MUSCARINIC ACETYLCHOLINE RECEPTOR SUBTYPE 4 IS ESSSENTIAL FOR CHOLINERGIC STIMULATION OF DUODENAL BICARBONATE SECRETION IN MICE - RELATIONSHIP TO D CELL/SOMATOSTATIN
INTRODUCTION
The secretion of HCO3– from surface epithelial cells plays an important role in protecting the duodenal mucosa against acid injury (1-4). The mechanisms that govern the secretion of HCO3– involve neuro-humoral factors, including prostaglandins (PGs), nitric oxide (NO), and acetylcholone (ACh), as well as capsaicin-sensitive afferent neurons (1, 3). The action of the former two substances is known to be mediated intracellularly by cyclic nucleotides, such as 3', 5'-cyclic adenosine monophosphate (cAMP) and 3', 5'-cyclic guanosine monophosphate (cGMP), while that of ACh is mediated by an increase in intracellular Ca2+ concentration via the activation of muscarinic acetylcholine receptors (mAChRs) (5-7). The stimulatory action of PGE2 was known to be mediated by the activation of EP3/EP4 receptors, while that of NO was eventually mediated by PGE2 (8-13). However, the types of mAChRs that are mainly responsible for the stimulatory effects of ACh on HCO3– secretion in the duodenum have not yet been established.
The mAChRs consist of five subtypes (M1-M5) and are widely expressed in many peripheral organs as well as the central nervous system in order to mediate diverse autonomic functions (14, 15). In the gastrointestinal tract, mAChRs mediate motility and glandular secretion, including acid, pepsin, and mucus, as well as HCO3– (16). These receptors are G protein-coupled receptors; M1, M3, and M5 receptors are coupled to the Gq protein, while M2 and M4 receptors are coupled to the Gi protein (17). It has generally been accepted that the acid stimulatory actions of carbachol (CCh), a muscarinic agonist, are mediated by the activation of M1 and M3 receptors. However, a recent study showed that the acid secretion in response to CCh was mediated by M3 and M5, but not M1 receptors in knockout (KO) mice lacking M1~M5 receptors (18). We also reported the involvement of M4 receptors in the cholinergic stimulation of acid secretion, in addition to M3 receptors (19). However, the role of mAChR subtypes in the cholinergic regulation of duodenal HCO3– secretion has not yet been elucidated.
Somatostatin, a peptide hormone, regulates many physiological functions and also plays as a mediator of circadian rhythms of the motor and secretary activity in the gastrointestinal tract (20, 21). This peptide is produced and secreted from D cells and inhibits acid secretion through a tonic inhibitory effect on both parietal and enterochramaffin-like cells via the activation of G protein-coupled SST2 receptors (22, 23). Odes et al. (24) reported that somatostatin-14 inhibited not only basal HCO3– secretion, but also CCh- and vasoactive intestinal peptide (VIP)-stimulated duodenal HCO3– secretion in guinea pigs. Chiba and Yamada (25) demonstrated that CCh inhibited basal and pentagastrin-stimulated somatostatin secretion in isolated canine D cells in a Gi protein/cAMP-dependent manner. We recently showed the involvement of somatostatin in the regulatory mechanism of CCh-stimulated acid secretion (19, 26). Endogenous somatostatin may also be involved in the mechanism underlying duodenal HCO3– secretion in response to cholinergic stimulation.
In the present study, we examined the effects of CCh on HCO3– secretion in the isolated mouse duodenum in vitro and investigated the mAChR subtypes responsible for the stimulatory action of CCh, using subtype-selective mAChR antagonists and mAChR KO mice lacking M1-M5 receptors. We demonstrated the importance of M4 receptors in the cholinergic stimulation of duodenal HCO3– secretion and showed that this secretion was modulated by the activation of M4 receptors, with a focus on the relationship to D cell/somatostatin.
MATERIALS AND METHODS
Animals
All experimental procedures used were carried out in accordance with the Helsinki Declaration and have been approved by the Committee for Animal Experimentation established by Kyoto Pharmaceutical University.
Wild-type (WT) and M1, M2, M3, M4 or M5 receptor KO age-matched C57BL/6J mice, weighing 25 ~ 30 g, were used. The generation and characterization of each subtype of mAChR KO mouse strain have been previously described (14, 27, 28). Animals were housed in plastic cages with hardwood chips in an air-conditioned room (25°C) and were given standard dry pellets, CA-1 (CLEA Japan, Tokyo, Japan), and water ad libitum.
Determination of HCO3– secretion
HCO3– secretion was measured in an in vitro preparation of a mouse duodenum, according to previously published methods (29, 30). Under deep diethyl ether anesthesia, either a WT or KO mouse lacking the M1, M2, M3, M4, or M5 receptor was killed, and the abdomen was opened by a midline incision. The proximal duodenum (8 mm distal from the pylorus) was removed and immediately placed in ice-cold HCO3– Ringer's solution containing indomethacin (10-7 M) to suppress the trauma-induced release of PGs. The duodenum was opened along the mesenteric attachment and striped from the muscular layers under a dissecting microscope (SZ-PT; Olympus, Japan). The tissue was mounted between two halves of an Ussing chamber, with the exposed area being 12.5 mm2, and bathed in unbuffered saline (mmol/L: Na+, 154; Cl-, 154) gassed with 100% O2 on the mucosal side and HCO3– Ringer's solution (mmol/L: Na+, 140; Cl–, 120; K+, 5.4; Mg2+, 1.2; Ca2+, 1.2; HPO42-, 1.4; H2PO4–, 2.4; HCO3–, 25; glucose, 10; indomethacin, 1 × 10-4) gassed with 95% O2 - 5% CO2 on the serosal side. These solutions were warmed at 37°C and continuously circulated using a gas-lift system (Fig. 1) (31). The osmolalities of both solutions were approximately 308 mOsm/kg. HCO3– secretion was measured by the pH-stat method (Comtite-980, Hiranuma Industries, Ibaraki, Japan) using 2 mmol/L HCl as the titrant to maintain the mucosal pH at 7.0. Measurements were made every 5 min starting at least 1 hour after mounting of the tissues. After the rate of secretion had stabilized for 30 min, CCh (1 × 10–5, 3 × 10–5 or 1 × 10–4 M) was applied to the serosal solution. Atropine (1 × 10–7, 1 × 10–6, 1 × 10–5 M), indomethacin (1 × 10–5 M), L-NAME (a non-selective inhibitor of nitric oxide (NO) synthase: 1 × 10–3 M), pirenzepine (an M1 ≥ M3 antagonist: 3 × 10–5 M), methoctramine (an M2 antagonist: 1 × 10–5 M) (32), 4-DAMP (an M3 > M1 antagonist: 3 × 10–6 M) (33), tropicamide (an M4 antagonist: 3 × 10–6 M) (33), and tetrodotoxin (a Na+ channel blocker: 1 × 10–5 M) was added to the serosal solution 30 min before the application of CCh. In some cases, the effects of octreotide (an analogue of somatostatin-14) on both basal and CCh-stimulated HCO3– secretion were examined in WT mice. Octreotide (1 × 10–6 M) was added to serosal solution 30 min after basal secretion had stabilized or 1 hour after the addition of CCh (1 × 10–5 M); in some cases CYN154806 (a somatostatin SST2 receptor antagonist: 1 × 10–5 M) (34) was added to the serosal solution 30 min before the application of octreotide. In another experiment, the effects of CYN154806 on the HCO3– response to CCh (1 × 10–4 M) was examined in M4 KO mice. CYN154806 (1 × 10–5 M) was added to the serosal solution 30 min before the application of CCh.
Analyses for gene expression of mAChR subtypes mRNAs in mouse duodenum
Whole duodenums and brains were collected from C57BL/6J mice, immediately frozen in liquid nitrogen, and stored at –80°C until use. Total RNA was extracted from tissue samples using Sepasol RNA I (Nacalai Tesque, Kyoto, Japan). Total RNA was reverse-transcribed with a first strand cDNA synthesis kit (ReverTra Ace alpha, TOYOBO, Osaka, Japan). The sequences of the sense and antisense primers for mouse M1-M5 receptors and GAPDH, and the sizes of the expected RT-PCR products are shown in Table 1. An aliquot of the RT reaction product served as a template in 35 cycles of PCR with 0.5 min of denaturation at 95°C and 1 min of extension at 68°C using the Advantage 2 polymerase mixture (CLONTECH, Mountain View, CA) in a thermal cycler (PC-806, ASTEC, Fukuoka, Japan). A portion of the PCR mixture was electrophoresed in 1.5% agarose gel in Tris-acetic acid-EDTA buffer (40 mM Tris, 20 mM acetic acid, and 2 mM EDTA; pH 8.1), and the gel was stained with ethidium bromide and then photographed (Bio Doc-It Imaging System; UVP, Upland, CA, USA). Images were analyzed with Image J (version 1.39), and semi-quantitative measurements of mRNA expression were presented as a ratio relative to GAPDH.

Morphological study
The expression of the mAChR M4 subtype and somatostatin was immuno-histochemically examined in the duodenal mucosa of WT mice. The duodenums were excised, rinsed with ice-cold PBS, and embedded in O.C.T. compound (Tissue-Tek, Sakura, Tokyo, Japan) iced with liquid CO2. Frozen samples were sectioned at a thickness of 10 µm at –20°C using a cryostat microtome (Leica Biosystems CM1510, Nussloch, Germany). These sections were then exposed to 3% bovine serum albumin solution for 1 hour to reduce the nonspecific binding of anti-sera. The sections were exposed to each primary antibody for 16 hours at 4°C, and incubated with the appropriate secondary antibody for 2 hours at a room temperature. The sections were mounted with VECTASHIELD mounting medium, including 4,6-diamidino-2-phenylindole (Vector Laboratories, Peterborough, UK). The preparations were observed using a fluorescence microscope (Olympus BX51, Tokyo, Japan) and photographed using an Olympus digital camera. The following primary antibodies were used: rabbit anti-mAChR M4 and goat anti-Somatostatin (Santa Cruz Biotechnology, Santa Cruz, CA). Alexa Fluor 488 conjugated donkey anti-rabbit IgG and Alexa Fluor 546 conjugated donkey anti-goat IgG (Molecular Probes, Eugene, OR) were used as secondary antibodies.
Preparation of drugs
The drugs used were carbamylcholine chloride (carbachol: CCh), atropine sulfate, pirenzepine dihydrochloride, methoctramine hydrate, 4-DAMP (4-diphenyl-acetoxy-N-methylpiperidine methiodide), tropicamide, indomethacin, verapamil, L-NAME (NG-nitro-L-arginine methylester), octreotide, CYN154806 (Sigma-Aldrich, St. Louis, MO), and tetrodotoxin (Nacalai tesque, Kyoto, Japan). All agents were dissolved in dimethyl sulfoxide (DMSO: Wako, Osaka, Japan) and diluted with distilled water to the desired concentrations. All agents were prepared before use and added to the serosal solution.
Statistical analysis
Data are presented as means ± S.E. Differences between two groups were evaluated with the Student's t-test. Differences between multiple groups were evaluated with an analysis of variance followed, where necessary, by Dunnett's multiple comparison test. Values of P < 0.05 were considered significant.
RESULTS
Effects of carbachol on duodenal HCO3– secretion in wild-type mice in vitro
The isolated duodenums of WT mice spontaneously secreted HCO3– at rates of 0.30 - 0.45 µEq/h as basal secretion in the absence or presence of 0.1% DMSO, a solvent for the agents used in the present study. The application of CCh (1 × 10–5, 3 × 10–5, and 1 × 10–4 M) to the serosal solution increased the secretion of HCO3– in a dose-dependent manner, and DHCO3– outputs were 0.042 ± 0.015 µEq/h, 0.072 ± 0.010 µEq/h, and 0.170 ± 0.025 µEq/h, respectively (Fig. 2A and 2B). The prior application of atropine (1 × 10–7 - 1 × 10–5 M), a nonselective mAchR antagonist, inhibited the HCO3– response to CCh (1 × 10–4 M) in a concentration-dependent manner (data not shown) and, at 1 × 10–6 M, almost completely attenuated this response, with the ΔHCO3– output being 0.012 ± 0.009 µEq/h, which were significantly lower than that obtained in the control duodenums given vehicle. The HCO3– stimulatory action of CCh (1 × 10–4 M) was also significantly mitigated by the pretreatment with verapamil (3 × 10-5 M), a Ca2+ channel blocker, with the ΔHCO3– output being 0.045 ± 0.017 µEq/hr, while neither indomethacin (1 ×10–5 M), a non-selective cyclooxygenase inhibitor, nor L-NAME (1 × 10–3 M), a non-selective NO synthase inhibitor, had any effect on this response (Fig. 3). In addition, the CCh-induced secretion of HCO3– was not significantly affected by the presence of tetrodotoxin (1 × 10–5 M).
 |
Fig. 1. Schematic illustration of the perfusion system and order of connections for measuring duodenal HCO3- secretion in vitro using an Ussing chamber preparation. A mouse duodenum was isolated and mounted on a chamber. This tissue was bathed in unbuffered Ringer solution gassed with 100% O2 on the mucosal side and HCO3- Ringer's solution gassed with 95% O2-5% CO2 on the serosal side, and these solutions were warmed at 35°C and continuously circulated by a gas-lift system. HCO3- secretion was measured at pH 7.4 using a pH-stat method by 2 mmol/L HCl. |
 |
Fig. 2. Effects of CCh on HCO3- secretion in isolated duodenums of WT mice. CCh (1 × 10–5-1 × 10–4 M) was applied to the serosal solution. In Fig. A, data are presented as the mean ± S.E. of values determined every 10 min from 4 ~ 7 mice. *Significantly different from the control, at P < 0.05. Fig. B shows the total net HCO3- output for 1 hour after the application of CCh, and data are presented as the mean ± S.E. from 4–7 mice. *Significantly different from the control, at P < 0.05. |
 |
Fig. 3. Effects of various drugs on the HCO3- response to CCh in isolated duodenums of WT mice. CCh (1 × 10-4 M) was applied to the serosal solution to stimulate HCO3- secretion. Atropine (1 × 10-6 M), verapamil (3 × 10-5 M), indomethacin (1 × 10-5 M), L-NAME (1 × 10-3 M), or tetrodotoxin (1 × 10-5 M) was added to the serosal solution 30 min before the application of CCh. Data show the total net HCO3- output for 1 hour after the application of CCh and are presented as the mean ± S.E. from 4–7 mice. Significantly different at P < 0.05; *from control; # from vehicle. |
Effects of various subtype-selective muscarinic acetylcholine receptors antagonists on carbachol-induced duodenal HCO3– secretion in wild-type mice in vitro
Since the HCO3– stimulatory action of CCh was almost completely inhibited by atropine, the nonselective antagonist of mAChR (Fig. 3), these results confirmed that CCh stimulated HCO3– secretion via the activation of mAChRs. We then investigated the subtype(s) of mAChRs mediating the CCh-stimulated secretion of HCO3– by examining the effects of various subtype-selective mAChR antagonists on the HCO3– response.
The CCh-stimulated secretion of HCO3– in the isolated duodenums of WT mice was significantly attenuated by pirenzepine (the M1 antagonist; 3 × 10–5 M) as well as 4-DAMP (the M3 antagonist; 3 × 10–6 M), with inhibition being 54.2% and 76.4%, respectively (Fig. 4). This response in WT mice was also potently inhibited by tropicamide (the M4 antagonist; 3 × 10–6 M), with inhibition being 65.8%, which was similar to that caused by 4-DAMP. However, methoctramine (the M2 antagonist; 1 × 10–5 M) had no effect on the HCO3– response induced by CCh.
 |
Fig. 4. Effects of various subtype-selective mAChR antagonists on the HCO3- response to CCh in isolated duodenums of WT mice. CCh (1 × 10–4 M) was applied to the serosal solution to stimulate HCO3- secretion. Pirenzepine (3 × 10–5 M), methoctramine (1 × 10-5 M), 4-DAMP (3 × 10–6 M), or tropicamide (3 × 10–6 M) was added to the serosal solution 30 min before the application of CCh. Data show the total net HCO3- output for 1 hour after the application of CCh and are presented as the mean ± S.E. from 4–7 mice. Significantly different at P < 0.05, *from control, # from vehicle. |
Effects of carbachol on duodednal HCO3– secretion in muscarinic acetylcholine receptors knockout mice in vitro
To further determine which subtype(s) of mAChRs was involved in the regulation of duodenal HCO3– secretion, we examined the effects of CCh on HCO3– secretion in the isolated duodenums of WT mice as well as those of mAChR KO mice lacking M1, M2, M3, M4, or M5 receptors. The duodenums of both WT and various mAChR KO mice consistently secreted 0.25 ~ 0.40 µEq/h of HCO3– as basal secretion, and no significant difference was observed in the basal rates of the duodenums of these animals. The stimulatory action of CCh (1 × 10–4 M) was similar between mAChR KO mice lacking M2 and M5 receptors and WT mice, with the ΔHCO3– outputs being 0.172 ± 0.030 µEq/h and 0.156 ± 0.029 µEq/h, respectively, which were almost equivalent to that (0.170 ± 0.032 µEq/h) in WT mice (Fig. 5). In contrast, the response to CCh was significantly decreased in M1 and M3 KO mice, with the ΔHCO3– outputs being 0.082 ± 0.018 µEq/h and 0.040 ± 0.012 µEq/h, respectively. CCh-induced HCO3– secretion was markedly decreased in M4 KO mice, with the ΔHCO3– output being 0.038 ± 0.009 µEq/h, which was significantly lower than that in WT mice.
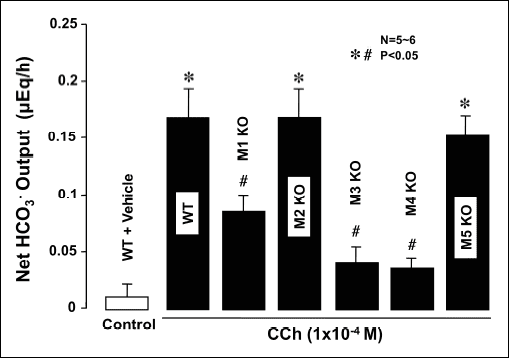 |
Fig. 5. Effects of CCh on HCO3- secretion in isolated duodenums of WT mice and those of KO animals lacking M1-M5 receptors. CCh (1 × 10–4 M) was applied to the serosal solution. Data show the total net HCO3- output for 1 hour after the application of CCh and are presented as the mean ± S.E. from 5–6 mice. Significantly different at P < 0.05, *from control (WT + vehicle), # from CCh in WT. |
Effects of somatostatin on duodenal HCO3– secretion in wild-type and muscarinic acetylcholine receptors-knockout mice in vitro
Odes et al. (24) previously reported that somatostatin inhibited both the basal and CCh-induced duodenal secretion of HCO3– in guinea pigs. Since the release of somatostatin from D cells is known to be mediated by a decrease in cAMP (20), and the activation of M4 receptors is coupled with Gi protein to inhibit adenylate cyclase (17), it is possible that the decreased HCO3– response observed in M4-KO mice may be associated with changes in somatostatin secretion. Therefore, we examined the effects of octreotide (an analogue of somatostatin) on duodenal HCO3– secretion under basal and CCh-stimulated conditions.
The basal secretion of HCO3– in the duodenum of WT mice was significantly decreased for approximately 40 min following the serosal addition of octreotide (1 × 10–6 M), and this was followed by a return to the levels observed prior to its application; the decrease in ΔHCO3– output was 0.047 ± 0.011 µEq/h (Fig. 6A and 6B). The duodenal HCO3– response induced by CCh (1 × 10–4 M) was also significantly reduced by the post-addition of octreotide (1 × 10–6 M); HCO3– secretion was decreased from 0.085 ± 0.003 µEq/10 min to the lowest value of 0.049 ± 0.002 µEq/10 min, and this effect persisted for 40 min (Fig. 7). This inhibitory effect of octreotide was almost completely abrogated when CYN154806 (1 × 10–5 M), a SST2 antagonist, was added 30 min before the application of octreotide.
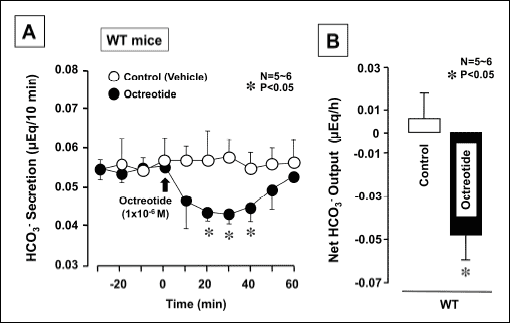 |
Fig. 6. Effects of octreotide on basal HCO3- secretion in isolated duodenums of WT mice. Octreotide (1 × 10–6 M), an analogue of somatostatin-14, was added to the serosal solution. In Fig. A, data are presented as the mean ± S.E. of values determined every 10 min from 5–6 mice. *Significantly different from control (vehicle), at P < 0.05. Fig. B shows the total net HCO3- output for 1 hour after the addition of octreotide, and data are presented as the mean ± S.E. from 5–6 mice. *Significantly different from control, at P < 0.05. |
 |
Fig. 7. Effects of octreotide on the HCO3- stimulatory action of CCh (1 × 10–4 M) in isolated duodenums of WT mice. CCh (1 × 10–4 M) was applied to the serosal solution. Octreotide (1 × 10–6 M) was added to the serosal solution 1 hour after the application of CCh, while CYN154806 (1 × 10–5 M) was added serosally 30 min before octreotide. Data are presented as the mean ± S.E. of values determined every 10 min from 5–6 mice. Significantly different at P < 0.05, *from control, #from CCh + vehicle, $from CCh + octreotide. |
Effects of the somatostatin SST2 receptor antagonist CYN154806 on duodenal HCO3– secretion in M4 konockout mice in vitro
We herein demonstrated that octreotide, an exogenous somatostatin analogue, significantly inhibited both the basal and CCh-stimulated secretion of HCO3– in the isolated duodenums of WT mice in vitro. Krempels et al. (35) reported that the surface epithelium of the rat duodenum strongly expressed the mRNA of the somatostatin SST2 receptor subtype. Therefore, to investigate the involvement of endogenous somatostatin in the decreased HCO3– response to CCh in M4 KO mice, we examined the effects of CYN154806, a SST2 receptor antagonist, on the CCh- stimulated HCO3– response in M4 KO mice.
The application of CCh (1 × 10–4 M) in the serosal solution did not significantly increase HCO3– secretion in the isolated duodenums of M4 KO mice; the net HCO3– output was 0.018 ± 0.003 µEq/h, which was almost equivalent to that in the control duodenums treated with saline (Fig. 8A and 8B). However, when the duodenums of M4 KO mice were pretreated with CYN154806 (1 × 10–6 M), the application of CCh significantly increased HCO3– secretion, with the net HCO3– output being 0.119 ± 0.035 µEq/h, which was significantly greater than that of vehicle-treated duodenums.
 |
Fig. 8. Effects of CYN154806 (1 × 10–6 M) on the HCO3- stimulatory action of CCh (1 × 10–4 M) in isolated duodenums of M4 KO mice. CCh (1 × 10–4 M) was applied to the serosal solution. CYN154806 was added to the serosal solution 30 min before the application of CCh. In Fig. A, data are presented as the mean ± S.E. of values determined every 10 min from 4–7 mice. Fig. B shows the total net HCO3- output for 1 hour after the application of CCh, and data are presented as the mean ± S.E. from 4–7 mice. * Significantly different from vehicle, at P < 0.05. |
Immunostaining of somatostatin and M4 receptors in wild-type mouse duodenum
To confirm the presence of M4 receptors in D cells, we performed immunostaining on the duodenal mucosa of WT mice with anti-somatostatin and anti-M4 receptor antibodies. As expected, the expression of somatostatin was apparently observed in the duodenum; it was stained red (Fig. 9). On the other hand, immunostaining of M4 receptors was also observed in the same area of the duodenum; a higher magnification revealed the co-existence of M4 receptors with somatostatin (a lower panel), suggesting the expression of M4 receptors on D cells.
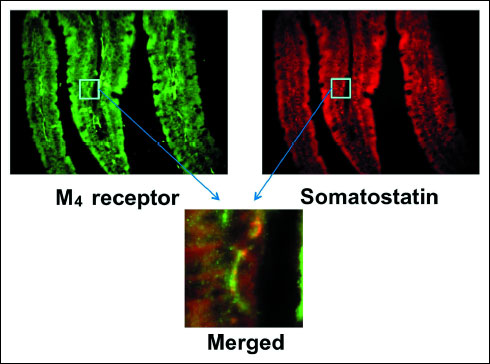 |
Fig. 9. Fluorescence immunochemical staining of the duodenal mucosa with anti-somatostatin and anti-M4 receptor antibodies in WT mice. Somatostatin stained red, while the M4 receptor stained green. The lower panel shows higher magnification to indicate the co-expression of M4 receptors with somatostatin on D cells as visualized by yellow in the merged image. |
Gene expression of muscarinic acetylcholine receptors subtypes in mouse duodenum
Since the M1, M3, and M4 receptors were found to be involved in the stimulatory effects of CCh on duodenal HCO3– secretion, we examined the gene expression of the mAChR subtypes, M1-M5, in the mouse duodenum. The expression of M1-M5 mRNAs was observed in the duodenums of WT mice, similar to the mouse brain, with the expression of the M3 and M4 receptors being markedly higher (Fig. 10). The expression of the mAChR subtypes was also observed in the duodenum of KO mice lacking M1-M5 receptors, except for those lacking the respective mAChR subtype in the corresponding KO mice.
 |
Fig. 10. Gene expressions of mAChR subtypes (M1-M5) in the mouse duodenum. A respective mAChR subtype was lacking in the corresponding KO mice lacking M1-M5 receptors in the duodenum. |
 |
Fig. 11. Working hypothesis for the involvement of M4 receptors in the regulatory mechanism underlying the cholinergic stimulation of duodenal HCO3- secretion. The cholinergic stimulation of HCO3- secretion is mediated by the activation of M1, M3, and M4 receptors. In this case, the activation of M1 and M3 receptors may directly affect HCO3- secretion from surface epithelial cells through a Gq protein/Ca2+ pathway, while the activation of M4 receptors inhibits endogenous somatostatin secretion from D cells and, by doing so, unmasks the stimulatory effects of CCh mediated by the activation of M1 and M3 receptors. |
DISCUSSION
The secretion of HCO3– from the surface epithelium is one of the mucosal defensive mechanisms and plays an important role in protecting the gastroduodenal mucosa against acid (1-4, 13). This secretion was previously shown to be regulated by both humoral and neuronal factors, including endogenous PGs, NO, and capsaicin-sensitive afferent neurons (1, 4, 10, 35, 36), and intracellularly mediated by cAMP and cGMP as well as Ca2+ (1, 5, 6, 9, 29, 38, 39). Unlike the cellular mechanisms involved in the HCO3– response to PGs and NO, the cholinergic regulation of duodenal HCO3– secretion currently remains unexplored, including the mAChR subtypes involved in this response. In the present study, we demonstrated for the first time the involvement of muscarinic M4 receptors in the regulatory mechanism underlying the cholinergic stimulation of duodenal HCO3– secretion.
We found that the application of CCh, a muscarinic agonist, to the serosal solution dose-dependently increased HCO3– secretion in in vitro preparations of isolated duodenums from WT mice, and this response was almost completely inhibited by the prior addition of atropine, but not by indomethacin or L-NAME. These results confirmed that CCh stimulated duodenal HCO3– secretion mediated by the cholinergic system via the activation of mAChR, and neither endogenous PGs nor NO are involved in this response. We also showed that CCh-stimulated duodenal HCO3– secretion was significantly abrogated by pirenzepine, 4-DAMP, and tropicamide, while methoctramine had no effect. We could not obtain any information on the involvement of M5 receptors because no antagonist for M5 receptors is currently available. These results suggested that CCh-induced duodenal HCO3– secretion occurred through the activation of M1, M3, and M4 receptors. Previous studies reported that peripheral M1 receptors were involved in the regulation of duodenal HCO3– secretion; however, these findings were not without controversy (5, 7, 40). Safsten et al. (7, 40) demonstrated that pirenzepine and telenzepine by themselves caused a dose-dependent increase in duodenal HCO3– secretion in rats, and this response was attenuated by vagotomy or a pretreatment with phentolamine, an a-adrenoceptor antagonist. Based on these results, they suggested that these M1 antagonists had stimulatory effects by antagonizing muscarinic M1 transmission at peripheral sympathetic ganglia and removing postsynaptic adrenergic inhibition (40). However, we herein showed that pirenzepine had no effect on basal HCO3– secretion in vitro because of the lack of extrinsic nerves in the isolated duodenal preparation, but significantly inhibited the HCO3– response to CCh in this preparation. Xie et al. (41) reported that gastric chief cells express both M1 and M3 receptors and that the presence of either M1 or M3 receptors is sufficient to allow for robust CCh-induced pepsinogen secretion. Other studies also indicated co-expression of M1 and M3 receptors in rat sublingual glands and pancreatic acinar cells (42, 43).
Consistent with the results obtained by subtype-selective muscarinic antagonists, we also found using KO mice lacking M1-M5 receptors that CCh stimulated HCO3– secretion in M2 and M5 KO animals as effectively as in WT mice; however, this stimulatory effect was significantly attenuated in M1, M3, and M4 KO mice. Therefore, we assumed that the cholinergic stimulation of duodenal HCO3– secretion was mediated by the activation of M1, M3, and M4 receptors. The most important finding in the present study was that M4 receptors were involved in the process of CCh-stimulated duodenal HCO3– secretion, in addition to M1 and M3 receptors.
Since M1 and M3 receptors have been shown to mediate various secretory functions in the gastrointestinal tract, such as the secretion of acid, pepsin, and saliva (16, 17), it is reasonable to assume that these subtypes of mAChR play a role in the regulatory mechanism underlying HCO3– secretion. These receptors are coupled to the Gq/11 protein to increase intracellular Ca2+ levels, which are known to mediate the secretion of HCO3– and acid as well as many hormones (17). The present results together with our previous findings (5) showed that HCO3– secretion in response to CCh was significantly attenuated by verapamil, a Ca2+ channel blocker, thereby supporting the involvement of M1 and M3 receptors in this response. Although information regarding the Ca2+ entry pathway in non-excitable cells, including the surface epithelium, that do not express voltage-dependent Ca2+ channels is limited, Simson and Silen (38) reported, using the isolated amphibian duodenum, that HCO3– secretion was increased by addition of the Ca2+ ionophore, A23187, to the serosal solution. On the other hand, it currently remains unknown how the activation of M4 receptors modulates (facilitates) the HCO3– response to CCh in the duodenum. Since M4 receptors are coupled to the Gi protein in order to decrease intracellular cAMP production (17), and duodenal HCO3– secretion is intracellularly mediated by cAMP, in addition to Ca2+ (1, 2, 38, 44), it is unlikely that M4 receptors are expressed in epithelial cells and directly mediate the stimulation of HCO3– secretion in response to CCh. Therefore, we assumed that these receptors were expressed in cells other than HCO3–-secreting cells (the surface epithelium) and indirectly affected the HCO3– response to cholinergic stimulation.
Somatostatin is synthesized in various organs of the mammalian body and almost ubiquitously exerts an inhibitory effect on various physiological processes mediated by SST2 receptors (20, 45). In the gastrointestinal tract, this peptide has been shown to inhibit motility and secretory functions and antagonize the actions of several hormones (6, 21, 24, 46). A previous study demonstrated that the duodenum abundantly expressed D cells, which are responsible for the release of somatostatin (47). A few in vivo and in vitro studies reported that somatostatin significantly influenced duodenal HCO3– secretion (24, 48), however, its effects remain controversial. Odes et al. (24) showed that somatostatin-14, given intravenously, inhibited basal and CCh-stimulated duodenal HCO3– secretion in guinea pigs, and its mechanism of action was not via the inhibition of adenylate cyclase activity in duodenal enterocytes. However, Lenz and Forquignon (48) reported that the cerebroventricular administration of somatostatin-28 stimulated duodenal HCO3– secretion in rats via vagal efferents by the release of and, in part, by a cholinergic pathway. We found that octreotide, an analogue of somatostatin-14, significantly suppressed basal and CCh-stimulated HCO3– secretion in the mouse duodenum in vitro.
Chiba and Yamada (25) reported that CCh inhibited both basal and pentagastrin-stimulated somatostatin secretion in a Gi protein/cAMP-dependent manner in isolated canine D cells. Five subtypes, SST1-SST5, of somatostatin receptors are currently known to exist, all of which are expressed in the gastrointestinal tract (35, 49, 50). Therefore, the activation of M4 receptors may inhibit the secretion of somatostatin secretion from D cells and, by doing so, indirectly affect HCO3– secretion. If this is the case, the following should be demonstrated; 1) somatostatin suppresses the HCO3– response to cholinergic stimulation such as CCh, 2) the decreased HCO3– response in M4 KO mice can be reversed by an SST2 antagonist, and 3) the release of somatostatin from D cells is increased in M4 KO mice. We found that the HCO3– response to CCh was markedly suppressed by octreotide, a somatostatin analogue. We also showed that the suppressed HCO3– response to CCh in M4 KO mice was significantly restored by the prior application of the somatostatin SST2 antagonist, CYN154806, and the total net HCO3– output was almost the same as that in WT mice. We previously reported that serum somatostatin levels were increased in M4 KO mice under basal conditions and that carbachol significantly decreased somatostatin levels in WT animals; this effect was completely negated in M4 KO mice (26). These results strongly support our hypothesis that a decrease in the HCO3– response in M4 KO mice is explained by the inhibitory effects of somatostatin mediated by SST2 receptors. Therefore, we assumed that the activation of M4 receptors inhibited somatostatin release from D cells and removed the negative influence of this peptide on HCO3– secretion, resulting in the potentiation of the HCO3– response to CCh (22, 24). Although the mechanism underlying the stimulation of somatostatin release currently remains unknown, this process may be positively affected by cholinergic stimulation via the activation of mAChR subtype(s) other than M4 receptors, in addition to gastrin (20).
It has yet to be established whether M4 receptors are actually expressed on D cells. We performed immunostaining of the duodenal mucosa with anti-somatostatin and anti-M4 receptor antibodies in WT mice. The results obtained clearly showed that M4 receptors were co-expressed with somatostatin, indicating the expression of M4 receptors on D cells. We confirmed that M4 receptors were not observed in the duodenums of M4 KO mice. These results strongly suggested that CCh inhibited somotostatin release from D cells via the activation of M4 receptors. We intend to measure the tissue levels of somatostatin before and after the addition of CCh, demonstrate that CCh significantly decreases somatostatin levels in WT mice, and show that this effect is negated in the presence of the M4 antagonist or in M4 KO mice.
The results of the present study indicated that the cholinergic stimulation of duodenal HCO3- secretion was mediated by the activation of M1/M3 receptors and modified indirectly by M4 receptors. We assumed that the activation of M4 receptors inhibited the release of somatostatin from D cells, thereby enhancing the HCO3– response by removing the negative influence of somatostatin via the activation of SST2 receptors (Fig. 11).
Acknowledgements: We are greatly indebted to Dr. Kikuko Amagase and the undergraduate students at the Department of Pharmacology and Experimental Therapeutics, Kyoto Pharmaceutical University for her valuable advice and their technical assistance, respectively.
Conflict of interests: None declared.
REFERENCES
- Flemstrom G, Garner A. Gastroduodenal HCO3– transport: characteristics and proposed role in acidity regulation and mucosal protection. Am J Physiol 1982; 242: G183-G193.
- Takeuchi K, Furukawa O, Tanaka H, Okabe S. A new model of duodenal ulcers induced in rats by indomethacin plus histamine. Gastroenterology 1986; 90: 636-645.
- Flemstrom G. Gastric and duodenal mucosal bicarbonate secretion. In: Physiology of the Gastrointestinal Tract, LR Johnson, J Cristensen, MI Grossman, ED Jacobson, SG Schultz (eds). New York, Raven Press, 1987, pp. 1011-1034.
- Allen A, Flemstrom G, Garner A, Kivilaakso E. Gastroduodenal mucosal protection. Physiol Rev 1993; 73: 823-857.
- Takeuchi K, Niida H, Okabe S. Characterization of alkaline secretion induced by cholinergic agents in the rat duodenum: Involvement of mucarinic M2 receptors and Ca2+-dependent process. J Pharmacol Exp Ther 1990; 254: 465-470.
- Odes HS, Muallem R, Reimer R, Beil W, Schwenk M, Sewing KF. Cholinergic regulation of guinea pig duodenal bicarbonate secretion. Am J Physiol 1993; 265: G270-G276.
- Safsten B, Jedstedt G, Flemstrom G. Cholinergic influence on duodenal mucosal bicarbonate secretion in the anesthetized rat. Am J Physiol 1994; 267: G10-G17.
- Takeuchi K, Yagi K, Kato S, Ukawa H. Roles of prostaglandin E-receptor subtypes in gastric and duodenal bicarbonate secretion in rats. Gastroenterology 1997; 113: 1553-1559.
- Furukawa O, Kitamura M, Sugamoto S, Takeuchi K. Stimulatory effect of nitric oxide on bicarbonate secretion in bullfrog duodenums in vitro. Digestion 1999; 60: 324-331.
- Sugamoto S, Kawauch S, Furukawa O, Mimaki TH, Takeuchi K. Role of endogenous nitric oxide and prostaglandin in duodenal bicarbonate response induced by mucosal acidification in rats. Dig Dis Sci 2001; 46: 1208-1216.
- Takeuchi K, Ukawa H, Furukawa O, et al. Prostaglandin EP receptor subtypes involved in gastroduodenal bicarbonate secretion. J Physiol Pharmacol 1999; 50: 155-168.
- Aihara E, Nomura Y, Sasaki Y, Ise F, Kita T, Takeuchi K. Involvement of prostaglandin E receptor EP3 subtype in duodenal bicarbonate secretion in rats. Life Sci 2007; 80: 2446-2453.
- Aihara E, Kagawa S, Hayashi M, Takeuchi K. ACE inhibitor and ATI antagonist stimulate duodenal HCO3–secretion mediated by a common pathway: involvement of PG, NO and bradykinin. J Physiol Pharmacol 2005; 56: 391-406.
- Matsui M, Motomura D, Karasawa H, et al. Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc Natl Acad Sci USA 2000; 97: 9579-9584.
- Matsui M, Yamada S, Oki T, Manabe T, Taketo MM, Ehlert FJ. Functional analysis of muscarinic acetylcholine receptors using knockout mice. Life Sci 2004; 75: 2971-2981.
- Tobin G, Giglio D, Lundgren O. Muscarinic receptor subtypes in the alimentary tract. J Physiol Pharmacol 2009; 60: 3-21.
- Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev 1998; 50: 279-290.
- Aihara T, Nakamura Y, Taketo MM, Natsui M, Okabe S. Cholinergically stimulated gastric acid secretion is mediated by M3 and M5 but not M1 muscarinic acetylcholine receptors in mice. Am J Physiol 2005; 288: G1199-G1207.
- Takahashi N, Yamazaki Y, Amagase K, Takeuchi K. Activation of muscarinic acetylcholine receptor subtype 4 is essential for carbachol-induced acid secretion in mice: relation to D cells/somatostatin. Gastroenterology 2009; 136: A163.
- Lucey MR, Yamada T. Biochemistry and physiology of gastrointestinal somatostatin. Dig Dis Sci 1989; 34 (Suppl. 3), 5S-13S.
- Konturek PC, Brzozowski T, Konturek SJ. Gut clock: implication of circadian rhythms in the gastrointestinal tract. J Physiol Pharmacol 2011; 62: 139-150.
- Warhurst G, Higgs NB, Fakhoury H, Warhurst AC, Garde J, Coy DH. Somatostatin receptor subtype 2 mediates somatostatin inhibition of ion secretion in rat distal colon. Gastroenterology 1996; 111: 325-333.
- Patel YC. Molecular pharmacology of somatostatin receptor subtypes. J Endocrinol Invest 1997; 20: 348-367.
- Odes HS, Muallem R, Reimer R, et al. Effect of somatostatin-14 on duodenal mucosal bicarbonate secretion in guinea pigs. Dig Dis Sci 1995; 40: 678-684.
- Chiba T, Yamada T. Mechanisms for muscarinic inhibition of somatostatin release from canine fundic D cells. Metabolism 1990; 39: 122-124.
- Takeuchi K, Hayashi S, Amagase K, Kato S, Okabe S, Matsui M. Activation of muscarinic acetylcholine receptor subtype 4 is essential for carbachol-induced acid secretion in mice: Relation to D cells/somatostatin. Ulcer Res 2012; 39: 10-12.
- Ohno-Shosaku T, Matsui M, Fukudome Y, et al. Postsynaptic M1 and M3 receptors are responsible for the muscarinic enhancement of retrograde endocannabinoid signalling in the hippocampus. Eur J Neurosci 2003; 18: 109-116.
- Nakamura T, Matsui M, Uchida K, et al. M3 muscarinic acetylcholine receptor plays a critical role in parasympathetic control of salivation in mice. J Physiol 2004; 558: 561-575.
- Takeuchi K, Ukawa H, Kato S, et al. Impaired duodenal bicarbonate secretion and mucosal integrity in mice lacking prostaglandin E receptor subtype EP3. Gastroenterology 1999; 117: 1128-1135.
- Hayashi M, Kita T, Ohashi Y, Aihara E, Takeuchi K. Phosphodiesterase isozymes involved in regulation of HCO3– secretion in isolated mouse duodenum in vitro. Biochem Pharmacol 2007; 74: 1507-1513.
- Tuo B, Riederer B, Wang Z, Colledge WH, Soleimani M, Seidler U. Involvement of the anion exchanger SLC26A6 in prostaglandin E2– but not forskolin- stimulated duodenal HCO3– secretion. Gastroenterology 2006; 130: 349-358.
- Jakubik J, Zimcik P, Randakova A, Fuksova K, El-Fakahany EE, Dolezal V. Molecular mechanisms of methoctramine binding and selectivity at muscarinic acetylcholine receptors. Mol Pharmacol 2014; 86: 180-192.
- Erosa-Rivero HB, Bata-Garcia JL, Alvarez-Cervera FJ, Heredia-Lopez FJ, Gongora-Alfaro JL. The potency and efficacy of anticholinergics to inhibit haloperidol-induced catalepsy in rats correlates with their rank order of affinities for the muscarinic receptor subtypes. Neuropharmacology 2014; 81: 176-187.
- Feniuk W1, Jarvie E, Luo J, Humphrey PP. Selective somatostatin sst (2) receptor blockade with the novel cyclic octapeptide, CYN-154806. Neuropharmacology 2000; 39: 1443-1450.
- Krempels K, Hunyady B, O'Carroll AM, Mezey E. Distribution of somatostatin receptor messenger RNAs in the rat gastrointestinal tract. Gastroenterology 1997; 112: 1948-1960.
- Heylings JR, Garner A, Flemstrom G. Regulation of gastroduodenal HCO3–transport by luminal acid in the frog in vitro. Am J Physiol 1984; 246: G235-G242.
- Takeuchi K, Matsumoto J, Ueshima K, Okabe S. Role of capsaicin-sensitive afferent neurons in alkaline secretory response to luminal acid in the rat duodenum. Gastroenterology 1991; 101: 954-961.
- Simson JN, Merhav A, Silen W. Alkaline secretion by amphibian duodenum. III. Effect of DBcAMP, theophylline, and prostaglandins. Am J Physiol 1981; 241: G528-G536.
- Seidler U, Blumenstein I, Kretz A, et al. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca2+-dependent HCO3–secretion. J Physiol 1997; 505: 411-423.
- Safsten B, Flemstrom G. Stimulatory effect of pirenzepine on mucosal bicarbonate secretion in rat duodenum in vivo. Acta Physiol Scand 1986; 127: 267-268.
- Xie G, Drachenberg C, Yamada M, Wess J, Raufman JP. Cholinergic agonist-induced pepsinogen secretion from murine gastric chief cells is mediated by M1 and M3 muscarinic receptors. Am J Physiol 2005; 289: G521-G529.
- de la Vega MT, Nunez A, Arias-Montano JA. Carbachol-induced inositol phosphate formation in rat striatum is mediated by both M1 and M3 muscarinic receptors. Neurosci Lett 1997; 233: 69-72.
- Schmid SW, Modlin IM, Tang LH, Stoch A, Rhee S, Nathanson MH, Scheele GA, Gorelick FS. Telenzepine-sensitive muscarinic receptors on rat pancreatic acinar cells. Am J Physiol 1998; 274: G734-G741.
- Hogan DL, Crombie DL, Isenberg JI, Svendsen P, Schaffalitzky de Muckadell OB, Ainsworth MA. CFTR mediates cAMP- and Ca2+-activated duodenal epithelial HCO3–secretion. Am J Physiol 1997; 272: G872-G878.
- Stengel A, Goebel M, Wang L, et al. Selective central activation of somatostatin receptor 2 increases food intake, grooming behavior and rectal temperature in rats. J Physiol Pharmacol 2010; 61: 399-407.
- Warhurst G, Turnberg LA, Higgs NB, Tonge A, Grundy J, Fogg KE. Multiple G-protein-dependent pathways mediate the antisecretory effects of somatostatin and clonidine in the HT29-19A colonic cell line. J Clin Invest 1993; 92: 603-611.
- Holle GE, Spann W, Eisenmenger W, Riedel J, Pradayrol L. Diffuse somatostatin-immunoreactive D-cell hyperplasia in the stomach and duodenum. Gastroenterology 1986; 91: 733-739.
- Lenz HJ, Forquignon I. Stimulation of duodenal bicarbonate secretion in conscious rats by cerebral somatostatin-28. Role of neurohumoral pathways. Gastroenterology 1990; 99: 340-344.
- Schafer J, Meyerhof W. sst1 mRNA is the prominent somatostatin receptor mRNA in the rat gastrointestinal tract: reverse transcription polymerase chain reaction and in situ-hybridization study. Neuropeptides 1999; 33: 457-463.
- Piqueras L, Martinez V. Role of somatostatin receptors on gastric acid secretion in wild-type and somatostatin receptor type 2 knockout mice. Naunyn Schmiedebergs Arch Pharmacol 2004; 370: 510-520.
A c c e p t e d : February 18, 2015