinase-mediated parallel activation of prostaglandin and constitutive nitric oxide synthase pathways. Inflammopharmacology 2008; 16: 76-82.
- Frank S, Stallmeyer B, Kampher H et al. Leptin enhances wound re-epithelization and constitutes a direct function of leptin in skin repair. J Clin Invest 2000; 106: 501-509.
- Wu-Wang CY, Wang SL, Lim C et al. Impairment by ethanol of prostaglandin production in rat salivary glands. Arch Oral Biol 1991; 36: 9-13.
- Slomiany BL, Piotrowski J, Slomiany A. Chronic alcohol ingestion enhances tumor necrosis factor-alpha expression and salivary gland apoptosis. Alcohol Clin Exp Res 1997; 21: 1530-1533.
- Slomiany BL, Piotrowski J, Slomiany A. Alterations in buccal mucosal endothelin-1 and nitric oxide synthase with chronic alcohol ingestion. Biochem Mol Biol Int 1998; 45: 681-688.
- Scott J, Burns J, Flower EA. Histological analysis of parotid and submandibular glands in chronic alcohol abuse: A necropsy study. J Clin Pathol 1988; 41: 837-840.
- Dutta SK, Dukenhart M, Narang A et al. Functional and structural changes in parotid glands in alcoholic cirrhotic patients. Gastroenterology 1989; 96: 510-518.
- Murakami M, Naraba H, Tanioka T et al. Regulation of prostaglandin E2 biosynthesis by inducible membrane-associated prostaglandin E2 synthase that acts in concert with cyclooxygenase-2. J Biol Chem 2000; 275: 32783-32792.
- Xu L, Han C, Wu T. Activation of cytosolic phospholipase A2alpha through nitric oxide-induced S-nitrosylation. Involvement of inducible nitric-oxide synthase and cyclooxygenase-2. J Biol Chem 2008; 283: 3077-3087.
- Hirabayshi T, Shimizu T. Localization and regulation of cytosolic phospholipase A2. Biochim Biophys Acta 2000; 1488: 124-138.
- Shirai Y, Balasinde J, Dennis EA. Localization and functional interrelationships among cytosolic Group IV, secreted Group V, and CA2+-independent Group IV phospholipase A2s in p388D1 macrophages using GFP/REP constructs. Biochim Biophys Acta 2005; 1735: 119-129.
- Kim GS, Hong JS, Kim SW et al. Leptin induces apoptosis via ERK/cPLA2/cytochrome c pathway in human bone marrow stromal cells. J Biol Chem 2003; 278: 21920-21929.
- Slomiany BL, Slomiany A. Leptin modulates the detrimental effect of Porphyromonas gingivalis lipopolysaccharide-induced cytosolic phospholipase A2 activation on salivary mucin synthesis via ERK-signal transduction. Inflammopharmacology 2006; 14: 250-255.
- Shida D, Kitayama J, Mori K et al. Transactivation of epidermal growth factor receptor is involved in leptin-induced activation of janus-activated kinase 2 and extracellular signal-regulated kinase1/2 in human gastric cancer cells. Cancer Res 2005; 65: 9159-9163.
- Singh AB, Sugimoto K, Harris RC. Juxtacrine activation of epidermal growth factor(EGF) receptor by membrane-anchored heparin-binding EGF-like growth factor protects epithelial cells from anoikis while maintaining an epithelial phenotype. J Biol Chem 2007; 282: 32890-32901.
- Shah BH, Farshori MP, Jambusaria A et al. Roles of Src and epidermal growth factor receptor transactivation in transient and sustained ERK1/2 responses to gonadotropin-releasing hormone receptor activation. J Biol Chem 2003; 278: 19118-19126.
- Lee CW, Lin CC, Lin WN et al. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3K/Akt cascade and promotion of NF-kB/p300 binding in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007; 292: L799-L812.
- Liu K, Gualano RC, Hibbs ML et al. Epidermal growth factor receptor signaling to Erk1/2 and STATs control the intensity of the epithelial inflammatory responses to rhinovirus infection. J Biol Chem 2008; 283: 9977-9985.
- Slomiany BL, Slomiany A. Platelet-activating factor mediates Porphyromonas gingivalis lipopolysaccharide interference with salivary mucin synthesis via phosphatidylinositol 3-kinase-dependent nitric-oxide synthase activation. J Physiol Pharmacol 2004; 55: 85-98.
- Kawanabe Y, Hashimoto N, Masaki T. Characterization of Ca2+ channels involved in ET-1-induced transactivation of EGF receptors. Am J Physiol Heart Circ Physiol 2002; 283: H2671-H2675.
- Fruhbeck G. Intracellular signaling pathways activated by leptin. Biochem J 2006; 393: 7-20.
- Proctor G, Shori DK. The effects of ethanol on salivary glands. In Alcohol and Gastrointestinal Tract, VR Preedy, RR Watson (eds). Boca Raton, CRC Press, 1995, pp.111-122.
- Slomiany BL, Murty VLN, Piotrowski J et al. Salivary mucin in oral mucosal defense. Gen Phrmacol 1996; 27: 761-771.
- Hur EM, Park YS, Lee BD et al. Sensitization of epidermal growth factor-induced signaling by bradykinin is mediated by cSrc. J Biol Chem 2004; 279: 5852-5860.
- Ren Y, Meng S, Mei L et al. Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J Biol Chem 2004; 279: 8497-8505.
- Roell S, Grosse R, Aigner A et al. Matrix metalloproteinases 2 and 9 mediate epidermal growth factor receptor transactivation by gonadotropin-releasing hormone. J Biol Chem 2003; 278: 47307-47318.
- Andl CD, Mizushima T, Oyama K et al. EGFR-induced migration is mediated predominantly by the JAK-STAT pathway in primary esophageal keratinocytes. Am J Physiol Gastrointest Liver Physiol 2004; 287: G1227-G1237.