UNDERLYING MECHANISM OF REGULATORY ACTIONS OF DICLOFENAC, A NONSTEROIDAL ANTI-INFLAMMATORY AGENT, ON NEURONAL POTASSIUM CHANNELS AND FIRING: AN EXPERIMENTAL AND THEORETICAL STUDY
INTRODUCTION
Diclofenac (DIC), 2-[(2,6-diclorophenyl)amino]phenylacetate, is a nonsteroidal anti-inflammatory drug (NSAID) with anti-inflammatory, antipyretic and analgesic activities. The treatment with this agent may be accompanied by deleterious effects including gastrointestinal damage and platelet dysfunction (1, 2). Notably, some of its pharmacological or toxicological effects might be linked to any modifications of ion channels caused by many types of NSAIDs. However, whether diclofenac produce any effects on ion currents inherent in neurons remains incompletely understood.
There is emerging evidence showing that diclofenac can be a direct regulator of ion channels, especially the potassium channels, and those actions may have pharmacological or toxicological implications. For example, diclofenac was reported to activate voltage-gated K+ currents (3-5), while this compound effectively blocks K+ currents expressed in leukocytes (6). It was also shown to increase anti-nociceptive efficacy possibly through activation of ATP-sensitive K+ channels (3). Several investigators have recently demonstrated that diclofenac can bind to and modulate the amplitude and kinetics of voltage-gated K+ (Kv) channel subtypes 7.2–7.5 (Kv7.2-7.5 or KCNQ2-5) (7, 8). The increased activity of Kv7.2/7.3 channels generates the M-type K+ current (IK(M)) which is a slowly activating and deactivating current suppressed by stimulation of muscarinic receptors (9, 10). Agents known to activate IK(M) were reported to exert anticonvulsant activity (11-13). Flupirtine, an activator of IK(M), was intriguingly noted to be efficacious in suppressing or preventing repetitive febrile seizures (14). However, although previous findings seem to validate KCNQ2/Q3 channels as targets for diclofenac, it should be noted that this agent or other openers of Kv7 channels may exert some actions on other types of Kv channels (15). These experimental results with regard to the effects of diclofenac on different types of ion currents, remain controversial and are required to be further studied. The NSC-34 cell is a hybridoma cell line derived from the fusion of neuroblastoma cells with mice spinal cord cells. These cells have currently attracted growing interest as a useful cell line for studying effects of potential neuroprotective compounds against different insults including excitotoxins, mitochondrial toxins, and oxidants (16-19). Previous observations in our laboratory have shown the ability of flupirtine, to suppress IK(DR) in a concentration- and state-dependent manner in the NSC-34 cells (15). Dorsal root ganglion (DRG) neurons, conveying somatic and visceral sensory information from peripheral tissues to the spinal cord, exhibit voltage-gated K+ currents including IK(DR) formed by Kv1-3 subunits, and IK(M) generated by KCNQ2-5 subunits (20). Whether diclofenac had any effects on IK(DR) or IK(M) in these cells is unclear.
Therefore, the goal of this study was to evaluate the effects of diclofenac on ionic currents in differentiated NSC-34 motor neuron-like cells and DRG neurons. We found diclofenac not only diminished the IK(DR) amplitude but also increased the time course of current inactivation. It stimulated the amplitude of IK(M) in these cells. Our simulation modeling was allowed to predict that both blockade of IK(DR) and stimulation of IK(M) caused by diclofenac may synergistically act to affect the functional activity of mammalian neurons in cell culture or in vivo.
MATERIALS AND METHODS
Drugs and solutions
Diazepam, diclofenac, flupirtine, linopirdine, glibenclamide, and naloxone were obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.), apamin, iberiotoxin and sea anemone toxin BDS-1 were from Alomone Labs. (Jerusalem, Israel). Midazolam was obtained from Nang Kuang Pharmaceutical Co. (Tainan City, Taiwan). All culture media, fetal bovine serum, L-glutamine, trypsin/EDTA, penicillin-streptomycin and amphotericin B were obtained from Invitrogen (Carlsbad, CA, U.S.A.). All other chemicals were obtained from regular commercial chemicals and of reagent grade. Reagent water obtained from a Milli-Q ultrapure water purification system (Millipore, Bedford, MA, U.S.A.) was used in all experiments. The composition of normal Tyrode's solution is as follows (in mM): NaCl 136.5, KCl 5.4, CaCl2 1.8, MgCl2 0.53, glucose 5.5, and HEPES-NaOH buffer 5.5 (pH 7.4). To record IK(DR), IK(M), or membrane potential, the patch pipette was filled with a solution (mM): K-aspartate 130, KCl 20, KH2PO4 1, MgCl2 1, Na2ATP 3, Na2GTP 0.1, EGTA 0.1, and HEPES-KOH buffer 5 (pH 7.2).
Cell preparation
NSC-34 neuronal cells were originally produced by fusion of motor neuron-enriched, embryonic mouse spinal cords with mouse neuroblastoma (16, 17). They were routinely maintained in 1:1 mixture of Dulbecco's Modified Eagle Medium (DMEM) and Ham's F12 medium supplemented with 10% (v/v) fetal bovine serum and 1% penicillin-streptomycin. Cultures were incubated at 37°C in a humidified environment of 5% CO2/95% air. The medium was replenished every 2–3 days for removal of non-adhering cells. In order to slow cell proliferation and enhance their maturation towards a differentiated state, before confluence, cells were grown in 1:1 DMEM plus Ham's F12 medium supplemented with 1% fetal bovine serum for 48 hours (16, 17, 19).
The rat dorsal root ganglion neuron was obtained from Lonza Walkersville, Inc. (R-DRG-505; Walkersville, MD, USA). Cells were maintained in the PNGMTM BulletKitTM (Lonza Walkersville, Inc.) and equilibrated in a humidified atmosphere of 5% CO2/95% air at 37°C. The fully supplemented media contained 2 mM L-glutamine, 50 mg/ml gentamicin, 37 ng/ml amphotericin, and 2% N-ethylmaleimide-sensitive fusion protein-1 (NSF-1). The experiments were performed using cells obtained between passages 2 and 4.
RNA isolation and reverse transcriptase-polymerase chain reaction (RT-PCR)
To detect the mRNA expression of Kv3.1 and M-type channel mRNA in differentiated NSC-34 cells, a semi-quantitative RT-PCR assay was performed. Total RNA samples were extracted from NSC-34 cells according to TRIzol reagent protocol (Invitrogen). First-strand cDNA was synthesis using GoScriptTM Reverse Transcriptase (Promega). The sequences of forward and reverse primers for kcnc1 were as follows: kcnc1-f, 5'- TGACTCTGGCTTTCACGTTG -3'; and kcnc1-r, 5'- CTCAGTGGTGGGAGGTTGTT -3'. Forward and reverse primers for kcnq2 were as follows: kcnq2-f, 5'- CCCTGAAAGTCCAAGAGCAG -3'; and kcnq2-r, 5'- AGGCCCCATAGGTTTGAGTT-3'. Forward and reverse primers for kcnq3 were as follows: kcnq3-f, 5'- GTGGCTTCAGCATCTCACAA-3'; and kcnq3-r, 5'- CTTGTTGGAAGGGGTCCATA -3'. Forward and reverse primers for kcnq4 were as follows: kcnq4-f, 5'- CGATCACACTGACGACCATT -3'; and kcnq4-r, 5'- GAGATTAGCTGCTGGCATCC-3'. Forward and reverse primers for kcnq5 were as follows: kcnq5-f, 5'-ACAGTTTTCAGGCAGCGAGT -3'; and kcnq5-r, 5'- AGATGACCGTGACCTTCCAG -3'. PCR cycling conditions were 35 cycles of 95°C for 2 min, 95°C for 20 s, 60°C for 20 s, and 72°C for 10 min. These PCR products were resolved on 2% agarose gels, analyzed on 1.5% (w/v) agarose gel containing ethidium bromide and then visualized under ultraviolet light. Optical densities of DNA bands were scanned and quantified by AlphaImager 2200 (ProteinSimple; Santa Clara, CA, U.S.A.).
Electrophysiological measurements
Cells were harvested with 1% trypsin/EDTA solution prior to the experiments and an aliquot of cell suspension was transferred to a recording chamber mounted on the stage of an inverted fluorescent microscope (CKX-41; Olympus, Tokyo, Japan). Cells were bathed at room temperature (20–25°C) in normal Tyrode's solution containing 1.8 mM CaCl2. Patch-clamp recordings in the whole-cell configuration were made with an RK-400 amplifier (Bio-Logic, Claix, France) (19, 21). Voltage-clamp protocols with either rectangular or ramp shapes were computer driven using an acquisition system (Digidata 1322A board; Molecular Device, Sunnyvale, CA, U.S.A.) and pClamp 9.2 (Molecular Devices). Patch pipettes with resistances of 3-5 ΩM were pulled from Kimax-51 glass capillaries (Kimble; Vineland, NJ, U.S.A.) on a PP-830 electrode puller (Narishige, Tokyo, Japan). The experiments were designed to evaluate the possible effects of diclofenac on ion currents and membrane potential in NSC-34 cells and DRG neurons.
Data recordings and analyses
The data were stored online in a TravelMate-6253 computer (Acer, Taipei, Taiwan) at 10 kHz through Digidata 1322A interface (Molecular Devices). The latter device was equipped with Adaptec SlimSCSI card (Milpitas, CA, U.S.A.) via PCMCIA slot and controlled by pCLAMP 9.2 (Molecular Devices). Current signals were low pass-filtered at 3 kHz. Ion currents were generally analyzed by using Origin 8.0 (OriginLab, Northampton, MA, U.S.A.) or custom-made macros built in the Excel 2007 spreadsheets under Windows 7. The duration of action potential (AP) in response to brief current injection recorded under current-clamp mode was measured at 90% of repolarization.
The concentration-response data for diclofenac-induced inhibition of IK(DR) measured from NSC-34 neuronal cells or dorsal root ganglion (DRG) neurons were adequately fitted using a modified form of the Hill equation. That is:
where y is the relative amplitude of IK(DR) measured at the end of depolarizing pulse; [C] is the DIC concentration; IC50 and nH are the concentration required for a 50% inhibition and the Hill coefficient, respectively. Maximal inhibition (i.e., 1–a) of IK(DR) during cell exposure to DIC was estimated. The inactivation time constants of IK(DR) with or without addition of DIC measured from the different levels of depolarizing voltages were determined by fitting the trajectory of each current trace with a single exponential. The nonlinear curve-fitting sets were performed using Origin 8.0 (OriginLab).
To evaluate the concentration-dependent effect of DIC on IK(M) in DRG neurons, the IK(M) was elicited by membrane hyperpolarization to –40 mV from –10 mV. The data obtained at the end of hyperpolarizing pulses were fitted to another modified Hill equation:
where y is the relative amplitude of IK(M) measured at the end of hyperpolarizing pulse; [C] is the DIC concentration; EC50 and nH are the concentration required for a 50% stimulation and the Hill coefficient, respectively. Maximal stimulation (i.e., 1–a) of IK(M) during cell exposure to DIC was estimated.
Values are provided as means ± standard error of the mean (S.E.M.) for n number of samples. The paired or unpaired Student's t-test and one-way ANOVA with a least-significance difference method for multiple comparisons were used for the statistical evaluation of differences among means. A value of P<0.05 was considered to be statistically significant.
Computational simulation
To mimic the effect of DIC on electrical behaviors of AP firing in central mammalian neurons, a theoretical model of hippocampal CA1 pyramidal neuron (22) was modified and implemented. In this model, rhythmic bursting was mathematically reconstructed from ionic processes that were formulated on the basis of the data experimentally obtained from CA1 pyramidal cells of brain slice preparation. The model mainly comprises a fast and transient Na+ current, a persistent Na+ current, a high-threshold Ca2+ current, two Ca2+-activated K+ currents, a transient outward K+ current, a delayed rectifier K+ current (IK(DR)) and an M-type K+ current (IK(M)). The nature and construction of this model framework is detailed previously (22) and the model formulations are available at http://senselab.med.yale.edu/senselab/modeldb.
Increasing the concentration of diclofenac not only reduced IK(DR) amplitude, but also enhanced the apparent inactivation. The inhibitory effect of DIC on IK(DR) can be explained by state-dependent block where it binds to the open state of the channel according to a minimal kinetic scheme (23, 24):

where α and β are the voltage-dependent rate constant for the opening and closing of the Kv channel, k+1 and k-1 are those for blocking and unblocking by DIC, and [B] is the DIC concentration. The unblock rate constant (k-1) and the block rate constant (k+) in the presence of DIC was estimated to be 0.00432 ms–1 and 0.113 ms–1 µM–1, respectively.
To model DIC-induced block of IK(DR), an inactivation variable h was included in simulated IK(DR) and the macroscopic current was then expressed as
Here, gK(DR) is the maximal conductance of IK(DR), and VK the K+ reversal potential. Because as DIC-induced block is a state-dependent process, the form of the equation describing the inactivation variables is then modified as follows:
In this study, the solution to the differential sets of ordinary differential equations in numerical simulations accompanied by studies of bifurcation diagrams was made using the simulation package XPPAUT (25, 26). Parts of numerical simulations were verified with Microsoft Excel (27). Numerical computations were generally run under a Hewlett Packard xw9300 workstation (Palo Alto, CA, U.S.A.). The conductance values used to solve the set of differential equations are summarized in Table 1.

RESULTS
The mRNA expression for Kv3.1 in differentiated NSC-34 cells
We detected the mRNA expression of Kv3.1 (kcnc1) and M-type channel (kcnq2-kcnq5) in NSC-34 cells by a semi-quantitative RT-PCR assay. Our RT-PCR analysis clearly presented the mRNA of kcnc1and kcnq2-kcnq5 in NSC-34 cells (Fig. 1).
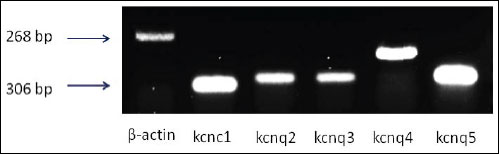 |
Fig. 1. The Kv3.1 (kcnc1) and M-channels (kcnq2-kcnq5) mRNA isolated from differentiated NSC-34 cells were studied. Total RNA was isolated and RT-PCR analysis was performed. The mRNA of kcnc1and kcnq2-kcnq5 was clearly present in NSC-34 cells. |
Inhibitory effect of diclofenac on IK(DR) in differentiated NSC-34 cells
The whole-cell recordings of the patch-clamp technique were first used to evaluate the effect of diclofenac on ionic currents in these cells. To record K+ outward currents and avoid any contamination of Ca2+-activated K+ currents, cells were bathed in Ca2+-free Tyrode's solution. When the cell examined was held at –50 mV and the depolarizing voltage pulses ranging from –80 to +40 mV in 10-mV increments were applied with a duration of 1 s, a family of outward currents accompanied by slight inactivation was elicited (Fig. 2). The threshold for elicitation of these outward currents was around –30 mV, a value that is higher than that of the classical IK(DR) described in the squid giant axons (28), and their current magnitudes became strongly greater with larger depolarization. This population of outwardly rectifying currents was identified as IK(DR) and noted to resemble the KV3.1-encoded currents (23, 29, 30). Notably, 2 min after exposure to diclofenac (100 µM), the IK(DR) amplitude was greatly reduced at the potentials between –20 and +40 mV. For example, when the depolarizing pulse from –50 to +40 mV was applied, diclofenac (100 µM) decreased the IK(DR) amplitude measured at the end of voltage pulses by 53±6 % from 963±158 to 457±107 pA (n = 10). After washout of DIC, current amplitude at +40 mV was returned to 567±126 pA (n = 6). Fig. 2B illustrates the I-V relationships of IK(DR) measured at the beginning and end of voltage pulses, as the data were obtained with or without addition of 100 µM DIC. To clarify whether the DIC-sensitive component of IK(DR) is mediated by Kv3.1, we applied the sea anemone toxin BDS-1, which is known to be an effective inhibitor of Kv3.1 (31). BDS-1 (10 nM) can significantly suppress the amplitude of IK(DR) in NSC-34 cells (data not shown). Moreover, as cells were exposed to Ca2+-free Tyrode's solution containing 1 mM BAPTA, the magnitude of DIC-mediated inhibition of IK(DR) in NSC-34 cells remained unaltered.
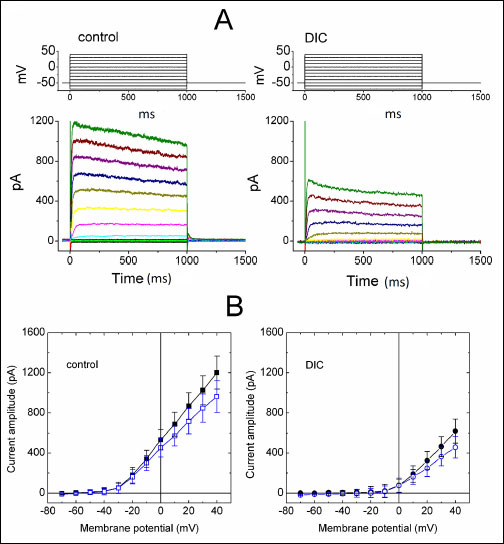 |
Fig. 2. Inhibitory effect of diclofenac (DIC) on IK(DR) in differentiated NSC-34 neuronal cells. In these experiments with whole-cell configuration, cells were bathed in Ca2+-free Tyrode's solution and the recording pipette was filled with K+-containing solution. (A) Superimposed current traces obtained in the absence (left) and presence (right) of 100 µM DIC. The upper part shown in each panel indicates the voltage protocol examined. (B) Average I-V relations for initial (filled symbols) and late components (open symbols) of IK(DR) obtained in the control (left; squares) and during the exposure to 100 µM DIC (right; circles) (mean ±S.E.M., n = 7–9 for each point. |
We further determined the relationship between the diclofenac concentration and the relative amplitude of IK(DR). As depicted in Fig. 3A, DIC ranging between 10 µM and 1 mM suppressed the IK(DR) amplitude in a concentration-dependent fashion. Half-maximal concentration required for the inhibitory effect of DIC (i.e., IC50 value) was measured to be 73 µM, and at a concentration of 1 mM, it almost fully suppressed current amplitude. Therefore, the experimental results indicate that similar to flupirtine (15), DIC exerts a depressant action on IK(DR) in these cells.
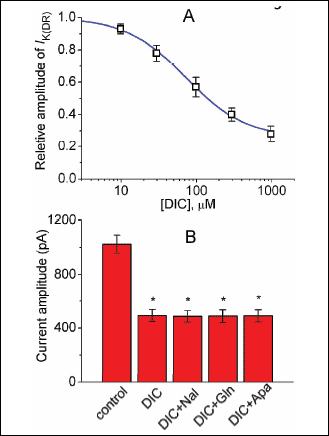 |
Fig. 3. Relationship between the diclofenac (DIC) concentration and the relative amplitude of IK(DR) (A) and effects of naloxone, glibenclamide and apamin on IK(DR) recorded from NSC-34 cells. Current amplitude with or without addition of diclofenac was measured at the end of depolarizing pulse from –50 to +40 mV. Smooth line shown in (A) represents best fit to the modified Hill equation as described in Methods. The values for IC50 and the Hill coefficient were 73 µM and 1.1, respectively. Each point represents the mean ±S.E.M. (n = 8–13). Note that diclofenac suppressed the IK(DR) amplitude in a concentration-dependent manner. (B) Bar graph showing the inability of naloxone, glibenclamide or apamin to alter DIC-induced inhibition of IK(DR) (mean ±S.E.M., n = 6–9 for each bar). DIC: 100 µM; Nal: 30 µM naloxone; Gln: 30 µM glibenclamide; Apa: 200 nM apamin. *Significantly different from control. |
Inability of naloxone, glibenclamide or apamin to reverse diclofenac-mediated inhibition of IK(DR) in differentiated NSC-34 cells
Earlier studies showed that diclofenac-induced analgesic action is mediated by endogenous opioids in certain nuclei of brain stem related to pain sensation (32). This compound is also thought to exert anti-nociceptive action through the activation of K+ currents (3, 4). For these reasons, further experiments were performed to see whether diclofenac-mediated inhibition of IK(DR) observed in NSC-34 cells can be altered by subsequent application of naloxone, glibenclamide or apamin. However, as shown in Fig. 3B, neither naloxone (30 µM), glibenclamide (30 µM) nor apamin (200 nM) produced notable effects on the reduction by DIC (100 µM) of IK(DR) in these cells. Moreover, iberiotoxin (200 nM) or apamin (200 nM) alone produced little or no effects on the IK(DR) amplitude, while naloxone (30 µM) or glibenclamide (30 µM) slightly decreased IK(DR) amplitude by about 10%. As evidenced by these findings, it seems unlikely that DIC-mediated block of IK(DR) in NSC-34 cells is mediated predominantly through activation of either ATP-sensitive K+ or calcium-activated K+ channels.
Diclofenac-induced elevation in the inactivation rate of IK(DR) in differentiated NSC-34 cells
The observed IK(DR) during cell exposure to diclofenac tends to exhibit a pronounced peak followed by an exponential decay to a steady-state level. We next evaluated voltage and time dependence of diclofenac-induced decrease in the inactivation time constant of IK(DR). As illustrated in Fig. 4A, the trajectories of current inactivation with or without addition of diclofenac (100 µM) were fitted by a monoexponential function to assess the inactivation time constants. As cells were exposed to diclofenac at a concentration of 100 µM, the inactivation time constant of IK(DR) elicited by membrane depolarization from –50 to +40 mV was significantly shortened to 311±21 ms (n = 8) from a control of 527±23 ms (n = 8). Furthermore, addition of diclofenac was found to accelerate the process of IK(DR) inactivation with a voltage-dependent property (Fig. 4B). However, during cell exposure to 100 µM DIC, the activation rate of IK(DR) in response to the same voltage protocol remained unaffected. No significant change in current amplitude at the holding potential of –50 mV was also demonstrated in the presence of diclofenac. Cell exposure to diclofenac did not alter the midpoint and the slope factor of IK(DR) inactivation curve. Moreover, subsequent application of linopirdine (10 µM), a selective inhibitor of IK(M) (15, 33), did not alter diclofenac-induced increase of current inactivation process (data not shown).
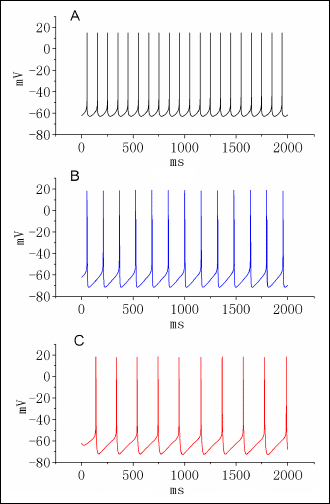 |
Fig. 4. Effect of diclofenac (DIC) on the inactivation time constant of IK(DR) in differentiated NSC-34 cells. In (A), the time courses of current inactivation obtained in the absence (1) and presence of 100 µM DIC (2) were adequately fitted by a single exponential as indicated by smooth lines. In (B), the relationship of inactivation time constant versus membrane potential was constructed (mean ±S.E.M., n = 6–8 for each point). |
Effect of diclofenac on M-type K+ current (IK(M)) in differentiated NSC-34 cells
Previous work has demonstrated the ability of diclofenac to activate KCNQ2/KCNQ3 currents (7, 34). Several studies have also reported the presence of IK(M) in another motor-neuron-like cells, namely, NG108-15 cells (35, 36). Therefore, we further evaluated whether diclofenac produces any effects on the amplitude of IK(M) expressed in NSC-34 cells. As illustrated in Fig. 5, when cells were hyperpolarized from –10 to –40 mV, the IK(M) amplitude were significantly greater in the presence of diclofenac at a concentration of 300 µM as compared with that in the control. In continued presence of diclofenac (300 µM), subsequent application of linopirdine (10 µM) reversed DIC-induced stimulation of IK(M). Similarly, addition of either flupirtine (10 µM) or meclofenamic acid (10 µM), known to be an activator of KCNQ2/Q3 channels (15, 33, 34), significantly elevated IK(M) amplitude (Fig. 5B). The data suggested that besides an inhibitor of IK(DR), diclofenac is capable of stimulating IK(M) in differentiated NSC-34 cells.
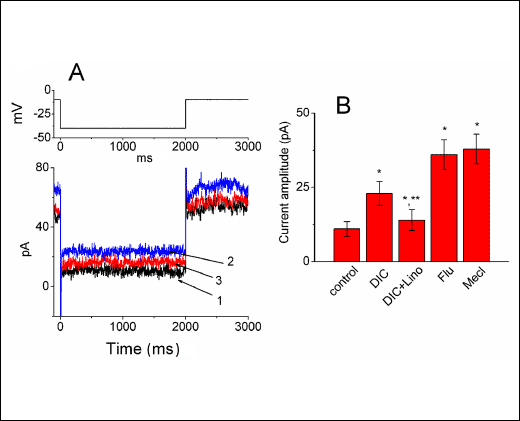 |
Fig. 5. Stimulatory effect of diclofenac (DIC) on IK(M) in NSC-34 cells. In these experiments, cells were bathed in Ca2+-free Tyrode's solution and the pipette was filled with a K+-containing solution. (A) Current traces obtained in the control (1) and during exposure to 300 µM of DIC (2) or to 300 µM of DIC plus 30 µM linopirdine (3). The upper part indicates the voltage protocol used. (B) Bar graph showing the effects of diclofenac, diclofenac plus linopirdine, flupirdine and meclofenamic acid on the IK(M) amplitude measured at –40 mV in NSC-34 cells (mean ±S.E.M., n = 7–11 for each bar). DIC: diclofenac (300 µM); Lino: linopirdine (10 µM); Flu: flupirtine (10 µM); Mecl: meclofenamic acid (10 µM). *Significantly different from control. **Significantly different from diclofenac (300 µM) alone group. |
Effects of diclofenac on IK(DR) and IK(M) in rat dorsal root ganglion neurons
The electric properties in NSC-34 cells could be distinguishable from those in other types of neurons. We also investigated the existence of IK(DR) and IK(M) in rat dorsal root ganglion (DRG) neurons. In this set of experiments, DRG neurons were bathed in Ca2+-free Tyrode's solution containing 1 µM tetrodotoxin. As shown in Fig. 6, under the same voltage profile used for NSC-34 neuronal cells, we were able to identify the properties of IK(M) and IK(DR) in these cells. As cells were exposed to diclofenac (100 µM), the amplitude of IK(M) was increased and the IK(DR) amplitude was diminished with a shortening of inactivation time constant. The relationships between the diclofenac concentration and the relative amplitude of IK(DR) or IK(M) were constructed and are illustrated in Fig. 6C and 6D, respectively.
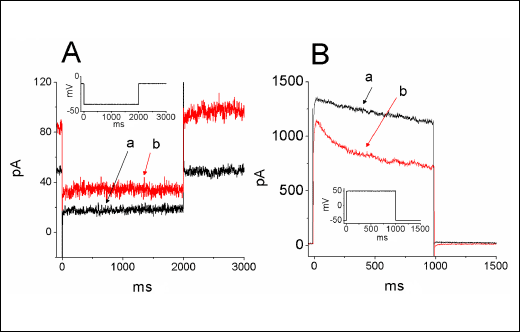 |
Fig. 6. Effect of diclofenac (DIC) on IK(M) (A) and IK(DR) (B) in rat dorsal root ganglion neurons. In these experiments, cells were bathed in Ca2+-free Tyrode's solution. Current traces labeled a in each panel are control and those labeled b were obtained after addition of diclofenac (100 µM). The inset in each panel indicates the voltage protocol used. (C) and (D) showed the relationship between the diclofenac concentration and the relative amplitude of IK(DR) and IK(M), respectively. The half-maximal concentrations required for diclofenac-induced inhibition of IK(DR) and stimulation of IK(M) in DRG neurons are 66 and 63 µM, respectively. |
Effect of varying gK(DR) on action potential (AP) firing frequency in modeled neuron
The next set of simulations was designed to determine the effects of IK(DR) blockade on the frequency of action potentials firing. Fig. 7 depicts the relationship of firing frequency versus varying stimuli. As shown in Fig. 8A, as the gK(DR) value increased from 1.0 to 5.0 mS/cm2, the firing frequency of action potentials was notably elevated. In consistent with findings described in Fig. 9 there was a notable increase in the duration of neuronal action potentials when gK(DR) was reduced. The oscillation frequency generated from bifurcation diagram is illustrated in Fig. 8B as a function of varying gK(M) values. When the gK(DR) value was lower than 10 mS/cm2, action potentials firing frequency was found to increase sharply with the increase of gK(DR), despite the lengthening of AP width. However, as the gK(DR) value was greater than 15 mS/cm2, the firing frequency of modeled neuron was gradually diminished with increasing gK(DR). Therefore, worthy of being noted was that a reversal of firing frequency from positive to negative relationship emerged between the gK(DR) values of 10 and 15 mS/cm2.
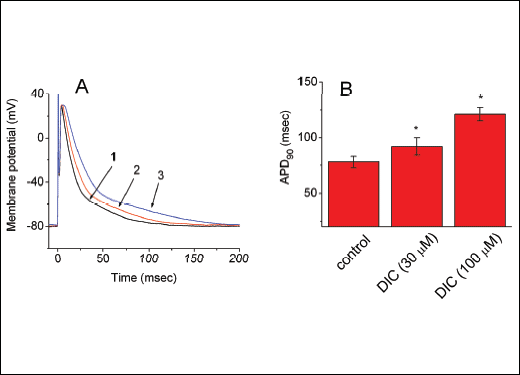 |
Fig. 7. Effect of diclofenac (DIC) on APs recorded from differentiated NSC-34 cells. In these experiments, cells were bathed in normal Tyrode's solution containing 1.8 mM CaCl2. The patch pipettes were filled with a K+-containing internal solution and changes in membrane potential were then measured under current-clamp configuration. The single spike was elicited in response to short brief stimuli. (A) Potential traces obtained in the absence (1) and presence of 10 µM DIC (2) or 30 µM DIC (3). Notably, addition of DIC produces an increase in AP duration. (B) Bar graphs showing the effects of diclofenac on AP duration at 90% repolarization (APD90). Each bar represents the mean ± S.E.M. (n=8). *Significantly different from control. |
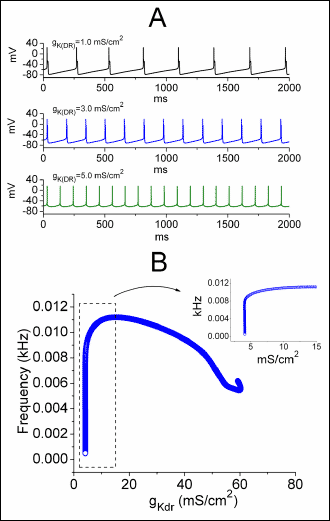 |
Fig. 8. Effect of varying gK(DR) on simulated firing of action potentials (APs). The model formulations are detailed in Methods and the default parameters shown in Table 1. (A) Simulated action potentials generated when the value of gK(DR) was set at 1.0, 3.0 and 5.0 mS/cm2. (B) Bifurcation diagram showing effect of varying gK(DR) on firing frequency of simulated action potentials. Notably, when the gK(DR) value is less than 10 mS/cm2, the firing frequency is elevated with the increasing gK(DR); however, as gK(DR) is greater than 15 mS/cm2, the frequency is progressively reduced. The inset in (B) indicates an expanded record corresponding to that shown in the dashed box. |
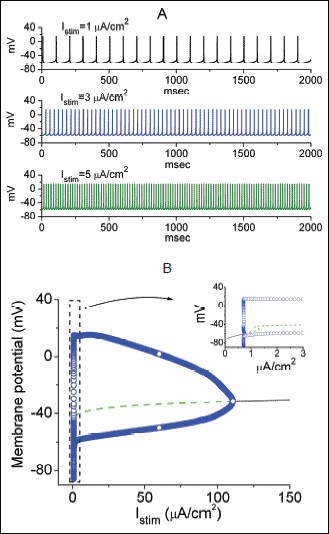 |
Fig. 9. Effect of current stimuli (Istim) on AP firing generated from the simulated model of hippocampal CA1 pyramidal neurons (A) and numerically computed bifurcation diagram for this model as the value of stimulus intensity (Istim) varies (B). The model description was detailed in Methods. (A) Simulated AP firing generated when the Istim value was arbitrarily set at 1, 3 and 5 µA/cm2. In (B), steady-state (curves) and periodic (circles) solutions were plotted against Istim. Notably, this type of bifurcation is called a Hopf bifurcation because the emergence of periodic solutions emanating from a fixed point is established from the Hopf bifurcation theorem. Stable fixed points are indicated as solid black line, and unstable are dashed line. The inset in (B) indicates an expanded record corresponding to that appearing in the dashed box. |
Effect of varying gK(M) on the repetitive firing of simulated action potentials
Because of the ability of diclofenac to increase IK(M) in NSC-34 cells, we further evaluate the possible effects of gK(M) on action potential firing. Fig. 10 illustrates a significant effect of gK(M) on the firing of simulated action potentials generated from this modeled neuron. In contrast to effects of varying gK(DR) on action potential firing, it can be seen that as the gK(M) value was raised from 0 to 1.5 mS/cm2, the firing frequency of simulated APs exponentially decreased with resultant changes in the IK(M) amplitude (Fig. 10). When gK(M) was removed, action potential firing frequency was reached to be about 80 Hz, while as the gK(M) value was arbitrarily increased to 1.5 mS/cm2, the frequency was declined to about 10 Hz. The frequency remained relatively unaltered as gK(M) was higher than 1.2 mS/cm2. As the gK(M) value was greater than 1.6 mS/cm2, action potential firing was totally suppressed. It also needs to be mentioned that the resting membrane potential became more hyperpolarized with the increasing value of gK(M) (Fig. 10A). The latter results basically support the notion that IK(M) is a non-inactivating, voltage-dependent K+ current that activates in a time- and voltage-dependent manner at around –60 mV, which is close to the resting membrane potential but more negative than that for IK(DR) activation. Therefore, the simulation results clearly indicate that unlike gK(DR), appropriate changes in gK(M) can virtually make a significant contribution to the resting membrane potential inherent in this modeled neuron because the activation range of IK(M) is far more negative than that of IK(DR) (9, 10).
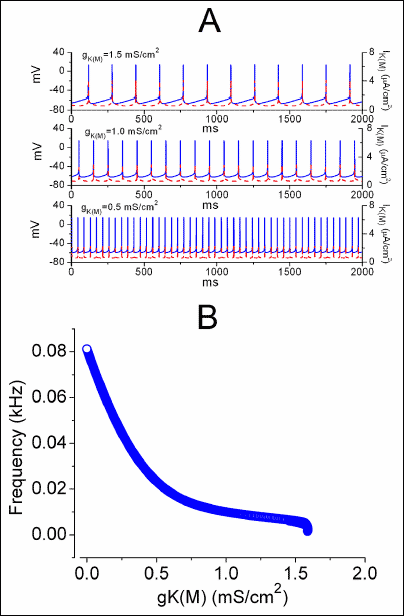 |
Fig. 10. Effect of varying gK(M) on simulated frequency of APs in modeled neuron. (A) Simulated APs (solid lines) and corresponding IK(M) (dashed lines) generated in the different value of gK(M) (i.e., 0.5, 1.0 and 1.5 mS/cm2). (B) Bifurcation diagram showing the relationship of firing frequency versus gK(M). Note that as gK(M) continues to increase, the oscillating frequency of sustained firing generated from modeled neuron is progressively decreased. |
Effects of increasing gK(M) and decreasing gK(M) on the firing of simulated action potentials
Because our experimental results made in differentiated NSC-34 neurons reflect that the depressant action of diclofenac on neuronal firing is mediated through both blockade of IK(DR) and activation of IK(M), we further evaluate how changes in gK(DR) and gK(DR) influence the firing frequency of simulated APs to mimic the effect of diclofenac. As the gK(M) was elevated by 50% (from 1 to 1.5 mS/cm2), the frequency of sustained firing as a function of Istim was progressively shifted in a downward direction (Fig. 11). However, to mimic the action of diclofenac on IK(DR) and IK(M), as gK(M) increased by 50% (from 1 to 1.5 mS/cm2) with the accompanied reduction of gK(DR) by 50% (from 6 to 3 mS/cm2), repetitive firing of simulated APs as a function of Istim was even more depressed. Based on simulations, it is reasonable to propose that attenuation of AP firing caused by increased gK(M) can be theoretically potentiated by a further reduction of gK(DR). In the model, the reduction of firing frequency was achieved by increasing gK(M) and decreasing gK(DR). As a corollary to these findings, the simulation results allow us to predict that modest changes in IK(M) and IK(DR) have sufficient leverage to mediate the effect of diclofenac on electrical activity in central neurons in vivo.
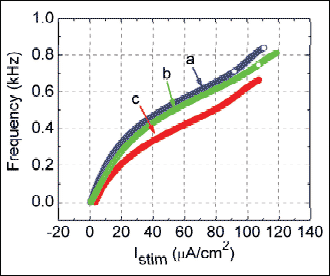 |
Fig. 11. Firing frequency versus Istim relationships of modeled neuron with different values of gK(DR) and gK(M). Bifurcation diagrams of frequency as a function of varying Istim were derived. (a): gK(DR)=6.0 mS/cm2, gK(M)=1.0 mS/cm2; (b): gK(DR)=6.0 mS/cm2, gK(M)=1.5 mS/cm2; (c): gK(DR)=3.0 mS/cm2, gK(M)=1.5 mS/cm2. As the Istim value is elevated, the frequency is increased. Notably, for mimicking the action of diclofenac on neuronal firing, the combination of reduced gK(DR) and increased gK(M) (c) depresses the relationship of oscillation frequency versus Istim in a downward direction to a greater extent as compared with that under the increase of gK(M) alone (b). |
Effect of diclofenac on spontaneous action potentials in a modeled neuron
In order to evaluate how diclofenac alters the discharge pattern of neurons, a simulation model, originally derived from Golomb et al. (22), was implemented (Fig. 12). In this modeled cell, as gK(DR) was decreased from 6 to 3 mS/cm2 and the block of IK(DR) by diclofenac at a concentration of 100 µM was simulated, the firing frequency of simulated APs was readily diminished. When gK(M) was further elevated to 1.5 mS/cm2 in continued presence of reduced gK(DR) and IK(DR) inactivation where the DIC action was mimicked, the spontaneous APs were further reduced along with membrane hyperpolarization. As a result, the reduced gK(DR), the increased decay of IK(DR) inactivation and the elevated gK(M), which mimics the DIC action, can combine to cause changes in the firing of simulated APs.
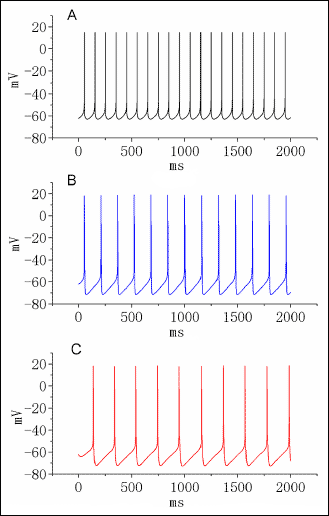 |
Fig. 12. Simulation modeling used to mimic diclofenac effects on repetitive firing of neuronal action potentials. The model was developed based on the electrophysiological properties of hypothalamic CA1 pyrmidal cells as described in materials and methods. In (A), the firing of spontaneous APs under control condition (i.e., gK(DR)= 6 mS/cm2 and gK(M)= 1 mS/cm2). In (B), when gK(DR) was decreased from 6 to 3 mS/cm2 and the diclofenac concentration was set at 100 µM, the AP firing was diminished together with membrane hyperpolarization. In (C), when gK(M) was elevated 1.5 mS/cm2 in the continued presence to reduced gK(DR) and increased inactivation rate of IK(DR) where the diclofenac (100 µM) action was mimicked, the spike discharge was further decreased. |
DISCUSSION
In our study, diclofenac has a complex and interesting profile in that it decreases action potentials firing via mechanisms other than an interaction at the M-type K+ channels. One of these mechanisms may involve IK(DR) (i.e., fast delayed rectifier), the biophysical properties of which are distinguishable from the Hodgkin-Huxley-type delayed rectifier current (23, 28, 36). In addition to a stimulatory effect of IK(M), diclofenac suppressed IK(DR) in a concentration- and time-dependent fashion. Given that the importance of IK(DR) (i.e., KV3.1-encoded current) in contributing to neuronal excitability and automaticity, it is conceivable that diclofenac-induced block of IK(DR) is involved in the alteration of neuronal firing, particularly at high-spiking neurons. We have also integrated experimental and theoretical approaches in order to study potential influence of this agent or its functionally related compounds on neuronal firing. The computer simulation with minimal binding scheme suggests that diclofenac may act as a state-dependent blocker of Kv channel.
The biophysical nature of IK(DR) in NSC-34 cells resembles the Kv3.1-encoded current because of positive mRNA detection of Kv3.1 and a great sensitivity to inhibition by BDS-1. NSC-34 cells differentiated with 1% low serum medium for 48 hours were found to increase the mRNA level of class III β-tubulin, a neuron-specific marker, as compared with that in normal cells. The Kv channels from the Kv3.1 type, which are the major determinant of IK(DR) in NSC-34 cells, another motor neuron-like cells (NG108-15), and DRG neurons (20), are thought to make little contribution to the resting membrane potential. However, due to their fast activating and deactivating properties, these channels are responsible for spike repolarization and after-hyperpolarization of neurons with high-frequency firing (24, 37). The inhibition of IK(DR) caused by diclofenac is able to retard the AP repolarization and slow recovery of INa inactivation, which may pose the neuron to decreased rhythmic firing of APs. However, Kv3.1 channels are well known to be regulated at both the transcriptional and posttranslational levels. When IK(DR) amplitude is considerably high, this effect may diminish neuronal hypo-excitability of IK(M) activation as predicted from the modeled neuron, while with a lower value of gK(DR) it can facilitate the inhibitory effects of IK(M) activation on AP firing. Based on our studies, it is indeed conceivable that the effects of Kv7-channel openers on AP firing rely on the pre-existing magnitude of IK(DR), despite no clear evidence to show that KV3 and Kv7 channels can co-assemble to form a protein complex.
The physiological importance of Kv7 channels has been recognized in a variety of brain regions including the hippocampus, neocortex and cerebellar cortex, where are key sites for neuronal network oscillations (9-11). The effect of diclofenac observed in NSC-34 neurons was noted to display biophysical or pharmacological features that were not merely confined to stimulation of IK(M). The IC50 value for the effect of this agent on IK(DR) was about 73 µM, a value that is close to the concentration required for its stimulatory action on Kv7 channels. Its effects on IK(DR) are most likely to be therapeutically or clinically relevant. Our study thus prompted us to propose that Kv3 and Kv7 channels have a considerable pharmacological overlap. It is tempting to speculate that diclofenac can exert an interaction at a similar binding site inherently existing on Kv7 and Kv3 channels.
It has been previously demonstrated that central analgesic action of diclofenac tends to be mediated by the release of endogenous opioids in certain brain stem nuclei which is linked to control of pain sensation (32). However, in this study, subsequent application of naloxone, a blocker of opioid receptors, did not reverse inhibition of IK(DR) caused by diclofenac. Moreover, when NSC-34 cells were further exposed to naloxone, diclofenac-induced widening of AP remained unaltered. Therefore, findings from our results reflect that the action of diclofenac on ionic currents observed in NSC-34 cells does not arise from the release of opioid-like substance(s) or the binding of this agent to opioid receptors.
Interestingly, it has been proposed that phospholipase C- and Ca2+/phosphatidylinositol 4,5-bisphosphate-mediated inhibition of IK(M) in sensory neurons may represent one of the general mechanisms underlying pain produced by inflammatory mediators (38). Our study suggests the potential IK(M)-modulating role of diclofenac on pain disorders which are produced by inflammatory mediators, in addition to its traditionally anticipated role as an anti-inflammatory agent.
It needs to be noted that the benzodiazepines such as midazolam and diazepam were also found to suppress IK(DR) and to accelerate the process of current inactivation in NSC-34 neurons (data not shown). The diclofenac-mediated reduction of IK(DR) in combination with attenuation of AP firing may be accentuated in continued presence of benzodiazepine agonists. Therefore, it is important to evaluate the extent to what IK(DR) blockade induced by these agents with concomitant activation of IK(M) contributes to the anticonvulsant activity of benzodiazepine agonists (39), as these latter agonists exert inhibitory effects on IK(DR) (40).
An intriguing consequence of our simulation findings is that the inhibitory effects of diclofenac on neuronal firing in vivo may result from activation of IK(M) and inhibition of IK(DR), especially during the lower value of gK(DR). It is clear that decreasing gK(DR) can facilitate whereas increasing gK(DR) counteracts the inhibition of oscillating frequency caused by increasing gK(M). The main reason for these results can be explained by the fact that the recovery of Na+ channels was hindered by decreased gK(DR) (28, 37), while the inactivation of Na+ channels was largely enhanced by the increase of gK(M) (22). Activation of IK(DR) is so fast as to influence the repolarization of single AP. Although decreased gK(DR) can prolong the duration of spike after depolarization and may subsequently facilitate to its escalation to a burst, the concomitant increase of gK(M) as numerically simulated here for the diclofenac action will consequently render neuronal hyperexcitability to be suppressed.
A recent study found rebamipide could attenuate NSAIDs including diclofenac's inducing lipid peroxidation in gastric epithelial cells by increasing the expression of manganese superoxide dismutase protein and decreasing superoxide anion leakage from mitochondria in both gastric and small intestinal epithelial cells. Whether this effect is related to the ionic mechanism described in our study is worth further studies (41).
In summary, diclofenac can directly block IK(DR) in a concentration-dependent manner and accelerate the decay of IK(DR) in differentiated NSC-34 and DRG neurons. Diclofenac-mediated reduction of neuronal AP firing might not be solely explained by the activation of IK(M), although one frequently cited explanation for the beneficial effect of this and other functionally similar compounds is that they are efficacious as an activator of IK(M) (7, 8, 12, 14, 33, 42, 43). Because IK(M) and IK(DR) tend to have reciprocal effects, it is difficult to experimentally determine their relative importance for the effects of these agents on action potential firing (44), including their subtypes (45). However, based on our simulations, it needs to speculate that, in terms of action potential firing, a combined approach using the activation of IK(M) and the inhibition of IK(DR) would be more useful than using the activator of IK(M) alone. Suppression of sustained firing by these agents may similarly involve both activation of IK(M) and inhibition of IK(DR). The contribution of IK(M) to the pattern and frequency of action potential firing may be accentuated under certain conditions in vivo as a result of either high expression of Kv3 channels or pharmacological modulation of Kv3 channels (e.g., diazepam or midazolam). If similar findings are experimentally made in neurons occurring in cell culture or in vivo to those described herein, the actions of DIC or its functionally related agents (e.g., flupirtine) will result in significant changes in neuronal excitability (12).
Acknowledements: The work in this laboratory was supported by grants from the National Science Council (NSC-101-2320-B-006-009, NSC-100-2314-B-006-002, and NSC-101-2314-B-006-059), Taiwan, and from the Aim for the Top University Project, National Cheng Kung University, Taiwan.
The authors acknowledge Hsien-Chin Huang who assisted in cell preparations used for this study.
Conflict of interests: None declared.
REFERENCES
- Sanchez-Hernandez MC, Delgado J, Navarro AM, Orta JC, Hernandez M, Conde J. Seizures induced NSAID. Allergy 1999; 54: 90-91.
- Ng SC, Chan FK. NSAID-induced gastrointestinal and cardiovascular injury. Curr Opin Gastroenterol 2010; 26: 611-617.
- Ortiz MI, Torres-Lopez JE, Castaneda-Hernandez G, Rosas R, Vidal-Cantu G, Granados-Soto V. Pharmacological evidence for the activation of K+ channels by diclofenac. Eur J Phamacol 2002; 438: 85-91.
- Liu LY, Fei XW, Li ZM, Zhang ZH, Mei YA. Diclofenac, a nonsteroidal anti-inflammatory drug, activates the transient outward K+ current in rat cerebellar granule cells. Neuropharmacology 2005; 48: 918-926.
- Miceli F, Soldovieri MV, Martire M, Taglialatela M. Molecular pharmacology and therapeutic potential of neuronal Kv7-modulating drugs. Curr Opin Pharmacol 2008; 8: 65-74.
- Villalonga N, David M, Bielanska J, et al. Immunomodulatory effects of diclofenac in leukocytes through the targeting of KV1.3 voltage-dependent potassium channels. Biochem Pharmacol 2010; 80: 858-866.
- Takahashi Y, Kaba H. Muscarinic receptor type 1 (M1) stimulation, probably through KCNQ/Kv7 channel closure, increases spontaneous GABA release at the dendrodendritic synapse in the mouse accessory olfactory bulb. Brain Res 2010; 1339: 26-40.
- Brueggemann LI, Mackie AR, Martin JL, Cribbs LL, Byron KL. Diclofenac distinguishes among homomeric and heteromeric potassium channels composed of KCNQ4 and KCNQ5 subunits. Mol Pharmacol 2011; 79: 10-23.
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol 2009; 156: 1185-1195.
- Soldovieri MV, Miceli F, Taglialatela M. Driving with no brakes: molecular pathophysiology of Kv7 potassium channels. Physiology 2011; 25: 365-376.
- Cooper EC. Potassium channels: how genetic studies of epileptic syndrome open paths to new therapeutic targets and drugs. Epilepsia 2001; 42(Suppl. 5): 49-54.
- Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 2012; 53: 412-424.
- Large CH, Sokal DM, Nehlig A, et al. The spectrum of anticonvulsant efficacy of retigabine (ezogabine) in animal models: implications for clinical use. Epilepsia 2012; 53: 425-436.
- Yu F, Liu Y, Wang Y, et al. Protective effect of the KCNQ activator flupirtine on a model of repetitive febrile seizures. Epilepsia Res 2011; 97: 64-72.
- Wu SN, Hsu MC, Liao YK, Wu FT, Jong YJ, Lo YC. Evidence for inhibitory effects of flupirtine, a centrally acting analgesic, on delayed rectifier K+ currents in motor neuron-like cells. Evid Based Complement Alternat Med 2012; 2012: 148403.
- Hsu SH, Lai MC, Er TK, et al. Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) regulates the level of SMN expression through ubiquitination in primary spinal muscular atrophy fibroblasts. Clin Chimica Acta 2010; 411: 1920-1928.
- Hsu YY, Chen CS, Wu SN, Jong YJ, Lo YC. Berberine activates Nrf2 nuclear translocation and protects against oxidative damage via a phosphatidylinositol 3-kinase/Akt-dependent mechanism in NSC34 motor neuron-like cells. Eur J Pharmaceut Sci 2012; 46: 415-425.
- Wu SN, Lo YC, Chen BS, So EC, Chen LT. Contribution of blocked potassium current conductance and increased conductance of persistent sodium current to the afterdischarge in myelinated neuron. Muscle Nerve 2012; 46: 297-299.
- Wu SN, Yeh CC, Huang HC, So EC, Lo YC. Electrophysiological characterization of sodium-activated potassium channels in NG108-15 and NSC-34 motor neuron-like cells. Acta Physiol 2012; 206: 120-134.
- Bocksteins E, Van de Vijver G, Van Bogaert PP, Snyders DJ. Kv3 channels contribute to the delayed rectifier current in small cultured mouse dorsal root ganglion neurons. Am J Physiol Cell Physiol 2012; 303: C406-C415.
- Huang CW, Wu SN, Cheng JT, Tsai JJ, Huang CC. Diazoxide reduces status epilepticus neuron damage in diabetes. Neurotox Res 2010; 17: 305-316.
- Golomb D, Yue C, Yaari Y. Contribution of persistent Na+ current and M-type K+ current to somatic bursting in CA1 pyramidal cells: combined experimental and modeling study. J Neurophysiol 2006; 96: 1912-1926.
- Lin MW, Wang YJ, Liu SI, Lin AA, Lo YC, Wu SN. Characterization of aconitine-induced block of delayed rectifier K+ current in differentiated NG108-15 neuronal cells. Neuropharmacology 2008; 54: 912-923.
- Wu SN, Chen BS, Lin MW, Liu YC. Contribution of slowly inactivating potassium current to delayed firing of action potentials in NG108-15 neuronal cells: experimental and theoretical studies. J Theor Biol 2008; 252: 711-721.
- Ermentrout GB. Simulating, Analyzing, and Animating Dynamical Systems: A Guide to XPPAUT for Researchers and Students. Philadelphia, Society for Industrial Mathematics (SIAM), 2002.
- Chen BS, Wu SN. Functional role of the activity of ATP-sensitive potassium channels in electrical behavior of hippocampal neurons: experimental and theoretical studies. J Theor Biol 2011; 272: 16-25.
- Wu SN. Simulations of the cardiac action potential based on the Hodgkin-Huxley kinetics with the use of Microsoft Excel spreadsheets. Chin J Physiol 2004; 47: 15-22.
- Baranauskas G. Ionic channel function in action potential generation: current perspective. Mol Neurobiol 2007; 35: 129-150.
- Wu SN, Lo YK, Chen H, Li HF, Chiang HT. Rutaecarpine-induced block of delayed rectifier K+ current in NG108-15 neuronal cells. Neuropharmacology 2001; 41: 834-843.
- Chen BS, Lo YC, Peng H, Hsu TI, Wu SN. Effects of ranolazine, a novel anti-anginal drug, on ion currents and membrane potential in pituitary tumor GH3 cells and NG108-15 neuronal cells. J Pharmacol Sci 2009; 110: 295-305.
- Yeung SY, Thompson D, Wang Z, Fedida D, Robertson B. Modulation of Kv3 subfamily potassium currents by the sea anemone toxin BDS: significance for CNS and biophysical studies. J Neurosci 2005; 25: 8735-8745.
- Björkman R. Central antinociceptive effects of non-steroidal anti-inflammatory drugs and paracetamol. Experimental studies in the rat. Acta Anaesthesiol Scand (Suppl) 1995; 103: 1-44.
- Xiong Q, Gao Z, Wang W, Li M. Activation of Kv7 (KCNQ) voltage-gated potassium channels by synthetic compounds. Trends Pharmacol Sci 2008; 29: 99-107.
- Peretz A, Degani N, Nachman R, et al. Meclofenamic acid and diclofenac, a novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties. Mol Pharmacol 2005; 67: 1053-1066.
- Meves H, Schwarz JR, Wulfsen I. Separation of M-like current and ERG current in NG108-15 cells. Br J Pharmacol 1999; 127: 1213-1223.
- Huang CW, Huang CC, Lin MW, Tsai JJ, Wu SN. The synergistic inhibitory actions of oxcarbazepine on voltage-gated sodium and potassium currents in differentiated NG108-15 neuronal cells and model neurons. Int J Neuropsychopharmacol 2008; 11: 597-610.
- Akemann W, Knopfel T. Interaction of Kv3 potassium channels and resurgent sodium current influences the rate of spontaneous firing of Purkinje neurons. J Neurosci 2006; 26: 4602-4612.
- Linley JE, Rose K, Patil M, Robertson B, Akopian AN, Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci 2008; 28: 11240-11249.
- De Sarro G, Di Paola ED, Conte G, Pasculli MP, De Sarro A. Influence of retigabine on the anticonvulsant activity of some antiepileptic drugs against audiogenic seizures in DBA/2 mice. Naunyn Schmiedebergs Arch Pharmacol 2001; 363: 330-336.
- Friederich P, Urban BW. Interaction of intravenous anesthetics with human neuronal potassium currents in relation to clinical concentrations. Anesthesiology 1999; 91: 1853-1860.
- Nagano Y, Matsui H, Shimokawa O, et al. Rebamipide attenuates nonsteroidal anti-inflammatory drugs (NSAID) induced lipid peroxidation by the manganese superoxide dismutase (MnSOD) overexpression in gastrointestinal epithelial cells. J Physiol Pharmacol 2012; 63: 137-142.
- Wu YJ, Dworetzky SI. Recent developments on KCNQ potassium channel openers. Curr Med Chem 2005; 12: 453-460.
- Pattnaik BR, Hughes BA. Effects of KCNQ channel modulators on the M-type potassium current in primate retinal pigment epithelium. Am J Physiol Cell Physiol 2012; 302: C821-833.
- Huang CW, Tsai JJ, Huang CC, Wu SN. Experimental and simulation studies on the mechanisms of levetiracetam-mediated inhibition of delayed-rectifier potassium current (KV3.1): contribution to the firing of action potentials. J Physiol Pharmacol 2009; 60: 37-47.
- Wang YJ, Lin MW, Lin AA, Peng H, Wu SN. Evidence for state-dependent block of DPI 201-106, a synthetic inhibitor of Na+ channel inactivation, on delayed-rectifier K+ current in pituitary tumor (GH3) cells. J Physiol Pharmacol 2008; 59: 409-423.
A c c e p t e d : June 27, 2013