INDOMETHACIN-INDUCED GENERATION OF REACTIVE OXYGEN SPECIES LEADS TO EPITHELIAL CELL INJURY BEFORE THE FORMATION OF INTESTINAL LESIONS IN MICE
INTRODUCTION
Nonsteroidal anti-inflammatory drugs (NSAIDs) such as aspirin and indomethacin are the most commonly prescribed drugs for arthritis, inflammation, and cardiovascular protection (1). However, they cause gastrointestinal complications such as ulcers and erosions (1). The pathophysiology of these complications has mostly been ascribed to NSAID's action on cyclooxygenase inhibition and subsequent prostaglandin (PG) deficiency (2). In the 1970s and 1980s, under the concept of cytoprotection, extensive research revealed the role of PG in the gastric mucosal defense system (3). According to these studies, an insufficiency of gastric mucosal mucin production caused by the inhibition of PG synthesis by NSAIDs is most likely the reason for the formation of these lesions. Because mucin is a very important protective factor, insufficient mucin production would be expected to cause an imbalance between aggressive factors (such as acid) and protective factors. In fact, some researchers have also shown that inhibition of acid secretion with histamine-H2 receptor antagonists or proton pump inhibitors significantly inhibited the formation of lesions (4-6).
Recent clinical studies have shed some light on NSAID-induced small intestinal mucosal injury. Capsule endoscopic and double-balloon endoscopic examinations have revealed that NSAID-induced mucosal damage in the small intestine, including erosion and ulceration, occurs more often than that previously expected (7, 8). There are no acid-secreting cells in the small intestine; therefore, the balance theory cannot apply in this case. A pathophysiological concept that would also explain NSAID-induced injury in the small intestine is desired, because this lesion has been reported to be fatal (9). However, the pathophysiology of NSAID-induced small intestinal injury is not as well understood as that of gastric injury.
The authors have previously reported that NSAID-derived mitochondrial reactive oxygen species (ROS) are directly involved in gastrointestinal (GI) cellular injury in vitro and that treatment with rebamipide, which is clinically prescribed for gastric injury patients, prevented this cellular injury by upregulating the expression of manganese superoxide dismutase (MnSOD) (10). To investigate the mechanisms underlying NSAID-induced intestinal injury, in this study, we performed live imaging of intestinal mucosa with a microscope during intestinal injury formation.
MATERIALS AND METHODS
Reagents
Indomethacin was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan); aminophenyl fluorescein (APF), from Sekisui Medical Co. Ltd. (Japan); propidium iodide (PI), from Sigma-Aldrich; and the Guava viaCount reagent, from EMD Millipore Co. (MA, USA). Rebamipide was kindly provided by Otsuka Pharmaceutical Co., Ltd. (Tokyo, Japan); the molecular structure of rebamipide is shown in Fig. 1.
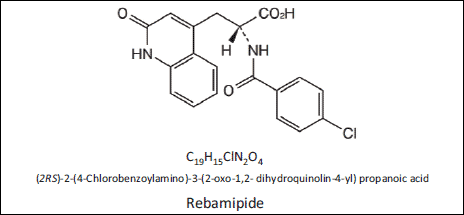 |
Fig. 1. Chemical structure of rebamipide. |
Ethics
For the animal experiments, all procedures and animal care were approved by the Committee on the Ethics of Animal Experimentation of the University of Tsukuba and were conducted according to the Guidelines for Animal Experimentation of the University of Tsukuba.
Live imaging of indomethacin-induced small intestinal injury in mice
Seven-week-old male ICR mice were purchased from SLC Co., Ltd. (Hamamatsu, Japan). Indomethacin or rebamipide was resuspended in 0.5% carboxyl methyl cellulose. Indomethacin (20 mg/kg) was administered p.o. to mice. In the rebamipide pretreatment group, rebamipide (30 mg/kg) was administered p.o. 10 min before the administration of indomethacin. The treated mice were kept for 12 hours without fasting and then subjected to live imaging. For live imaging, mice were anesthetized with urethane (1.75 g/kg, i.s.), and the jejunum was picked out through a small incision of the peritoneum. The mucosa of jejunum was visualized by making an incision approximately 2 cm long along the long axis for microscopic observation. Twenty microliters of the fluorescent ROS indicator aminophenyl fluorescein (APF) (11) (50 µmol/L) was poured on the mucosa after wiping away the intraluminal contents with a painting brush to remove detached cells. Fifteen minutes after the addition of APF, the formation of ROS on the mucosa was observed by fluorescence microscopy (Optiphoto Nikon, Tokyo, Japan). Then, 90 min after adding APF, propidium iodide (PI) (10 µL; 5 µg/mL) was applied to the mucosal surface to visualize dead cells. The fluorescence intensity of both APF and PI images was measured at randomly chosen observation fields by using NIH ImageJ software, and averages and standard error were calculated.
Flow cytometric analysis of detaching cells
Following live imaging of indomethacin-induced small intestinal injury as described above, cells that were detached or in the process of detaching from the mucosa were obtained using a No. 2 round-type painting brush (Sakura Color Products Co., Japan). The mucosa was brushed ten times in the same direction, and then the brush was washed in 1 mL of saline to collect the detached cells. This procedure was performed three times. The number of the detached cells was counted using a flow cytometer after staining them with the Guava viaCount Reagent. Flow cytometric analysis was carried out using the Guava EasyCyte Mini system (Guava Technologies, Hayward, CA).
Detection of intestinal injury by Evans blue staining
Small intestinal injury was induced by indomethacin treatment without prior rebamipide administration. Intestinal injury detection was performed as previously described (12, 13). Briefly, 0.5 mL of 1% Evans blue was bolus-injected into the tail vain. After 15 min, the mouse was killed with an pentobarbital overdose. The small intestine of the mouse was removed from the body and then incised longitudinally along the antimesenteric border. Lesions in the small intestinal mucosa as revealed by leakage of Evans blue were observed macroscopically.
Statistical analysis
The statistical significance of the data was evaluated using analysis of variance (ANOVA) followed by Tukey method. A P-value of <0.05 was considered significant.
RESULTS
Indomethacin-induced small intestinal injury in the mouse
At 12 huors after oral administration of indomethacin, mice had serious ulcers in the ileum (the lower part of the intestine; Fig. 2a), and the ulcers could be visualized with Evans blue (Fig. 2b). However, Evans blue was not detected in the jejunum (the upper part of the small intestine), which is usually empty. These observations suggest that the ulcers were caused by mechanical damage from the contents in the intestine because the area of the ulcer was almost the same size as the area that had been covered by intestinal contents.
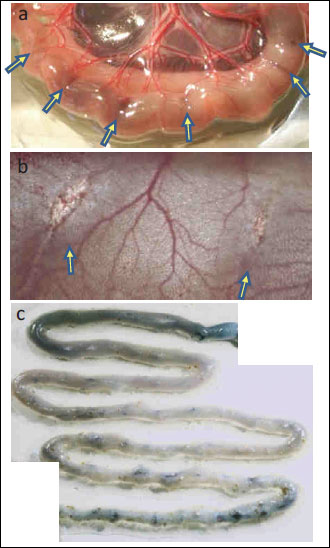 |
Fig. 2. Images of small intestinal mucosa 12 hours after indomethacin administration. (a) The ulcer in the lower intestine commonly developed in association with intestinal contents. Arrows indicate the perforations confirmed outside the intestinal tract. (b) Perforations were observed in the intestinal inner cavity. (c) Image of staining with Evans blue. |
Live imaging of the jejunum
Injury prior to ulcer development was detected in the jejunum. Twelve hours after oral administration of indomethacin, mucosal damage in the jejunum could not be seen in bright-field images; however, fluorescence from APF, an ROS indicator, could be observed in the mucosal epithelial cells (representative images are shown in Fig. 3a, 3d, 3g). APF fluorescence in the control mice, which had been administered the vehicle instead of indomethacin, was almost undetectable and was weaker in rebamipide-pretreated mice than in indomethacin-treated mice (representative images are shown in Fig. 3b, 3e, 3h).
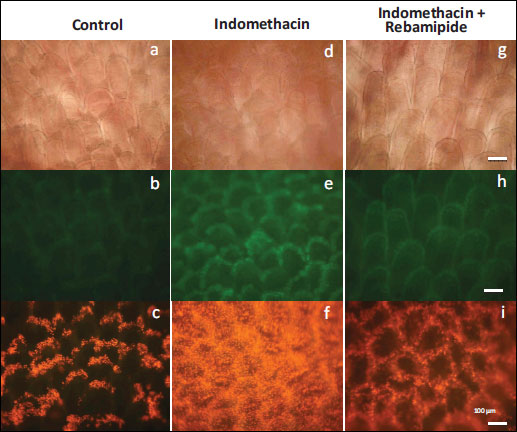 |
Fig. 3. Representative images of live imaging of mouse small intestine (jejunum). (a), (d), (g): Bright-field images 12 hours after administration p.o. of indomethacin or vehicle. (b), (e), (h): Fluorescence images after APF application on the mucosa for 15 min. ROS producing cells were observed. (c), (f), (i): PI fluorescence images taken 90 min after the initial mucosal clean up. Fluorescent dots are dead cells. Scale bar: 100 µm; magnification of each image is same. |
Ninety minutes after removing intestinal contents such as digested foods, we treated villi with 10 µL of PI (5 µg/mL). Because the compromised cellular membranes of dead cells allow the membrane-impermeant PI dye to enter and stain nuclei, dead cells can be easily detected as PI-positive cells (14). In the intestines of indomethacin-treated mice, many PI-positive cells could be seen: they covered almost all the intestinal mucosa. In contrast, only a few PI-positive cells could be detected on the summit of each villus in the control mice. Moreover, there were fewer PI-positive cells in the intestines of rebamipide-treated mice than in those of indomethacin-treated mice. Fluorescence intensity was the highest in indomethacin-treated mice, followed by that in rebamipide and indomethacin-treated mice, and that in the control mice (representative images are shown in Fig. 3c, 3f, 3i).
In additional, when cells that had detached from the mucosa were analyzed by flow cytometry, the same trend in the intensity of APF fluorescence was observed (Fig. 5). These results indicate that indomethacin induces cellular injury in the small intestinal mucosa through oxidative stress.
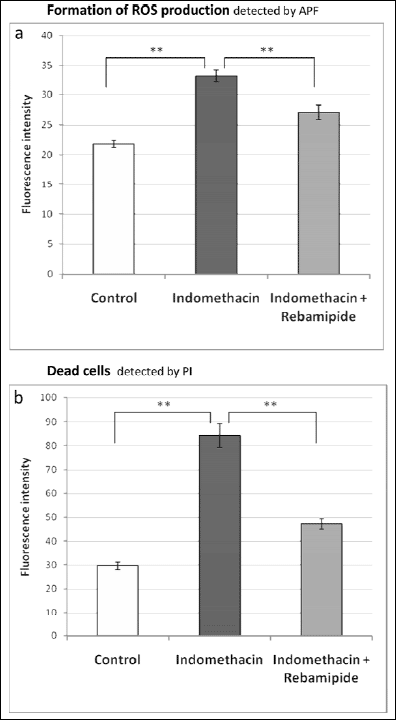 |
Fig. 4. The amount of ROS production and number of dead epithelial cells in areas of the small intestine mucosa (jejunum) that macroscopically appeared to be uninjured. Fluorescence intensities were measured from images taken with the same settings as Fig. 3. (A) ROS production calculated from the APF fluorescence images. (B) The number of dead cells calculated from PI fluorescence images. n=6, mean ± S.E. ** p<0.05, ANOVA followed by the Tukey method. |
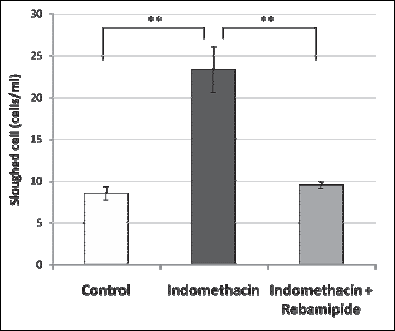 |
Fig. 5. Determination of the number of detaching cells. Indomethacin or vehicle was administered p.o. to mice, and mice were kept for 12 hours. The intestine was exposed through a small incision of the peritoneum. Detaching cells were obtained by wiping the small intestinal mucosa with a painting brush. Numbers of the detaching cells were counted by flow cytometry after staining with the Guava ViaCount Reagent. n=4, mean ± S.E. ** p<0.01, ANOVA followed by the Tukey method. |
DISCUSSION
In this study, we found for the first time that indomethacin treatment causes the death of intestinal cells by inducing a high concentration of ROS prior to the formation of lesions.
Intestinal cells are produced at the area close to the crypt (located at the base of the villus). The cells age as they move to the summit of the villus, where they are sloughed off after their death. In our study, representative of this cell-kinetic phenomenon, dead cells, which were stained with PI, could be observed at the summit of each villus in control mice. In contrast, a larger number of dead cells were observed in the intestines from indomethacin-treated mice. An obvious increase in dead cells was also observed in areas that macroscopically appeared to be free of lesions. Therefore, we conclude that the formation of macroscopic lesions such as ulcers and erosions are involved physical force such as that caused by the movement of foods.
ROS are known to be involved in GI mucosal injury. Kwiecien et al. reported that peroxidation of gastric mucosal tissue is involved in gastric lesion formations in rats with water-immersion restraint stress (WRS), a model of gastric mucosal injury due to stress (15). In their model, tissue SOD and GSH production are reduced by stress. Moreover, the calcitonin gene-related peptide (CGRP)-sensitive sensory nerves are involved in the formation of ROS scavengers. The two known primary sources of cellular ROS generation are NADPH oxidase (NOX) in cell membranes and the mitochondrial respiratory chain. Proinflammatory cytokines induce ROS formation via activation of NOX, which triggers prostaglandin production (16). Meanwhile, ROS leakage to the cytoplasm owing to mitochondrial dysfunction triggers cellular events such as apoptosis. We have previously reported that cellular stress by exposure to hypertonic salt directly generates ROS from the mitochondria of gastric mucosal cells (17).
ROS from mitochondria are known to play an important role in NSAIDs-induced GI injury (18, 19). The mitochondrial respiratory chain is the major source of ROS, which are mainly generated at Complex I and III of the respiratory chain (20). The mitochondrial respiratory chain is also an important target for the damaging effects of ROS. ROS from mitochondria play an important role in the release of cytochrome c and other pro-apoptotic proteins, which can trigger caspase activation and apoptosis (21). NSAIDs inhibit oxidative phosphorylation to involve both ROS production and the liberation of cytochrome c from mitochondrial intermembranous space into cytosol, phenomena which cause caspase 9 and caspase 3 activation and cellular lipid peroxidation, leading to apoptosis (22-25). The uncoupling of mitochondria also decreases the concentration of intracellular ATP and causes Ca2+ to leak out of the mitochondria, leading to cellular osmotic imbalance and a loss of control over intracellular junctions and resulting in increased permeability and subsequent mucosal damage (26).
In the intestines of indomethacin-treated mice, distinctly fluorescent villi were observed after application of APF. In contrast, little fluorescence was observed in the villi of control mice. Moreover, the intestinal villi of mice that were pretreated with rebamipide before the indomethacin treatment were less fluorescent, and a smaller number of dead cells were observed in this group than in the indomethacin-treated mice. We have previously reported that rebamipide treatment upregulates the expression of MnSOD (10). MnSOD has been reported to scavenge mitochondria-specific O2-, thus reducing ROS involved in tissue injury, and it has also been reported that cells overexpressing MnSOD are resistant to oxidative stresses (27-31). Therefore, we propose that the APF fluorescence observed in our mouse model is likely to represent the presence of mitochondrial ROS after indomethacin treatment. Consequently, treatment with a mitochondrial ROS scavenger such as rebamipide should be considered as a preventive therapy for NSAID-induced intestinal injury.
In conclusion, indomethacin treatment causes intestinal cellular injury from mitochondrial ROS in areas both with and without macroscopic lesions. The MnSOD-inducing reagent rebamipide was shown to be effective at preventing this injury.
Conflict of interests: None declared.
REFERENCES
- Arora G, Singh G, Triadafilopoulos G. Proton pump inhibitors for gastroduodenal damage related to nonsteroidal anti-inflammatory drugs or aspirin: twelve important questions for clinical practice. Clin Gastroenterol Hepatol 2009; 7: 725-735.
- Graham DY, Agrawal NM, Roth SH. Prevention of NSAID-induced gastric ulcer with misoprostol: multicentre, double-blind, placebo-controlled trial. Lancet 1988; 332(8623): 1277-1280.
- Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology 2008; 135: 41-60.
- Guth PH, Aures D, Paulsen G. Topical aspirin plus HCl gastric lesions in the rat. Cytoprotective effect of prostaglandin, cimetidine, and probanthine. Gastroenterology 1979; 76: 88-93.
- Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology 2008; 135: 41-60.
- Higuchi K, Yoda Y, Amagase K, et al. Prevention of NSAID-induced small intestinal mucosal injury: prophylactic potential of lansoprazole. J Clin Biochem Nutr 2009; 45: 125-130.
- Tajima A. Non-steroidal anti-inflammatory drug (NSAID) - induced small intestinal injury. Pharm Anal Acta 2014; 5: 282
- Higuchi K, Umegaki E, Watanabe T, et al. Present status and strategy of NSAIDs-induced small bowel injury. J Gastroenterol 2009; 44: 879-888.
- Lanas A, Garcia-Rodríguez LA, Polo-Tomas M, et al. Time trends and impact of upper and lower gastrointestinal bleeding and perforation in clinical practice. Am J Gastroenterol 2009; 104: 1633-1641.
- Nagano Y, Matsui H, Shimokawa O, et al. Rebamipide attenuates nonsteroidal anti-inflammatory drugs (NSAID) induced lipid peroxidation by the manganese superoxide dismutase (MnSOD) overexpression in gastrointestinal epithelial cells. J Physiol Pharmacol 2012; 63: 137-142.
- Setsukinai K, Urano Y, Kakinuma K, Majima HJ, Nagano T. Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J Biol Chem 2003; 278: 3170-3175.
- Takagi T, Naito Y, Yoshikawa T, et al. Identification of dihalogenated proteins in rat intestinal mucosa injured by indomethacin. J Clin Biochem Nutr 2011; 48: 178-182.
- Amagase K, Yoshida Y, Hara D, Murakami T, Takeuchi K. Prophylactic effect of egualen sodium, a stable azulene derivative, on gastrointestinal damage induced by ischemia/reperfusion, double antiplatelet therapy and loxoprofen in rats. J Physiol Pharmacol 2013; 64: 65-75.
- Moore A, Donahue CJ, Bauer KD, Mather JP, et al. Simultaneous measurement of cell cycle and apoptotic cell death. Methods Cell Biol 1998; 57: 265-278.
- Kwiecien S, Konturek PC, Sliwowski T, et al. Interaction between selectiove cyclooxygenase inhibitors and capsaicin-sensitive afferent sensory nerves in pathogenesis of stress-induced gastric lesions. Role of oxidative stress. J Physiol Pharmacol 2012; 63: 143-151.
- Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D. The effect of reactiove oxygen species on the synthesis of prostanoids from arachidonic acid. J Physiol Pharmacol 2013; 64: 409-421.
- Tamura M, Matsui H, Nagano YN, et al. Salt is an oxidative stressor for gastric epithelial cells. J Physiol Pharmacol 2013; 64: 89-94.
- Somasundaram S, Sigthorsson G, Simpson RJ, et al. Uncoupling of intestinal mitochondrial oxidative phosphorylation and inhibition of cyclooxygenase are required for the development of NSAID-enteropathy in the rat. Aliment Pharmacol Ther 2000; 14: 639-650.
- Matsui H, Shimokawa O, Kaneko T, Nagano Y, Rai K, Hyodo I. The pathophysiology of non-steroidal anti-inflammatory drug (NSAID)-induced mucosal injuries in stomach and small intestine. J Clin Biochem Nutr 2011; 48: 107-111.
- Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000; 404: 787-790.
- Ott M, Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis 2007; 12: 913-922.
- Brand MD, Affourtit C, Esteves TC, et al. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 2004; 37: 755-767.
- Brzozowski T, Konturek PC, Konturek SJ, et al. Role of gastric acid secretion in progression of acute gastric erosions induced by ischemia-reperfusion into gastric ulcers. Eur J Pharmacol 2000; 398: 147-158.
- Tsutsumi S, Tomisato W, Takano T, Rokutan K, Tsuchiya T, Mizushima T. Gastric irritant-induced apoptosis in guinea pig gastric mucosal cells in primary culture. Biochim Biophys Acta 2002; 1589: 168-180.
- Nagano Y, Matsui H, Muramatsu M, et al. Rebamipide significantly inhibits indomethacin-induced mitochondrial damage, lipid peroxidation, and apoptosis in gastric epithelial RGM-1 cells. Dig Dis Sci 2005; 50 (Suppl. 1): S76-S83.
- Somasundaram S, Hayllar H, Rafi S, Wrigglesworth JM, Macpherson AJ, Bjarnason I. The biochemical basis of non-steroidal anti-inflammatory drug-induced damage to the gastrointestinal tract: a review and a hypothesis. Scand J Gastroenterol 1995; 30: 289-299.
- Indo HP, Inanami O, Koumura T, et al. Roles of mitochondria-generated reactive oxygen species on X-ray-induced apoptosis in a human hepatocellular carcinoma cell line, HLE. Free Radic Res 2012; 46: 1029-1043.
- Majima HJ, Matsui H, Ozawa T, et al. Mitochondria as source of free radicals. Free Radic Biol 2011; 29: 12-22.
- Indo HP, Davidson M, Yen HC, et al. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007; 7: 106-118.
- Majima HJ, Oberley TD, Furukawa K, et al. Prevention of mitochondrial injury by manganese superoxide dismutase reveals a primary mechanism for alkaline-induced cell death. J Biol Chem 1998; 273: 8217-8224.
- Majima HJ, Matsui H, Suenaga S, Matsui H, Yen HC, Ozawa T. Mitochondria as possible pharmaceutical targets for the effects of vitamin E and its homologues in oxidative stress-related diseases. Curr Pharm Des 2011; 17: 2190-2195.
A c c e p t e d : April 18, 2014