THE INFLUENCE OF ANGIOTENSIN-(1-7) PEPTIDOMIMETIC (AVE 0991)
AND NEBIVOLOL ON ANGIOTENSIN I METABOLISM IN AORTA
OF apoE-KNOCKOUT MICE
INTRODUCTION
The detrimental role of over activation of renin-angiotensin system (RAS) in atherogenesis is widely recognized and drugs that inhibit RAS, irrespective of their mode of action, were shown to reverse endothelial dysfunction and prevent or delay progression of atherosclerosis (1-4). Over the last two decades, early concepts of RAS, focused mainly on the pathways involved in generation of pro-atherogenic angiotensin II (Ang II) in plasma have been upgraded by studies showing robust, local tissue formation of various angiotensins, e.g. endogenous functional Ang II antagonist - Ang-(1-7) (Fig. 1A) (5-7). Thus, the current view of drugs interfering with RAS encompass not only their inhibitory effect on Ang II formation, but also their influence on Ang-(1-7) generation (8).
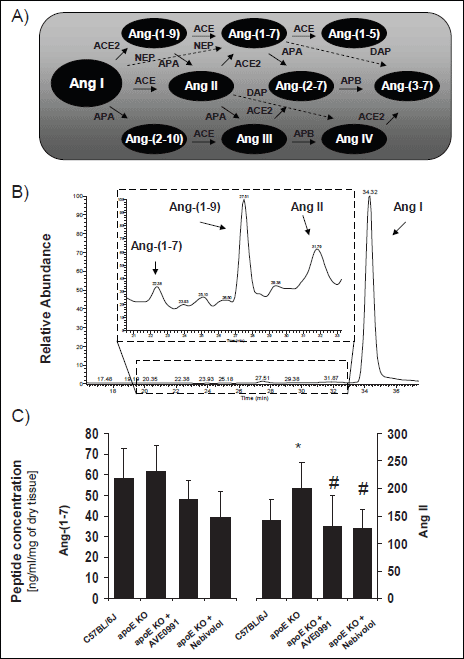 |
Fig. 1. (A) main pathways of Ang I metabolism. (B) representative chromato-gram of products of ex vivo Ang I conversion by the aorta of C57BJ6 mice; peaks represent relative abundance (Ang I = 100%), Insert: magnifications of chromatogram fragment. (C) quantitative data (pmoles/mg of dry tissue) showing the formation of Ang II and Ang-(1-7) in organ bath of aorta of C57BJ6 mice, apoE-knockout mice, apoE-knockout mice treated with AVE 0991 or apoE-knockout mice treated with nebivolol. * p<0.05 vs. wild type C57BL/6J mice; # p<0.05 vs. apoE KO mice |
The nitric oxide (NO) system seems to be mutually connected to RAS in regulation of vessel wall homeostasis. It has been shown that Ang II-elicited reactive oxygen species (ROS) are responsible for decrease of endothelial NO generation/bioavailability (9,10). Other way around, several lines of evidence point to the inhibitory influence of NO on activity of RAS in vessels (10, 11). Specifically, stimulation of Ang-(1-7) Mas receptor, via Akt kinase-dependent phosphorylation of endothelial NO synthase (NOS) has been demonstrated to abrogate detrimental action of Ang II (6).
Recently, we have demonstrated that Ang-(1-7) peptidomimetic AVE0991, as well as known beta-adrenolytic agent nebivolol, exert anti-atherogenic actions in mouse model of atherosclerosis - apoE-knockout mice (12-15). Importantly, both drugs were shown to increase endothelial NO production (16, 17). Here, using ex vivo system, we tested whether prolonged treatment of apoE-knockout mice by these drugs influences RAS in aorta of apoE-knockout mice in regard to generation of most active metabolites of Ang I-Ang II and Ang-(1-7).
MATERIALS AND METHODS
Animals and treatment
Female apoE-knockout mice on the C57BL/6J background were obtained from Taconic (Ejby, Denmark). Mice were maintained on 12/12-h dark/light cycles in air-conditioned rooms (22.5±0.5°C, 50±5% humidity) and access to diet and water ad libitum in Animal House of Chair of Immunology of Jagiellonian University Medical College. At the age of 8 weeks mice were put on chow diet made by Ssniff (Soest, Germany) for 4 months. Two experimental groups (in each n=4) received the same diet as a control group, mixed with: (A) AVE 0991 (a kind gift from Sanofi-Aventis Deutschland GmbH, Frankfurt am Main, Germany) at a dose 0.58 µmol per kg of body weight per day, or (B) nebivolol (a kind gift from Janssen Pharmaceutica, Geel, Belgium) at a dose 2.0 µmol per kg of body weight per day.
The procedure of isolation of aorta was described previously (13, 14, 18). Briefly, the age of 6 months 1000 UI of fraxiparine (Sanofi-Synthelabo, France) was injected into the peritoneum and after 10 min mice were killed in a chamber filled with carbon dioxide. Then, right atrium was incised and the heart was perfused by PBS through the apex of the left ventricle at a constant pressure of 100 mm Hg. Next, the whole thoracic aorta were dissected, washed cold with standard Krebs-Henseleit solution, cleaned of thrombi and tissue remnants, cut into a suitable number of rings and opened flat.
Ex vivo angiotensin I metabolism
Tissue incubation was performed according the principles described previously (19, 20). Briefly, tissue fragments were incubated for 30 minutes at 37°C in Eppendorf tubes in 550 µl of Krebs-Henseleit solution and continuously bubbled with 95%O2/5%CO2. Sample of 50 µl of buffer was removed to provide information on background production of angiotensin metabolites. Then, Ang I was added to a final concentration of 1 µM. Samples of 50 µl of buffer were removed after another 15 min of incubation. Each sample was promptly frozen at -70°C until further analysis. Tissue pieces were dried overnight at 60°C to allow estimation of angiotensin metabolites' production per mg of dry tissue.
Liquid chromatography - mass spectrometry (LC-MS) assessment of angiotensin peptides
Samples were purified and concentrated using Ultra-Micro Spin C-18 column (Harvard Apparatus, USA). Separation of angiotensin peptides was performed on a reversed-phase, high performance liquid chromatography (HPLC) system Ultimate 3000 (Dionex, USA) as described previously (21). Concentrations of angiotensins (Ang II, Ang 1-7) were calculated using the standard calibration curves, constructed by linear regression analysis by plotting of peak area versus angiotensin concentration and calculated as pmol/mg dry tissue.
Chemicals
Angiotensins: I (Ang I), III (Ang III), IV (Ang IV) and angiotensin fragments 1-9 (Ang-(1-9)) and 1-5 (Ang-(1-5)) were purchased from Bachem (USA). Angiotensin II (Ang II) and angiotensin fragment 1-7 (Ang-(1-7)) were purchased from Sigma Chemicals (USA). Formic acid (99%), trifluoroacetic acid (TFA) and ammonium formate were purchased from Fluka (USA). Acetonitrile (J.T. Baker, USA), and water (Rathburn, Scotland) were of HPLC grade.
Statistics
Concentrations of angiotensins were expressed as in pmol/mg dry tissue. All values in the figures and text are expressed as mean ±S.D. of n observations. Statistical comparisons between peptide concentrations were made by using Student’s t-test. P<0.05 was considered statistically significant.
RESULTS
Both, in the case of wild type (C57BJ6) and apoE-knockout mice incubation of Ang I with mouse aorta resulted in formation of high amounts of Ang-(1-9) and Ang II as well as two-fold lower production of Ang-(1-7) (Fig. 1B). There was increased generation of Ang II in aorta of apoE-knockout mice, as compared to wild type animals, while the formation of Ang-(1-7) did not differ between both groups (Fig. 1C). Either treatment with AVE0991 or nebivolol resulted in significant attenuation of Ang II production in aorta of apoE-knockout mice (Fig. 1C). Both compounds tended to decrease formation of Ang-(1-7), however this effect did not reach statistical significance.
DISCUSSION
In the "classical" view of RAS angiotensin converting enzyme (ACE)-derived Ang II is regarded as a major effector peptide in plasma, whereas "non-classical" view includes many peptides (e.g. Ang-(1-7), Ang-(1-9), Ang IV) and enzymes, e.g. angiotensin converting enzyme 2 (ACE2), neutral endopeptidase (NEP), expressed locally in tissues (5, 22). Recent studies have established a new regulatory axis in RAS (6). In this axis, Ang-(1-7) produced from Ang I (or Ang II) by the catalytic activity of ACE2, via stimulation of Mas receptor opposes in tissues detrimental actions of ACE-derived Ang II, executed via AT1 receptors. Here we demonstrated that ex vivo system of assessment of angiotensin conversion could be successfully applied to quantitative measurements of formation of two main Ang I metabolites (Ang II and Ang-(1-7)) in aorta of mouse model of atherosclerosis, namely in apoE-knockout mice. Our results revealed that aortic tissue isolated from atherosclerotic mice showed increased ability to synthesize of Ang II, as compared to wild type animals. This is in keeping with mainstream of current knowledge, strongly supporting the role of Ang II over production by ACE in atherogenesis (3). On the other hand, our finding is in contradiction to that of Weiss et al. (23), who showed using genetic models on the background of apoE-knockout mice that expression of ACE in vessel wall is not required for atherosclerotic lesion formation Noteworthy, our results, for the first time directly show increased ability of atherosclerotic vessel wall to produce Ang II. However, we are aware that to estimate the mechanism(s) of this phenomenon, our measurements should be expanded to contain detailed pharmacological analysis (e.g. estimation of ex vivo influence of selective ACE inhibitor perindoprilat on Ang II formation) and molecular studies (e.g. immunoblotting of ACE protein in mouse aorta).
Recently, we have shown that Ang-(1-7) peptidomimetic - AVE0991, as well as beta-adrenolytic agent nebivolol, exert anti-atherogenic action in mouse model of atherosclerosis - apoE-knockout mice (12-15). Here we demonstrated that for both drugs such action was associated with inhibitory influence on generation of Ang II in aorta of apoE-knockout mice. It has been shown previously, that stimulation of Mas receptor may inhibit tissue expression of ACE and AT1 receptors (24, 25). Interestingly, recent reports may point to the similar, vascular action of nebivolol (26, 27). Our results directly show the ability of both drugs to inhibit aortic Ang II generation. Such property could represent attractive new modality of action of clinically recognized beta-adrenolytic agent nebivolol, however such interference with RAS in patients treated with nebivolol remains to be tested.
Our study was not aimed to assess the mechanism(s) of influence of AVE0991 and nebivolol on aortic angiotensin I metabolism. Strikingly, both drugs are known to act as stimulators of endothelial NO release (17, 28). We hypothesize that there is close relationship between this property and ability of both drugs to decrease Ang II formation in aortic tissue, although it is not easy to delineate the cause and effect in such interaction. It has been shown in various animal models that ACE inhibition results in increase of NO release from endothelium (29, 30) and other way around, the inhibition of NOS by L-NAME is associated with increase of ACE activity (31-33). Clearly, further studies are required to clarify the nature of this mutual interaction.
In conclusion, here for the first time we directly demonstrated that aortic tissue of apoE knockout mice , generates Ang II and that such effect could be efficiently attenuated by treatment of nebivolol and Ang-(1-7) peptidomimetic - AVE0991. In the case of nebivolol such action could represent attractive new modality of action of this clinically widely recognized beta-adrenolytic agent. However, exact mechanism(s) responsible for its interference with RAS require further investigation.
Abbreviations: ACE, angiotensin converting enzyme; ACE2, angiotensin converting enzyme type 2; APA, aminopeptidase A; APB, aminopeptidase B; DAP, aminopeptidase D; NEP, neutral endopeptidase.
Acknowledgements: This study was supported by NCN grants No N402479937 (nr K/PBW/000544), No 2011/01/N/NZ4/03752 and No 2012/05/B/NZ4/02743.
Conflict of interests: None declared.
REFERENCES
- Lu H, Balakrishnan A, Howatt DA, et al. Comparative effects of different modes of renin angiotensin system inhibition on hypercholesterolaemia-induced atherosclerosis. Br J Pharmacol 2012; 165: 2000-2008.
- Patarroyo Aponte MM, Francis GS. Effect of angiotensin-converting enzyme inhibitors and angiotensin receptor antagonists in atherosclerosis prevention. Curr Cardiol Rep 2012; 14: 433-442.
- Durante A, Peretto G, Laricchia A, et al. Role of the renin-angiotensin-aldosterone system in the pathogenesis of atherosclerosis. Curr Pharm Des 2012; 18: 981-1004.
- Montero-Vega MT. The inflammatory process underlying atherosclerosis. Crit Rev Immunol 2012; 32: 373-462.
- Fyhrquist F, Saijonmaa O. Renin-angiotensin system revisited. J Intern Med 2008; 264: 224-236.
- Iwai M, Horiuchi M. Devil and angel in the renin-angiotensin system: ACE-angiotensin II-AT1 receptor axis vs. ACE2-angiotensin-(1-7)-Mas receptor axis. Hypertens Res 2009; 32: 533-536.
- Krskova K, Filipcik P, Zilka N, et al. Angiotensinogen and angiotensin-converting enzyme mRNA decrease and AT1 receptor mRNA and protein increase in epididymal fat tissue accompany age-induced elevation of adiposity and reductions in expression of GLUT4 and peroxisome proliferator-activated receptor (PPARgamma). J Physiol Pharmacol 2011; 62: 403-410.
- Epstein BJ, Leonard PT, Shah NK. The evolving landscape of RAAS inhibition: from ACE inhibitors to ARBs, to DRIs and beyond. Expert Rev Cardiovasc Ther 2012; 10: 713-725.
- Jugdutt BI. Nitric oxide and cardiovascular protection. Heart Fail Rev 2003; 8: 29-34.
- Patel KP, Schultz HD. Angiotensin peptides and nitric oxide in cardiovascular disease. Antioxid Redox Signal 2012; May 21: (epub. ahead of print)
- Gwathmey TM, Alzayadneh EM, Pendergrass KD, Chappell MC. Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am J Physiol Regul Integr Comp Physiol 2012; 302: R518-R530.
- Jawien J, Toton-Zuranska J, Kus K, Pawlowska M, Olszanecki R, Korbut R. The effect of AVE 0991, nebivolol and doxycycline on inflammatory mediators in an apoE-knockout mouse model of atherosclerosis. Med Sci Monit 2012; 18: BR389-BR393.
- Jawien J, Toton-Zuranska J, Gajda M, et al. Angiotensin-(1-7) receptor Mas agonist ameliorates progress of atherosclerosis in apoE-knockout mice. J Physiol Pharmacol 2012; 63: 77-85.
- Kus K, Gajda M, Pyka-Fosciak G, et al. The effect of nebivolol on atherogenesis in apoE-knockout mice. J Physiol Pharmacol 2009; 60:163-165.
- Toton-Zuranska J, Gajda M, Pyka-Fosciak G, et al. AVE 0991-angiotensin-(1-7) receptor agonist, inhibits atherogenesis in apoE-knockout mice. J Physiol Pharmacol 2010; 61: 181-183.
- Rath G, Balligand JL, Dessy C. Vasodilatory mechanisms of beta receptor blockade. Curr Hypertens Rep 2012; 14: 310-317.
- Santos RA, Ferreira AJ. Pharmacological effects of AVE 0991, a nonpeptide angiotensin-(1-7) receptor agonist. Cardiovasc Drug Rev 2006; 24: 239-246.
- Pawlowska M, Gajda M, Pyka-Fosciak G, et al. The effect of doxycycline on atherogenesis in apoE-knockout mice. J Physiol Pharmacol 2011; 62: 247-250.
- Bujak-Gizycka B, Olszanecki R, Suski M, Madek J, Stachowicz A, Korbut R. Angiotensinogen metabolism in rat aorta: robust formation of proangiotensin-12. J Physiol Pharmacol 2010; 61: 679-682.
- Olszanecki R, Bujak-Gizycka B, Madej J, et al. Kaempferol, but not resveratrol inhibits angiotensin converting enzyme. J Physiol Pharmacol 2008; 59: 387-392.
- Suski M, Gebska A, Olszanecki R, et al. Influence of atorvastatin on angiotensin I metabolism in resting and TNF-alpha -activated rat vascular smooth muscle cells. J Renin Angiotensin Aldosterone Syst 2013; Feb. 6 (epub ahead of print)
- Nguyen Dinh CA, Touyz RM. A new look at the renin-angiotensin system - focusing on the vascular system. Peptides 2011; 32: 2141-2150.
- Weiss D, Bernstein KE, Fuchs S, Adams J, Synetos A, Taylor WR. Vascular wall ACE is not required for atherogenesis in ApoE(-/-) mice. Atherosclerosis 2010; 209: 352-358.
- Sui YB, Chang JR, Chen WJ, Zhao L, Zhang BH, Yu YR et al. Angiotensin-(1-7) inhibits vascular calcification in rats. Peptides 2013; 42C: 25-34. doi: 10.1016/j.peptides. 2012.12.023.:25-34.
- Guang C, Phillips RD, Jiang B, Milani F. Three key proteases - angiotensin-I-converting enzyme (ACE), ACE2 and renin - within and beyond the renin-angiotensin system. Arch Cardiovasc Dis 2012; 105: 373-385.
- Varagic J, Ahmad S, Voncannon JL, et al. Nebivolol reduces cardiac angiotensin II, associated oxidative stress and fibrosis but not arterial pressure in salt-loaded spontaneously hypertensive rats. J Hypertens 2012; 30: 1766-1774.
- Yan W, Sheng ZM, Yu L. Nebivolol treatment improves resistant arterial function and reduces ventricular hypertrophy and angiotensin II in spontaneously hypertension rats. J Renin Angiotensin Aldosterone Syst 2013; 14: 146-155.
- Gao Y, Vanhoutte PM. Nebivolol: an endothelium-friendly selective beta1-adrenoceptor blocker. J Cardiovasc Pharmacol 2012; 59: 16-21.
- Comini L, Bachetti T, Cargnoni A, et al. Therapeutic modulation of the nitric oxide: all ace inhibitors are not equivalent. Pharmacol Res 2007; 56: 42-48.
- Oak JH, Cai H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes 2007; 56: 118-126.
- Korystova AF, Emelyanov MO, Kublik LN, et al. Distribution of the activity of the angiotensin-converting enzyme in the rat aorta and changes in the activity with aging and by the action of L-NAME. Age (Dordr) 2012; 34: 821-830.
- Sharifi AM, Akbarloo N, Darabi R. Investigation of local ACE activity and structural alterations during development of L-NAME-induced hypertension. Pharmacol Res 2005; 52: 438-444.
- Linardi A, Panunto PC, Ferro ES, Hyslop S. Peptidase activities in rats treated chronically with N(omega)-nitro-L-arginine methyl ester (L-NAME). Biochem Pharmacol 2004; 68: 205-214.
A c c e p t e d : June 21, 2013