Sepsis-induced acute lung injury has been associated with significant morbidity and mortality (1, 2). An important feature of acute lung injury is the mismatch of ventilation and perfusion leading to intrapulmonary shunting and systemic hypoxemia. This is caused by an impairment of hypoxic pulmonary vasoconstriction (HPV), a mechanism that diverts regional blood flow away from poorly ventilated toward better ventilated lung regions to preserve oxygenation. Although the mechanisms of HPV have been incompletely understood, several lines of evidence have implicated nitric oxide (NO) to be involved in the development of sepsis-induced pulmonary and systemic vascular hyporesponsiveness (3) We have previously shown that acute pharmacological inhibition of endogenous NO production at 18 h after LPS injection restored the blunted HPV in part, whereas congenital NOS-deficiency completely protected mice from impairment of HPV after LPS injection (4). These data have suggested that NO may counteract HPV not only by its abundant production in the later course of endotoxemia as being a vasodilator, but may also affect the mechanism of HPV itself at an earlier stage of endotoxemia.
NO which is produced excessively in response to lipopolysaccharides (LPS) or cytokines by the inducible isoform of nitric oxide synthase (NOS2) (5) acts mainly by stimulating soluble guanylate cyclase (sGC) to produce guanosine cyclic 3',5'-monophosphate (cGMP) from guanosine triphosphate (GTP). The second messenger cGMP activates protein kinase which phosphorylates several intracellular targets resulting in smooth muscle relaxation (6). Many of the actions, including vasodilation and platelet actions, are mediated by this NO-cGMP pathway (7). In addition, there are cGMP-independent mechanisms by which NO may cause vasodilation. Peroxynitrite, for example, which can be produced from NO and superoxide, may induce vasodilation by activating membrane potassium channels (8, 9). Moreover, NO or peroxynitrite can directly activate cyclooxygenases and therefore promote the production of vasoactive prostaglandins (10).
The present study was therefore designed to determine whether the effects of
NO on the development of HPV during endotoxemia are mediated
via the
NO-cGMP-pathway. We hypothesized that NO causes an impairment of HPV during
endotoxemia mainly by activation of the NO-cGMP pathway that may, therefore,
be counteracted by administration of 1
H-(1,2,4)oxadiazole(4,3-
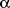
)quinoxaline-1-one
(ODQ), a specific antagonist of sGC (11), early in the course of endotoxemia.
MATERIAL AND METHODS
All experiments were approved by the governmental animal care committee of Baden-Wurttemberg,
Germany. Male C57BL/6 mice (body weight 20-35 g) were obtained from Charles
River GmbH, Sulzfeld, Germany.
Inhibition of guanylate cyclase after LPS challenge on HPV
Eighteen hours before lung perfusion experiments, mice were injected intraperitoneally
with
E. coli 0111:B4 lipopolysaccharide (Difco Lab., Detroit, USA) at
a dose of 20 mg/kg. Untreated mice served as controls.
To study the effects of inhibition of soluble guanylate cyclase after LPS challenge
on HPV, three hours after LPS challenge, mice were injected intraperitoneally
with the selective guanylate cyclase inhibitor 1
H-(1,2,4)oxadiazole(4,3-
)quinoxaline-1-one
(ODQ) solved in dimethyl sulfoxide (DMSO) 30%, at a dose of 2, 10, and 20 mg/kg,
respectively. Previous studies have suggested that inhibition of soluble guanylate
cyclase may have beneficial effects on vasopressor response (12) and survival
(13) if administered in an early stage of endotoxemia.
Data for
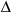
PAP
and P/Q relationships in mice treated with ODQ and LPS were compared with data
from untreated and LPS treated mice in the absence of ODQ (total of five groups
with n = 7 each).
In addition, to study the effect of ODQ on lungs of mice not treated with LPS, one group (n=5) of animals received 20 mg/kg ODQ 15 hours before the experiments.
Isolated perfused mouse lung
The isolated perfused mouse lung model was used as described previously (4).
Briefly, mice were euthanized by an intraperitoneal injection of pentobarbital
sodium (200 mg/kg body weight) and placed in a 37°C water-jacketed chamber (Isolated
perfused lung size I, Hugo-Sachs-Elektronik, March-Hugstetten, Germany). After
tracheostomy, volume-controlled ventilation (MiniVent 845, Hugo-Sachs-Elektronik,
March-Hugstetten, Germany) was initiated using an inspired gas mixture of 21%
O
2, 5% CO
2 and
74% N
2 (Messer Griesheim GmbH, Ludwigshafen,
Germany). A respiratory rate of 90/min, a tidal volume of 10 ml/kg body weight
and a positive end-expiratory pressure of 2 cmH
2O
were used. Lungs were exposed
via a midline thoracotomy, 10 IU heparin
were injected into the right ventricle, and the pulmonary artery was cannulated.
The left atrium was cannulated
via the apex of the left ventricle in
order to drain pulmonary venous effluent. Left atrial pressure was maintained
at 2 mmHg. Lungs were perfused at a constant flow of 50 ml/kg body weight/min
using a roller pump (Ismatec Laboratoriumstechnik GmbH, Wertheim-Monfeld, Germany)
in a non-recirculating system at 37 °C. The perfusate contained Hanks' Balanced
Salt Solution (Life Technologies Ltd., Paisley, Scotland) with 5% bovine serum
albumin (Serva, Heidelberg, Germany) 5% and dextran (Sigma-Aldrich Chemie GmbH,
Deisenhofen, Germany) added to prevent pulmonary edema (4, 14). To inhibit endogenous
prostaglandin and nitric oxide synthesis, 30 mM of indomethacin (Sigma-Aldrich
Chemie GmbH, Deisenhofen, Germany) and 1 mM of the non-selective nitric oxide
synthase (NOS) inhibitor, N
G-nitro-L -arginine
methyl esther (L-NAME, Sigma-Aldrich Chemie GmbH, Deisenhofen, Germany) were
added to the perfusate (4, 14). Perfusate flow was measured by a flowmeter (T106,
Transonic Systems Inc., Ithaca, NY, USA). The partial pressure of oxygen of
the perfusate was measured before and immediately after each experiment and
was found to range between 120 and 155 mmHg. Lungs were included in this study
if they had a homogenous white appearance with no signs of atelectasis, hemostasis
or edema and perfusion pressure was stable and below 10 mmHg during the first
five minutes of perfusion. Consequently, approximately 15% of lung preparations
in each group were discarded prior to further measurements.
Pulmonary artery pressure (PAP) and left atrial pressure (LAP) were measured using saline-filled membrane pressure transducers (Medex Medical GmbH & Co KG, Klein-Winterheim, Germany). Data were recorded at 150 Hz per channel on a personal computer using a data acquisition software system (DI-220/222, Dataq Instruments, Akron, OH, USA).
Quantification of HPV responsiveness and pulmonary vascular pressure-flow curves
After equilibration of the system, lungs were perfused with a flow of 25, 50,
75, and 100 ml/kg body weight/min in randomized order for 30 seconds each to
generate a pressure-flow (P-Q) curve under normoxic conditions (10). LAP was
readjusted to 2 mmHg at each flow step, and PAP was measured at the end of each
period. After return to baseline perfusion (50 ml/kg body weight/min for three
minutes), lungs were ventilated with a hypoxic gas mixture containing 1% O
2,
5% CO
2 and 94% N
2
(Messer Griesheim GmbH, Ludwigshafen, Germany). The hypoxic pulmonary vasoconstrictor
response (
PAP)
was defined as the increase in PAP 6 minutes after initiation of hypoxic ventilation
in percent of baseline PAP as described previously (4). A second P-Q curve was
generated during hypoxia as described above.
Analysis of pulmonary vascular P/Q curves
Pulmonary vascular pressure-flow (P-Q) relationships during normoxic and hypoxic
ventilation were analyzed for each experiment (Statistica for Windows, StatSoft
Inc., Tulsa, OK, USA) based on the non-linear regression model proposed by Linehan
et al. (15) as described previously (4). Briefly, according to this model,
the properties of the pulmonary vasculature are described by 'static' component
(R0, intrinsic vascular resistance) and a 'dynamic' component (a, vessel distensibility).
R
0 describes the pulmonary vascular resistance
that would exist if the vessels were at their respective diameter at zero vascular
pressure, and
is the vascular 'distensibility factor'.
Inhibition of guanylate cyclase after LPS challenge on HPV
Eighteen hours before lung perfusion experiments, mice were injected intraperitoneally
with
E. coli 0111:B4 lipopolysaccharide (Difco Lab., Detroit, USA) at
a dose of 20 mg/kg. Untreated mice served as controls.
To study the effects of inhibition of soluble guanylate cyclase after LPS challenge
on HPV, three hours after LPS challenge, mice were injected intraperitoneally
with the selective guanylate cyclase inhibitor 1
H-(1,2,4)oxadiazole(4,3-
)quinoxaline-1-one
(ODQ) solved in dimethyl sulfoxide (DMSO) 30%, at a dose of 2, 10, and 20 mg/kg,
respectively. Previous studies have suggested that inhibition of soluble guanylate
cyclase may have beneficial effects on vasopressor response (12) and survival
(13) if administered in an early stage of endotoxemia.
Data for
PAP
and P/Q relationships in mice treated with ODQ and LPS were compared with data
from untreated and LPS treated mice in the absence of ODQ (total of five groups
with n = 7 each).
In addition, to study the effect of ODQ on lungs of mice not treated with LPS, one group (n=5) of animals received 20 mg/kg ODQ 15 hours before the experiments.
Lung wet/dry weight ratio
At the end of all experiments, lungs excluding their hilar structures were excised and immediately weighed. Lungs were dried in an oven at 100 °C overnight, and then re-weighed. Lung wet-to-dry weight ratios were calculated as described previously (14).
Statistical analysis
All data are reported as means ± standard de
viation (SD). To compare
groups, a two-way ANOVA followed by an appropriate post hoc comparison test
was used. When significant differences were detected by ANOVA, a
post hoc
least significance difference (LSD) test for planned comparisons was used (Statistica
for Windows, StatSoft Inc., Tulsa, OK, USA). Statistical significance was assumed
at
p < 0.05.
RESULTS
Three hours after LPS injection, when ODQ or saline was injected, animals appeared clinically unchanged. Eighteen hours after LPS injection, both untreated mice and mice treated with ODQ showed lethargy, piloerection and diarrhea in equal measure. The mortality rate 18 hours after LPS injection was approximately 15% in all groups. Lung wet-to-dry weight ratios measured after completion of lung perfusion experiments did not differ between any of the studied groups (data not shown).
Pulmonary vascular response after LPS challenge
During normoxic ventilation LPS pretreatment did not change baseline perfusion
pressures (LPS: 7.7±0.7 mmHg vs. control: 7.5±0.8 mmHg). In contrast, LPS pretreatment
caused a significant increase in both intrinsic vascular resistance R0 (1.40±0.66
vs. 0.37±0.19 mmHg·ml
-1·kg
-1·min
-1,
p < 0.05) and vessel distensibility a (0.13±0.06
vs. 0.07±0.03 mmHg
-1,
p < 0.05) compared to untreated mice under normoxic conditions (
Fig.
2 and
3).
Hypoxic ventilation significantly increased PAP (
PAP
133.7±37.3 %) and R
0 (2.32±0.84 mmHg·ml
-1·kg
-1·min
-1
vs. 0.37±0.19 mmHg·ml
-1·kg
-1·min
-1,
p<0.05), in untreated mice, while
did not change significantly compared to normoxic ventilation (0.06±0.04
vs.
0.07±0.03 mmHg
-1). In LPS challenged mice, however,
the hypoxia-induced increase in PAP was significantly reduced (
PAP
26.4±27.1 %) as compared to untreated mice (
PAP
133.7±37.3 %,
p < 0.05). LPS treatment abolished the increase in R
0
during hypoxic ventilation (0.47±0.26
vs. 2.32±0.84 mmHg·ml
-1·kg
-1·min
-1),
while did not change significantly during hypoxia compared to untreated mice
(0.05±0.03
vs. 0.06±0.04 mmHg
-1).
Effects of ODQ on pulmonary vascular response
Inhibition of cGMP by 20 mg/kg ODQ did not change baseline PAP (6.9±0.4
vs.
7.5±0.8 mmHg) in untreated mice. In LPS treated animals, ODQ at a doses of 2,
10, and 20 mg/kg did not affect baseline PAP (
Fig. 1). Pretreatment with
20 mg/kg ODQ attenuated the increase in PAP during hypoxia in untreated mice
(
PAP 98.1±20.9
%) compared to animals not pretreated with ODQ (133.7±37.3 %, p < 0.05).
 |
Fig. 1. Pulmonary artery pressure
(PAP) during normoxic (nx) and hypoxic ventilation (hx) in isolated, perfused
lungs of lipopolysaccharide-treated (LPS) and untreated (control) mice
after treatment with different doses of 1 H-(1,2,4)oxadiazole(4,3-)quinoxaline-1-one
(ODQ). p < 0.05 versus control. |
Inhibition of cGMP by 20 mg/kg ODQ caused no significant change in R
0
or a during normoxia or hypoxia in untreated controls. In LPS-challenged mice,
however, ODQ at a dose of 2, 10, and 20 mg/kg abolished the LPS-induced increase
in R
0 and
during normoxic ventilation (
Fig. 2 and
3).
 |
Fig. 2. Static resistance
(R0) during normoxic (nx) and hypoxic
ventilation (hx) in isolated, perfused lungs of lipopolysaccharide-treated
(LPS) and untreated (control) mice after treatment with different doses
of 1 H-(1,2,4)oxadiazole(4,3-)quinoxaline-1-one
(ODQ). *p < 0.05 versus 0 mg/kg ODQ. p < 0.05 versus control. |
 |
Fig. 3. Pulmonary vessel distensibility
() during
normoxic (nx) and hypoxic ventilation (hx) in isolated, perfused lungs
of lipopolysaccharide-treated (LPS) and untreated (control) mice after
treatment with different doses of 1 -(1,2,4)oxadiazole(4,3-)quinoxaline-1-one
(ODQ). *p < 0.05 versus 0 mg/kg ODQ. p < 0.05 versus control. |
ODQ treatment at a dose of 20 mg/kg, but not at a dose of 2 or 10 mg/kg three
hours following LPS injection augmented
PAP
(
Table 1). PAP during hypoxic ventilation in LPS challenged mice treated
with 20 mg/kg ODQ was significantly higher than in mice not treated with ODQ
(79.9±13.9
vs. 26.4±27.1 %, p < 0.05,
Fig. 1) and did not differ
from controls treated with 20 mg/kg ODQ (79.9±13.9
vs. 98.1±20.9 %).
During hypoxic ventilation in LPS-challenged mice, ODQ at a dose of 2, 10, and
20 mg/kg caused no significant change in R
0
and compared to endotoxemic mice not treated with ODQ (
Fig. 2 and
3).
| Table 1. Changes
in pulmonary vasoconstrictor response (PAP),
static resistance (R0),
and vessel distensibility ()
during hypoxic ventilation compared to normoxia. LPS = lipopolysaccharide,
ODQ = 1 H-(1,2,4)oxadiazole(4,3-)quinoxaline-1-one.
*p < 0.05 versus 0 mg/kg ODQ. p < 0.05 versus control. |
 |
DISCUSSION
The present study demonstrates that selective inhibition of guanylate cyclase restores the blunted HPV responsiveness in endotoxemic mice. In addition, our results suggest that inhibition of guanylate cyclase counteracts lipopolysaccharide-induced alterations of pulmonary vascular properties under normoxic conditions.
We used an isolated, perfused lung model to study the effect of ODQ on pulmonary vasoreactivity in response to alveolar hypoxia in mice challenged with a single dose of intraperitoneal LPS. This model is highly reproducible in yielding systemic inflammation and has been shown to be sensitive for proving impaired reactivity of the pulmonary vasculature in response to vasoactive drugs (14) and hypoxic stimuli (4, 16). In addition, in contrast to
in vivo models, the isolated, perfused lung model allows to generate and study P/Q relationships that may provide more detailed information regarding the properties of the pulmonary vasculature that cannot be obtained by studying pulmonary artery pressures at a single given flow (11).
To prevent cGMP-independent mechanisms possibly interfering with HPV, we added indomethacin to the perfusate. The role of cyclooxygenase inhibitors in modulating HPV, however, is not clear without ambiguity. In a murine model, indomethacin treatment did not change HPV compared to untreated animals (17). In contrast, in a canine model of oleic acid-induced acute lung injury, indomethacin increased HPV (18), whereas in sheep isolated pulmonary veins cyclooxygenase inhibition with indomethacin reduced HPV (19).
To study the role of the endogenous NO-cGMP pathway 18 hours after LPS treatment, we perfused lungs with L-NAME. L-NAME is an inhibitor of non-selective NO synthesis and counteracts excessive NO production causing systemic hypotension during septic shock (20). Previously, we have shown that congenital deficiency of NOS2, but not treatment of wild type mice with L-NAME restored HPV during endotoxemia suggesting that mechanisms other than simply pulmonary vasodilation by NOS2-derived NO overproduction be responsible for impaired HPV during endotoxemia (4). We injected ODQ as a single dose 3 h after LPS pretreatment, because NO synthase 2 (NOS2) activity has been shown to vary over the course of endotoxemia with induction of mRNA in rat lungs being present as early as 15 minutes after LPS injection (21) and showing a maximal expression in murine lungs 6 h after LPS (22). In addition, inhibition of sGC by ODQ 4 h after LPS injection significantly improved survival at 24-144 h in mice (13).
Specific inhibition of soluble guanylate cyclase by ODQ had no effect on baseline
perfusion pressure in control or LPS-pretreated mice in our study. This is consistent
with the findings of other studies that showed no change in baseline perfusion
pressure by nonspecific NOS inhibition by N
G-nitro-L-arginine
methyl ester (L-NAME) or in mice with congenital deficiency of NOS2 (4, 23-25).
In addition, as L-NAME ODQ had no effect on intrinsic vascular resistance R
0
and vessel distensibility a under normoxic conditions in controls.
We did not find an augmentation of HPV by ODQ in untreated animals. In our study,
untreated mice showed a moderate attenuation in HPV after treatment with ODQ
without significant alterations of intrinsic vascular resistance R
0
and vessel distensibility
.
Although ODQ has been shown to potentiate HPV in the perfused rabbit (26) and
salt-perfused rat lung (27, 28), this effect was lost under conditions of preblocked
lung NO synthesis by L-NAME (26). In our experiments, the effect of L-NAME which
we used to prevent acute vasodilatory effects of NO during lung perfusion in
both ODQ-treated and ODQ-untreated mice, may have outweighed a potential augmentation
of HPV by ODQ. Moreover, in contrast to the abovementioned studies, we administered
a single dose of ODQ early in the course of endotoxemia, so that at the time
of lung preparation for perfusion 15 hours later tissue levels may not have
been sufficient to block guanylate cyclise activation by endogenous NO. Our
data are further supported by a study of Fernandes
et al. showing beneficial
effect of ODQ on restoration of vascular responsiveness to be most pronounced
at early stages of endotoxemia (12, 29).
This study shows prevention of the loss of HPV during endotoxemia by inhibition
of guanylate cyclase. Consistently with previous studies (4, 30), we found that
LPS-pretreatment in mice caused a significant attenuation of HPV that was accompanied
by an increase in R
0 and
under normoxia. ODQ at a dose of 20 mg/kg, but not at a dose of 2 mg/kg or 10
mg/kg, restored HPV after LPS challenge. In addition, even low doses of ODQ
(2 mg/kg) abolished the increase in R
0 and
in LPS-pretreated mice under normoxia, whereas ODQ at any dose had no effect
on R
0 and
under hypoxia in LPS-challenged mice. These results suggest that ODQ primarily
restores basal pulmonary vascular properties under normoxic conditions on the
basis of which hypoxic ventilation elicits vasoconstriction in endotoxemic mice.
These effects of ODQ were only detectable by analyzing pulmonary P-Q relations
since LPS treatment did not change baseline perfusion pressure significantly.
In line with our results, Zacharowski
et al. showed in a rat model that
ODQ at a low dose (2 mg/kg) was sufficient to reduce lung injury as assessed
by light microscopy of lung tissue whereas it had no positive effect on the
decrease in blood pressure caused by LPS (31). These and our data suggest that
there may be an additional mechanism that leads to the restoration of HPV at
higher doses of ODQ which we were not able to detect by analyzing P-Q relations.
In contrast to our results, administration of ODQ which was added to the recirculating
perfusate did not restore the blunted HPV in isolated lungs from rats with cirrhosis
(32). Based on our previous observations that suggested a role of NO in the
development of attenuation of HPV (4) and a potential role of sGC inhibitors
at an early stage of endotoxemia (12), however, we considered that an effect
of ODQ on HPV was most likely to occur if ODQ was administered at a time point
during endotoxemia when LPS-induced iNOS-activity would reach its peak,
i.e.
at 3 hours after the LPS-challenge.
It is unclear if early administration of ODQ can also limit the morphological lung injury after LPS challenge. Histological microscopic changes in lung tissue may not reflect the functional integrity of the pulmonary vasculature. Whereas the histological degree of lung injury was reduced six hours after LPS challenge in rats that had received ODQ two hours before LPS (31), inhalation of ODQ before inhalation of LPS caused a further increase in total cell number and protein concentration in the bronchoalveolar fluid of 12 and 24 hours after LPS treatment in mice (33). These studies, however, did not assess parameters of lung function. Lung dry/wet ratios in our experiments did not indicate a difference in lung edema between mice treated with ODQ compared to untreated mice. Further studies are needed to elucidate the effect of ODQ on lung integrity after endotoxemic lung injury.
Inhibition of sGC may be an effective way to block the effect of excessive NO production that is suggested to contribute to the impairment of pulmonary vascular responsiveness in patients with sepsis. Compared to specific inhibitors of inducible NOS, an inhibitor of sGC may be superior in terms of potential immunosuppressive effects, as NO-related antimicrobial effects are independent of the cGMP pathway (27). Methylene blue, an unspecific inhibitor of guanylate cyclase, has been used in clinical studies that showed a neutral (34) or even adverse effect (35) on arterial oxygenation. Since methylene blue, however, lacks specifity (36), may have only short-lasting effects (37) and has the capacity to generate free radicals (38), the use of a specific inhibitor of sGC in a clinical setting would help to determine if the positive effect of ODQ on HPV shown in the present study translate into a clinically relevant benefit.
In summary, the present study shows that inhibition of guanylate cyclase by ODQ restores impaired HPV in endotoxemic mice. Our data suggest that ODQ facilitates pulmonary vasoconstriction during hypoxia mainly by restoring pulmonary vascular properties during normoxia.
Acknowledgements:
This study was supported by grant WE 2114/4-2 by the Deutsche Forschungsgemeinschaft
to JW. We cordially thank Professor Martha M. Gebhard for her support during
our studies in her facilities.
Conflict of interests: None declared.
REFERENCES
- Astiz ME, Rackow EC. Septic shock. Lancet 1998; 351: 1501-1505.
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J
Med 2000; 342: 1334-1349.
- Mehta S. The effects of nitric oxide in acute lung injury. Vascul Pharmacol
2005; 43: 390-403.
- Spohr F, Cornelissen AJ, Busch C et al. Role of endogenous nitric
oxide in endotoxin-induced alteration of hypoxic pulmonary vasoconstriction
in mice. Am J Physiol Heart Circ Physiol 2005; 289: H823-H831.
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology,
and pharmacology. Pharmacol Rev 1991; 43: 109-142.
- Lincoln TM, Cornwell TL. Intracellular cyclic GMP receptor proteins. FASEB
J 1993; 7: 328-338.
- Moro MA, Russel RJ, Cellek S et al. cGMP mediates the vascular
and platelet actions of nitric oxide: confirmation using an inhibitor of
the soluble guanylyl cyclase. Proc Natl Acad Sci USA 1996; 93: 1480-1485.
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide
directly activates calcium-dependent potassium channels in vascular smooth
muscle. Nature 1994; 368: 850-853.
- Wei EP, Kontos HA, Beckman JS. Mechanisms of cerebral vasodilation by
superoxide, hydrogen peroxide, and peroxynitrite. Am J Physiol 1996; 271:
H1262-H1266.
- Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ. Peroxynitrite,
the coupling product of nitric oxide and superoxide, activates prostaglandin
biosynthesis. Proc Natl Acad Sci USA 1996; 93: 15069-15074.
- Garthwaite J, Southam E, Boulton CL, Nielsen EB, Schmidt K, Mayer B. Potent
and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-]quinoxalin-1-one.
Mol Pharmacol 1995; 48: 184-188.
- Fernandes D, da Silva-Santos JE, Duma D, Villela CG, Barja-Fidalgo C,
Assreuy J. Nitric oxide-dependent reduction in soluble guanylate cyclase
functionality accounts for early lipopolysaccharide-induced changes in vascular
reactivity. Mol Pharmacol 2006; 69: 983-990.
- Zingarelli B, Hasko G, Salzman AL, Szabo C. Effects of a novel guanylyl
cyclase inhibitor on the vascular actions of nitric oxide and peroxynitrite
in immunostimulated smooth muscle cells and in endotoxic shock. Crit Care
Med 1999; 27: 1701-1707.
- Weimann J, Bloch KD, Takata M, Steudel W, Zapol WM. Congenital NOS2 deficiency
protects mice from LPS-induced hyporesponsiveness to inhaled nitric oxide.
Anesthesiology 1999; 91: 1744-1753.
- Linehan JH, Haworth ST, Nelin LD, Krenz GS, Dawson CA. A simple distensible
vessel model for interpreting pulmonary vascular pressure-flow curves. J
Appl Physiol 1992; 73: 987-994.
- Ullrich R, Bloch KD, Ichinose F, Steudel W, Zapol WM. Hypoxic pulmonary
blood flow redistribution and arterial oxygenation in endotoxin-challenged
NOS2-deficient mice. J Clin Invest 1999; 104: 1421-1429.
- Ichinose F, Ullrich R, Sapirstein A et al. Cytosolic phospholipase
A(2) in hypoxic pulmonary vasoconstriction. J Clin Invest 2002; 109: 1493-1500.
- Leeman M, de Beyl VZ, Biarent D, Maggiorini M, Melot C, Naeije R. Inhibition
of cyclooxygenase and nitric oxide synthase in hypoxic vasoconstriction
and oleic acid-induced lung injury. Am J Respir Crit Care Med 1999; 159:
1383-1390.
- Uzun O, Demiryurek AT. Role of NO and prostaglandins in acute hypoxic
vasoconstriction in sheep pulmonary veins. Pharmacology 2006; 77: 122-129.
- Petros A, Bennett D, Vallance P. Effect of nitric oxide synthase inhibitors
on hypotension in patients with septic shock. Lancet 1991; 338: 1557-1558.
- Gebska A, Olszanecki R, Korbut R. Endotoxaemia in rats: role of leukocyte
sequestration in rapid pulmonary nitric oxide synthase-2 expression. J Physiol
Pharmacol 2005; 56: 299-311.
- Kristof AS, Goldberg P, Laubach V, Hussain SN. Role of inducible nitric
oxide synthase in endotoxin-induced acute lung injury. Am J Respir Crit
Care Med 1998; 158: 1883-1889.
- Sprague RS, Stephenson AH, Dimmitt RA et al. Effect of L-NAME on
pressure-flow relationships in isolated rabbit lungs: role of red blood
cells. Am J Physiol 1995; 269: H1941-H1948.
- Uncles DR, Daugherty MO, Frank DU, Roos CM, Rich GF. Nitric oxide modulation
of pulmonary vascular resistance is red blood cell dependent in isolated
rat lungs. Anesth Analg 1996; 83: 1212-1217.
- Weissmann N, Akkayagil E, Quanz K et al. Basic features of hypoxic
pulmonary vasoconstriction in mice. Respir Physiol Neurobiol 2004 ;139:
191-202.
- Weissmann N, Voswinckel R, Tadic A et al. Nitric oxide (NO)-dependent
but not NO-independent guanylate cyclase activation attenuates hypoxic vasoconstriction
in rabbit lungs. Am J Respir Cell Mol Biol 2000; 23: 222-227.
- Fouty B, Komalavilas P, Muramatsu M et al. Protein kinase G is
not essential to NO-cGMP modulation of basal tone in rat pulmonary circulation.
Am J Physiol 1998; 274: H672-H678.
- Gupte SA, Okada T, McMurtry IF, Oka M. Role of pentose phosphate pathway-derived
NADPH in hypoxic pulmonary vasoconstriction. Pulm Pharmacol Ther 2006; 19:
303-309.
- da Silva-Santos JE, Terluk MR, Assreuy J. Differential involvement of
guanylate cyclase and potassium channels in nitric oxide-induced hyporesponsiveness
to phenylephrine in endotoxemic rats. Shock 2002; 17: 70-76.
- Spohr F, Busch CJ, Reich C et al. 4-Aminopyridine restores impaired
hypoxic pulmonary vasoconstriction in endotoxemic mice. Anesthesiology 2007;
107: 597-604.
- Zacharowski K, Berkels R, Olbrich A et al. The selective guanylate
cyclase inhibitor ODQ reduces multiple organ injury in rodent models of
Gram-positive and Gram-negative shock. Crit Care Med 2001; 29: 1599-1608.
- Carter EP, Sato K, Morio Y, McMurtry IF. Inhibition of K(Ca) channels
restores blunted hypoxic pulmonary vasoconstriction in rats with cirrhosis.
Am J Physiol Lung Cell Mol Physiol 2000; 279: L903-L910.
- Glynos C, Kotanidou A, Orfanos SE et al. Soluble guanylyl cyclase
expression is reduced in LPS-induced lung injury. Am J Physiol Regul Integr
Comp Physiol 2007; 292: R1448-R1455.
- Andresen M, Dougnac A, Diaz O et al. Use of methylene blue in patients
with refractory septic shock: impact on hemodynamics and gas exchange. J
Crit Care 1998; 13: 164-168.
- Gachot B, Bedos JP, Veber B, Wolff M, Regnier B. Short-term effects of
methylene blue on hemodynamics and gas exchange in humans with septic shock.
Intensive Care Med 1995; 21: 1027-1031.
- Mayer B, Brunner F, Schmidt K. Inhibition of nitric oxide synthesis by
methylene blue. Biochem Pharmacol 1993; 45: 367-374.
- Keaney JF Jr., Puyana JC, Francis S, Loscalzo JF, Stamler JS, Loscalzo
J. Methylene blue reverses endotoxin-induced hypotension. Circ Res 1994;
74: 1121-1125.
- Marczin N, Tekeres M, Salzman AL, Szabo C. Methylene blue infusion in
septic shock. Crit Care Med 1995; 23 :1936-1938.