ATORVASTATIN-INDUCED ENDOTHELIAL NITRIC OXIDE SYNTHASE EXPRESSION IN ENDOTHELIAL CELLS IS MEDIATED BY ENDOGLIN
INTRODUCTION
Endoglin (CD105, TGF-β receptor III, ENG), is a 180-kDa transmembrane glycoprotein, which serves as an accessory receptor for transforming growth factor β (TGF-β). Endoglin is proposed to be a multipotent protein associated with vascular vital/physiological and pathophysiological processes such as angiogenesis (1) and inflammation (2). Increased endoglin expression has been observed in proliferating endothelial cells in the tumor vasculature (3) and importantly in microvessels of advanced carotid atherosclerotic lesions (4), in aortic endothelium of non-atherosclerotic (5) and atherosclerotic aorta in mice (6). Of note atherosclerosis is considered as a chronic inflammatory disease of arteries and it is characterized by increased levels of pro-inflammatory cytokines including for example TNF-α (7).
Endoglin expression is strongly related to expression and activity of endothelial nitric oxide synthase (eNOS) (8), a key enzyme for the proper function of the vascular endothelium (9). Indeed, altered expression of eNOS is involved in endothelial dysfunction and atherogenesis (10). Endoglin regulates eNOS expression/activity, thus modulating endothelial-dependent vascular tone (8, 11). In this context, endoglin-dependent regulation of eNOS involves the phosphorylation and activation of Smad transcription factors, in particular Smad2, which is involved in the expression of eNOS in endothelial cells (12).
In various pathological conditions, including preeclampsia, atherosclerosis and diabetes mellitus, increased levels of
a soluble form of endoglin, a blood biomarker related to endothelial dysfunction, are present in plasma (13, 14). Soluble endoglin is generated by the cleavage of the extracellular domain from the intact membrane endoglin by matrix metalloproteinase-14 (MMP-14) (15, 16), a proteolytic process that can be inhibited by statin treatment as shown in a mouse model of atherosclerosis (17).
Statins, competitive inhibitors of HMG-CoA reductase, are used predominantly for hypercholesterolemia treatment. The underlying mechanism of statin action includes not only the regulation of plasma lipoproteins, but also effects on plaque cellular components and their stability, as well as the development of thrombosis and inflammation. Moreover, a role for statins in endothelial function, beyond the lipid lowering effect, has been described, including the statins-induced increase of eNOS expression (18-20). It is of interest to mention even negative effects of statin treatment in metabolic syndrome (21) and at the risk of statin-induced type II diabetes manifestation (22). Our previous studies in mouse atherosclerotic models showed that endoglin is strongly co-expressed with eNOS in vascular aortic endothelium covering atherosclerotic plaques and that atorvastatin (ATV) is able to increase both endoglin and eNOS expression and reduce plaque size beyond its lipid lowering effects (23). However, the mechanisms, by which ATV increases eNOS expression, with potential involvement of endoglin in this process, remain to be elucidated. In this study, we hypothesized whether inflammatory conditions modulate ATV induction of endoglin and eNOS expression in endothelial cells and whether ATV-induced eNOS expression is regulated via endoglin.
MATERIALS AND METHODS
Reagents
The majority of reagents was purchased from Sigma-Aldrich Co. (St. Louis, USA). The stock solution of 10 mM atorvastatin (calcium salt trihydrate) in dimethylsulfoxide (DMSO) was prepared and aliquots were stored at –20°C. The volume of DMSO in culture medium never exceeded 0.1% (v/v). The stock solution of 10 µg/mL human recombinant tumor necrosis factor-alpha (TNF-α) was prepared in PBS buffer. The stock solutions of 10 mM SB431542 (hydrate), an inhibitor of the TGF-β receptor type I ALK5, and 10 mM H2DCF-DA (2´,7´-dichlorodihydrofluorescein diacetate) were prepared in DMSO.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were grown in EBM-2 medium containing 10% fetal bovine serum (FBS) (PAA Lab., Yeovil, UK) and supplemented with EGM-2 SingleQuots (Lonza, Walkersville, USA). For starving conditions, HUVECs were serum starved for 3 hours in the same medium, but containing 0.3% FBS. In all experiments, HUVECs were cultured in gelatin-coated flasks. All experiments were performed with HUVECs at passages between 5 and 9.
Experimental groups
Several experimental groups were prepared including:
- In TNF-α time-dependent experiments, cells were treated with 10 ng/mL TNF-α (culture medium with DMSO 0.1% v/v) for 2, 6 or 16 hours or with medium with DMSO 0.1% v/v for 16 hours.
- In ATV time-dependent experiments, cells were treated with 5 µM ATV (culture medium with DMSO 0.1% v/v) for 4, 16 or 24 hours (4ATV, 16ATV, 24ATV, respectively), or medium with DMSO 0.1% v/v for 24 hours.
- In ATV concentration-dependent experiments, cells were treated with different concentrations of ATV (ATV 1 µM, ATV 3 µM, ATV 5 µM) for 24 hours (culture medium with DMSO 0.1% v/v) or medium with DMSO 0.1% v/v for 24 hours.
- Control cells (culture medium with DMSO 0.1% v/v, incubation for 40 hours) for TNF-α and ATV pretreatment experiments (Fig. 2).
- ATV (5 µM ATV, cell incubation for 24 hours, culture medium with DMSO 0.1% v/v, followed by culturing for 16 hours in medium with DMSO 0.1% v/v).
- TNF-α (24 hours, culture medium with DMSO 0.1% v/v, followed by 10 ng/mL TNF-α treatment for 16 hours, culture medium with DMSO 0.1% v/v).
- ATV 5 µM/TNF-α (5 µM ATV, cell pretreatment for 24 hours, followed by 10 ng/mL TNF-α treatment for 16 hours, culture medium with DMSO 0.1% v/v).
- Control GFP (cells were transfected only with pmaxGFP Vector, culture medium with DMSO 0.1% v/v for 24 hours).
- siRNA (cells were transfected with pmaxGFP Vector and siRNAs, culture medium with DMSO 0.1% v/v for 24 hours).
- siRNA - ATV 5 µM (cells were transfected with pmaxGFP Vector and siRNAs; ATV at 5 µM, cell incubation for 24 hours, culture medium with DMSO 0.1% v/v).
- Control SB431542 (SB431542 cell pretreatment for 30 minutes at 10 µM, followed by 24 hours and 16 hours SB431542 treatment).
- ATV 5 µM - SB431542 (SB431542 cell pretreatment for 30 minutes at 10 µM, followed by cell incubation for 24 hours with ATV at 5 µM together with SB431542 treatment, and 16 hours SB431542 treatment).
- In Smad phosphorylation studies, cells were treated with ATV 5 µM (0.3% FBS culture medium with DMSO 0.1% v/v) for 30 minutes or 0.3% FBS culture medium with DMSO 0,1% v/v for 30 minutes.
Immunofluorescence flow cytometry
HUVEC monolayers were rinsed with PBS, detached by trypsin and then incubated with the desired monoclonal antibody for 1 hour at 4°C, followed by incubation with the compatible secondary antibody (30 min, at 4°C in the dark). Primary antibodies included anti-human endoglin P4A4 mouse monoclonal antibody (Developmental Studies Hybridoma Bank, University of Iowa, USA), anti-human eNOS (ab76198, Abcam plc, Cambridge, UK) and anti-human VCAM-1 (BBA5, R&D Systems, Minneapolis, USA). Secondary antibodies were Alexa Fluor® 488 goat anti-mouse and Alexa Fluor® 647 goat anti-mouse (Invitrogen, Life Technologies Co., California, USA). Endoglin and VCAM-1 were detected without fixation or permeabilization. For eNOS detection, cells were fixed with 4% paraformaldehyde and permeabilized with 0.1% Tween in PBS prior immunostaining. The determination of protein expression was performed using the cell staining protocol for flow cytometry on BD Accuri C6 Flow Cytometer (BD Biosciences, San Jose, USA) and Gallios Cytometer (Beckman Coulter Inc., California, USA). A total number of 5,000 events was analyzed per sample to calculate the number of positive cells and the mean level of protein expression. Data were analyzed by Kaluza software from Beckman Coulter Inc. (California, USA). Results are presented as relative expression index, calculated as the number of positive cells of the target sample multiplied by the mean fluorescence intensity, related to control sample as previously described (24).
Western blot analysis
Samples were homogenized in RIPA lysis buffer containing proteases and phosphatases inhibitors. Aliquots of cell lysates (20 µg of total protein) were separated by SDS-PAGE and electrotransferred to a polyvinylidene difluoride (PVDF) membrane (Sigma-Aldrich Co., St. Louis, USA). The membranes were blocked for 1 hour with 5% nonfat dry milk in Tris buffered saline containing 0.1% Tween-20 (TBST), and then incubated overnight at 4°C with primary antibodies including anti-human endoglin (sc-20632, Santa Cruz Biotech., California, USA) and anti-human eNOS (sc-654, Santa Cruz Biotech., California, USA), both at 1:500 dilution, anti-human Smad2 (3122, Cell Signaling Technology, Inc., Boston, USA) at 1:1000 dilution and anti-human p-Smad2 (3108, Cell Signaling Technology, Inc., Boston, USA) at 1:100 dilution. Equal loading of total proteins was confirmed by immunodetection with anti-human β-actin (A5316, Sigma-Aldrich Co., St. Louis, USA) at 1:10,000 dilution. As secondary antibodies, horseradish peroxidase-linked goat anti-rabbit IgG-(Fab’)2 (ab6112, Abcam plc, Cambridge, UK) at 1:2000 dilution for anti-endoglin and anti-eNOS antibodies, at 1:3000 dilution for anti-Smad2 antibody, and at 1:1000 for anti-p-Smad2 antibody, and horseradish peroxidase-conjugated goat anti-mouse IgG (Sigma-Aldrich, USA) at 1:10,000 dilution for anti-β-actin antibody, were used. After washing with TBST buffer, membranes were developed using an enhanced chemiluminescent reagent (GE Healthcare, Prague, Czech Republic). Quantification of immunoreactive bands on the exposed films was performed by image analysis software NIS (Laboratory Imaging, Prague, Czech Republic).
ELISA
Levels of soluble endoglin in culture supernatant were measured by means of Quantikine Human CD105 kit (R&D Systems, Minneapolis, USA) following the manufacturer´s instructions.
Reactive oxygen species measurement
Cellular reactive oxygen species (ROS) generation was measured using H2DCFDA, which is a general indicator of ROS production and can detect hydrogen peroxide, hydroxyl radicals, and peroxynitrites, among others (25). At 10 µM H2DCF-DA diffuses passively through the cellular membranes and is then oxidized by ROS, resulting in 2´,7´-dichlorofluorescein (DCF), which is a highly fluorescent substance. HUVECs were seeded in 96-well plates and subjected to different treatments. Then, cells were washed twice with ADS buffer (pH 7.4; 116 mM NaCl, 5.3 mM KCl, 1.2 mM MgSO4, 1.13 mM NaH2PO4, 20 mM HEPES, 5 mM glucose, 1 mM CaCl2) and loaded with 10 µM H2DCF-DA in ADS buffer (loading buffer). All solutions were pre-warmed to 37°C. The fluorescence intensity was measured at the same temperature within a 30 min period using a microplate spectrophotometer TECAN Infinite 200 M (TECAN, Grodig, Austria) at λex = 485 ± 9 nm and λem = 525 ± 20 nm.
Transfection of small interfering RNA
HUVECs were transfected with pre-designed siRNAs (Ambion, Life Technologies Co., California, USA) specific for human endoglin (s4679 and s4677) by using an Amaxa HUVEC Nucleofector kit (Lonza, Walkersville, USA), in accordance with the manufacturer’s protocol. s4677 siRNA sequence (5´›3´): sense UGACCUGUCUGGUUGCACAtt, antisense UGUGCAACCAGACAGGUCAgg. s4679 siRNA sequence (5´›3´): sense CAAGUAUGAUCAGCAAUGAtt, antisense UCAUUGCUGAUCAUACUUGct. As a positive control, cells were co-transfected with a green fluorescent protein (GFP) expression vector. After 48 hours, transfection efficiency was monitored by means of fluorescence microscopy for GFP. Then, cells were incubated with 5 µM atorvastatin for 24 hours and endoglin and eNOS expressions were measured by flow cytometry, as described above.
Quantitative real-time RT-PCR
Gene expression of endoglin was examined by quantitative real-time RT-PCR as described previously (26) on 7500 HT Fast real-time PCR system (Life Technologies, Foster City, USA). Total RNA from HUVECs was isolated using TRI reagent (Sigma-Aldrich) according to the manufacturer’s protocol and directly converted into cDNA via a high capacity cDNA reverse transcription kit (Life Technologies, Foster City, USA). Thirty ng of cDNA was loaded into the reaction and experiments were performed in triplicate. The amplifications were run using TaqMan® Fast Universal PCR Master Mix and pre-designed Taq-Man® Gene Expression Assay kits for the following genes: endoglin (Hs00923996_m1) and GAPDH (Hs02758991_g1) all provided by Life Technologies (Foster City, USA). The time - temperature profile used in the ‘fast’ mode was as follows: 95°C for 3 min; 40 times: 95°C for 7 s, 60°C for 25 s. The relative expression ratio was calculated as described previously (26). The glyceraldehyde 3-phosphate dehydrogenase gene (GAPDH) was used as a reference for normalizing data.
Statistical analysis
Results are expressed as mean ± standard error of the mean (S.E.M.). All multiple comparison data were analyzed using ANOVA with post-hoc Dunn’s multiple comparisons test and direct group-group comparisons were carried out using the Mann-Whitney test. qRT-PCR results were analyzed using ANOVA, followed by multiple comparisons in Newman-Keuls post hoc test. For the above-mentioned statistical analysis, the GraphPad Prism 6.0 software (GraphPad Software, Inc., San Diego, USA) was used. P values < 0.05 were considered statistically significant.
RESULTS
Inflammation reduces endoglin and endothelial nitric oxide synthase protein expression in endothelial cells
Inflammatory conditions in HUVECs were mimicked by TNF-α treatment (10 ng/mL). Total protein expression in cells was determined using Western blot analysis after cell homogenization. Flow cytometry analysis showed no changes in endoglin expression levels after 2 or 6 hours of TNF-α treatment (data not shown). On the contrary, Western blot analysis showed that TNF-α treatment for 16 hours decreased total endoglin (Fig. 1A) and eNOS (Fig. 1B) protein expression by 41% and 46%, respectively, when compared to control samples treated with vehicle (0.1% DMSO). As a proof of an effective treatment with TNF-α, inflammation was confirmed by increased VCAM-1 expression as compared to untreated HUVECs (133.8-fold; Fig. 1C).
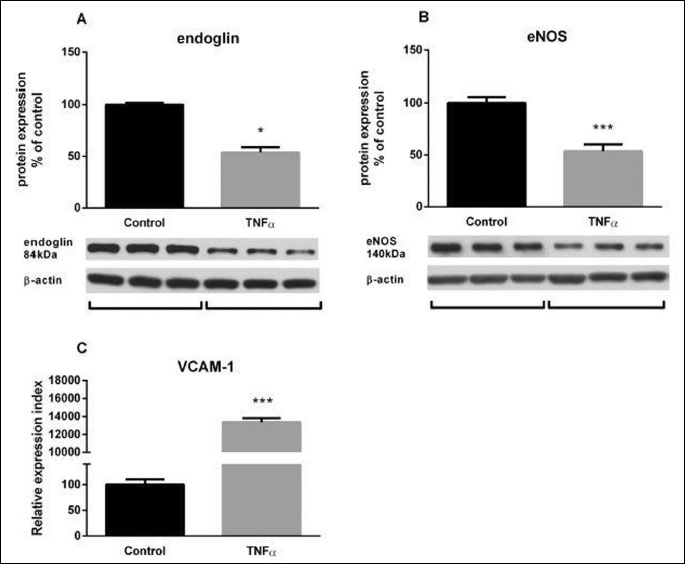
Atorvastatin increases endoglin and endothelial nitric oxide synthase expression and prevents tumor necrosis factor-α-induced endoglin and endothelial nitric oxide synthase down-regulation
HUVECs were exposed to ATV concentrations ranging from 1 µM to 5 µM for 24-hour based on previously published papers (27-29). This treatment led to a concentration-dependent increase in endoglin expression with a maximal effect occurring at 5 µM, as determined by immunofluorescence flow cytometry (Fig. 2A). Therefore, 5 µM ATV concentration was used in the following experiments in order to proof the concept of this study. To assess the kinetic expression of endoglin during ATV treatment, gene expression of endoglin at 4, 12 and 24 hours was examined (Fig. 2B). In 4ATV and 16ATV group, there was a significant increase in the endoglin expression (1.8-fold and 1.4-fold, respectively), whereas after 24 hours of ATV treatment, a decrease in the expression of endoglin was observed (0.7-fold) compared to untreated HUVECs (Fig. 2B).
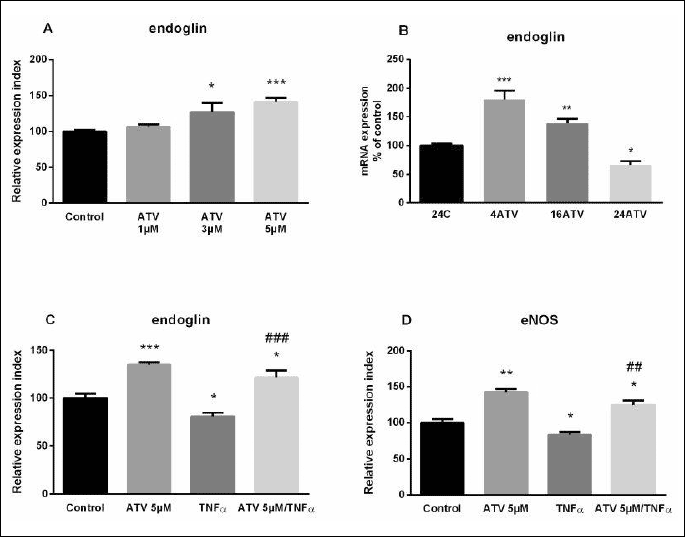
At the protein level, atorvastatin treatment resulted in a significant increase of endoglin (1.3-fold; Fig. 2C) and eNOS (1.4-fold; Fig. 2D) protein expression, as compared to control samples treated with vehicle (0.1% DMSO). TNF-α treatment for 16 hours caused a significant suppression of endoglin expression in HUVECs, when compared to control samples (0.8-fold; Fig. 2C). Similarly, to endoglin, eNOS expression was significantly decreased after TNF-α treatment for 16 hours (0.8-fold; Fig. 2D). In order to elucidate whether ATV was able to regulate endoglin and eNOS expression during inflammation, we used a pretreatment model in HUVECs, as previously described (30, 31). Cells were treated with 5 µM ATV for 24 hours, rinsed with PBS and cultured in 10 ng/mL TNF-α for 16 hours. As determined by immunofluorescence flow cytometry, ATV pretreatment significantly prevented the TNF-α-mediated decrease of endoglin and eNOS protein expression levels in comparison with cells treated only with TNF-α (both 1.5-fold; Fig. 2C and 2D).
Cooperative effects of atorvastatin and tumor necrosis factor-α on soluble endoglin levels
In order to reveal the changes of soluble endoglin levels in culture supernatants of HUVECs treated with different combinations of ATV and TNF-α, ELISA experiments were carried out. As shown in Fig. 3, atorvastatin treatment for 24 hours significantly increased soluble endoglin levels, compared to control samples treated with vehicle (1.032 ± 0.146 ng/mL vs. 1.646 ± 0.328 ng/mL) (1.6-fold; Fig. 3). Addition of TNF-α resulted in a significant increase of soluble endoglin, compared to control samples treated with vehicle (1.032 ± 0.146 ng/mL vs. 1.777 ± 0.205 ng/mL) (1.7-fold; Fig. 3). Interestingly, these values of soluble endoglin levels were even further increased upon atorvastatin pretreatment, followed by 16 hours treatment with TNF-α, when compared to samples treated only with TNF-α (1.777 ± 0.205 ng/ml vs. 2.848 ± 0.238 ng/mL) (1.6-fold; Fig. 3).
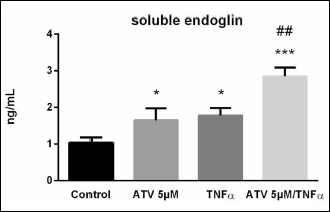 |
Fig. 3. Effect of atorvastatin and TNF-α treatment on soluble endoglin levels. ELISA analysis in culture supernatants from ATV-treated cells (5 µM ATV for 24 hours, medium for 16 hours), TNF-α-treated cells (medium for 24 hours, 10 ng/mL TNF-α for 16 hours) and ATV-pretreated cells (5 µM ATV for 24 hours, followed by 10 ng/mL TNF-α treatment for 16 hours). The culture medium in the control group contained 0.1% (v/v) of DMSO. The results are presented as concentration in ng/mL. Data are expressed as mean ± S.E.M. Each bar represents data from two independent experiments with n = 6 per experiment. *P < 0.05, ***P < 0.001 versus control samples, ##P < 0.01 versus TNF-α treated samples, analyzed by Mann-Whitney test. |
Atorvastatin prevents tumor necrosis factor-α-induced reactive oxygen species production
To test, whether ATV treatment was exerting its expected endothelial protective action, the atorvastatin effect on ROS production was assessed in HUVECs using the H2DCF-DA assay. This method revealed lower ROS levels in atorvastatin-treated cells, in comparison with control samples treated with 0.1% DMSO (mean fluorescence intensity: 189.3 ± 4.6 vs. 168.6 ± 5.9). In contrast, TNF-α treatment for 16 hours resulted in a significant increase of ROS levels when compared to control samples (mean fluorescence intensity: 189.3 ± 4.6 vs. 206.9 ± 6.0). Of note, atorvastatin pretreatment significantly prevented TNF-α-induced ROS production (mean fluorescence intensity: 206.9 ± 6.0 vs. 162.7 ± 8.0), in comparison with cells exposed only to TNF-α (0.8-fold; Fig. 4).
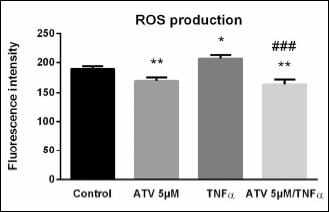 |
Fig. 4. Atorvastatin and TNF-α effect on ROS production. ROS production, measured by DCF assay, in ATV-treated cells (5 µM ATV for 24 hours, medium for 16 hours), TNF-α-treated cells (medium for 24 hours, 10 ng/mL TNF-α for 16 hours) and ATV-pretreated cells (5 µM ATV for 24 hours, followed by 10 ng/mL TNF-α treatment for 16 hours) in comparison with control cells treated with 0.1% DMSO. The results are presented as fluorescence intensity values. Data are expressed as mean ± S.E.M. Each bar represents data from two independent experiments with n = 8 per experiment. *P < 0.05, **P < 0.01 versus control samples, ###P < 0.001 versus TNF-α samples, analyzed by Mann-Whitney test. |
Suppression of endoglin, but not inhibition of transforming growth factor-β signaling, abrogates atorvastatin-induced endothelial nitric oxide synthase expression
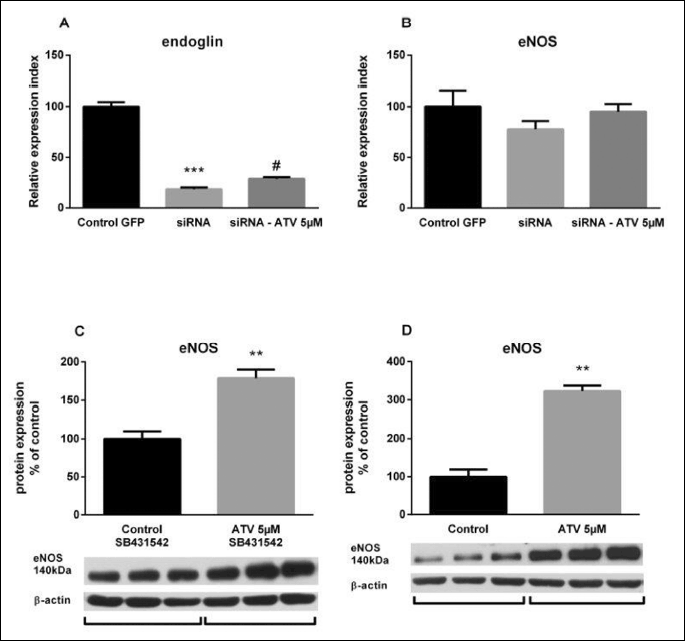
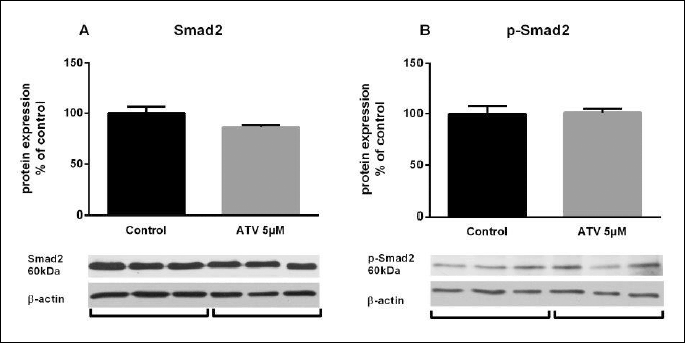
To assess the role of endoglin on the ATV-induced upregulation of eNOS, endoglin suppression experiments were carried out. A significant decrease of endoglin expression after specific silencing (siRNA column) was demonstrated, as compared to cells transfected just with the GFP vector (Control GFP column) (0.2-fold; Fig. 5A). Results are presented as relative expression index (number of positive cells for both GFP and endoglin/eNOS multiplied by fluorescence intensity mean of endoglin/eNOS). Twenty four-hour ATV treatment significantly upregulated endoglin expression in endoglin siRNAs cells (siRNA-ATV column), in comparison with siRNAs cells treated only with 0.1% DMSO (1.6-fold; Fig. 5A). Moreover, eNOS expression levels showed a mild decrease in the presence of endoglin siRNAs cells. More important was the fact that atorvastatin treatment did not result in significant increase of eNOS in endoglin siRNAs cells suggesting the importance of proper endoglin expression on atorvastatin-induced eNOS expression (Fig. 5B). Remarkably, Western blot analysis showed that 24-hour ATV treatment significantly increased eNOS expression by 74% in HUVECs incubated with SB431542 inhibitor in comparison with cells treated only with SB431542 inhibitor (Fig. 5C). Indeed twenty four-hour ATV treatment in the absence of SB431542, resulted in significant eNOS protein expression increase by 218%, when compared to control samples treated with DMSO 0.1% (Fig. 5D). In addition, 30 minutes of ATV treatment under starving conditions did not change the expression of either Smad2 (Fig. 6A) or phosphorylated Smad2 proteins (Fig. 6B).
DISCUSSION
Endoglin is a type III TGF-β receptor that modulates TGF-β signaling during various physiological and pathological conditions (32, 33). Its expression has been detected in various cell types that are considered to play important roles in the atherosclerotic process, a subject that has been reviewed recently (34). Endoglin regulates the expression and activity of eNOS via Smad2 in endothelial cells (12). Our previous papers demonstrated endoglin, eNOS and p-Smad2 co-expression in aortic endothelial cells in mouse models of atherosclerosis (6). In addition, we also demonstrated that atorvastatin is able to increase endoglin, eNOS and p-Smad2 expression in the aortic endothelium and to reduce plaque size beyond its lipid lowering effects (23). We proposed that endoglin might be involved in ATV-induced eNOS increase; however, this hypothesis could not be addressed in that model due to limitations of the in vivo experiments in mouse aorta. Thus, we hypothesized here whether endoglin expression determines ATV-induced eNOS expression under normal and inflammatory conditions in endothelial cells. Because atherosclerosis is considered an inflammatory disease that presents with high levels of inflammatory cytokines (7), we mimicked inflammatory conditions in vitro by using TNF-α. Immunofluorescence flow cytometry analysis, that measures surface expression of the extracellular domain of endoglin, showed that 16 hours of TNF-α treatment significantly reduces endoglin expression in HUVECs. This is in line with previously published data by Li et al. (2) and Rossi et al. (35). Rossi et al. showed a decrease in endoglin expression after TNF-α treatment measured by flow cytometry and an increase in the levels of endoglin at cell-cell contacts (35). On the other hand, our study showed also a decrease in total protein expression of endoglin (measured by Western blot analysis), suggesting that inflammation may not only change the localization of endoglin in the cell membrane, but may also reduce total endoglin content in the cells. TNF-α treatment also reduced eNOS expression and, as expected, markedly increased pro-inflammatory VCAM-1 expression.
It was demonstrated that membrane bound endoglin is cleaved from the intact membrane by MMP-14 in preeclampsia (36) and cancer (15). In addition, soluble endoglin might be an indicator of hypertension and diabetes-associated vascular pathologies like endothelial dysfunction and cardiovascular damage (14). Progression of atherosclerosis and hypercholesterolemia involves changes in levels of soluble endoglin in mice (37). In this study, TNF-α treatment increased levels of soluble endoglin in supernatants from cultured HUVECs. This is in line with a previously published report showing that TNF-α, and hydrogen peroxide increase soluble endoglin levels in HUVECs culture medium (38). Of note, Li et al. (2) and Rossi et al. (35) did not evaluate any changes of soluble endoglin after TNF-α treatment.
Taken together, these results suggest that inflammation reduces membrane-bound endoglin and endothelial-protective eNOS expression, and increases soluble endoglin levels in culture medium. This is actually in line with our previous in vivo study using a mouse model of atherosclerosis, where we demonstrated that hypercholesterolemia and progression of atherosclerosis lead to decreased expression of endoglin in atherosclerotic aorta and increased levels of soluble endoglin in blood (37). These results would suggest an increased endoglin shedding under inflammation. Supporting this view, the upregulation of MMP-14 mediated by TNF-α has been reported (39).
Several studies indicated that statins are able to modulate the expression of endoglin. Atorvastatin diminished the induction of endoglin in cardiac fibroblasts (40). On the contrary, atorvastatin increased endoglin expression together with tacrolimus, macrolytic agent with immunosuppressant activity in HUVECs but not in vascular smooth muscle cells (41). These data suggest that statin effects could be different in various cell lines and/or individual tissues. Our previous in vivo studies demonstrated endoglin expression in endothelium of atherosclerotic and non-atherosclerotic aorta in apoE/LDLr-deficient mice (6, 42). Moreover, atorvastatin increased endoglin expression in various experimental conditions in aorta of apoE/LDLr-deficient mice, together with hypolipidemic effects of atorvastatin (42) or beyond the lipid lowering effects of atorvastatin (23). Moreover, eNOS expression was also increased with reduced atherosclerosis (23). In this report, flow cytometry analysis clearly demonstrated a concentration dependent increase in endoglin expression after atorvastatin treatment. Time-dependent atorvastatin treatment showed a maximum increase of endoglin mRNA level after 4 hours treatment, followed by a decline in mRNA levels after 16 and 24 hours of treatment, respectively. Because of the long half-life of endoglin, estimated in ~17 hours in cultured HUVECs (43), we decided to study atorvastatin effect on transmembrane protein level of endoglin after 24 hours. Thus, 5 µM ATV treatment for 24 hours significantly increased both endoglin and eNOS expression in HUVECs, supporting our in vivo data. In addition, atorvastatin treatment surprisingly increased levels of soluble endoglin in culture supernatants. Although we did not show changes in MMP-14 levels upon atorvastatin treatment, however it appears that different mechanisms are involved since the simultaneous treatment of TNF-α and atorvastatin resulted in almost double increase of soluble endoglin when compared to each individual treatment. We might hypothesize that soluble endoglin release is dependent on the total amount of endoglin protein, on one hand, and on the other hand on the amount and activity of MMP14 (or other MMPs proteases) active in endoglin membrane shedding. ATV increases the total amount of membrane bound endoglin, while TNF-α decreases it. However, the mechanisms of soluble endoglin release are most likely different: while ATV increases the amount of substrate for the MMPs, TNF-α stimulates the rate of shedding by increasing MMP14 (and likely other MMPs), which, in turn, cleave membrane bound endoglin. The exact mechanisms underlying the effects of TNF-α and ATV on soluble endoglin levels deserve further investigation.
An important question that remained to be answered is whether atorvastatin is able to increase endoglin and eNOS expression also during inflammation. In our hands, ATV pretreatment significantly prevented TNF-α-mediated reduction in endoglin and eNOS expressions, a finding compatible with the statin-induced increase of eNOS expression through various mechanisms in endothelial cells (18, 44). In addition, more recently atorvastatin was demonstrated to enhance nitric oxide release and reduce blood pressure in hypertensive diabetic rats (45) and to have acute anti-fibrillating effects in the heart (46) showing clear benefit of atorvastatin treatment both in vitro and in vivo. It is of interest to mention that ATV effects are independent of its hypolipidemic effects, suggesting that statins are able to affect endoglin expression beyond its lipid-lowering effects, which is in line with our previously published study in mice (23). In addition, we found that ATV lowered TNF-α-induced ROS production. Because increased expression of eNOS together with reduced levels of ROS are considered protective for endothelial cells (47), we propose that atorvastatin-induced endoglin and eNOS expression might represent an endothelial-protective mechanism.
Santibanez et al. showed a potential mechanism of how endoglin upregulates eNOS expression and function. They showed that endoglin increased Smad2 protein levels, Smad2 phosphorylation and Smad2 stability, which resulted in an increased eNOS expression both in the absence and/or in the presence of exogenous TGF-β1 in endothelial cells (12). Thus, it was of interest to elucidate whether endoglin or Smad2 are crucial for atorvastatin-mediated eNOS upregulation. Accordingly, SB-431542, an inhibitor of activin receptor-like kinase (ALK-5, TGF-β type I receptor), which also inhibits TGF-β- and activin-induced phosphorylation of Smad2, was used in this study (48). Atorvastatin treatment resulted in a significant increase of eNOS despite p-Smad2 inhibition, suggesting that Smad2 activation is not involved in atorvastatin-induced eNOS upregulation. Supporting this view, we found that atorvastatin did not affect phosphorylation levels of Smad2 in Western blot studies. Our previous in vivo results showed an increase in p-Smad2 expression in atherosclerotic aorta together with endoglin and eNOS upregulation (23, 42). Despite the fact that p-Smad2 is co-localized with endoglin and eNOS in aortic endothelium, we cannot rule out the possibility that statin-induced p-Smad2 expression might have other benefits with respect to atherogenesis. For example, it was demonstrated that Smad2 expression and signaling are related to anti-inflammatory and plaque stabilizing effects (49, 50).
In order to evaluate whether endoglin itself may control an atorvastatin-mediated eNOS increase, we used short-interfering (si) RNA for silencing the endoglin gene. The endoglin siRNA showed substantially reduced endoglin protein expression in HUVECs, when compared to cells transfected only with GFP vector. In the same experiment, eNOS levels did not change in the presence of endoglin siRNA, suggesting that endoglin does not regulate eNOS expression under basal conditions. However, upon treatment with ATV, endoglin suppression inhibited the ATV-dependent increase of eNOS expression. The interdependence between endoglin and eNOS expression is likely related to the physical and functional association of these two proteins within caveolae (8), a subcellular structure closely linked to cholesterol composition, and therefore sensitive to ATV treatment. Thus, we propose that a proper endoglin expression is critical for ATV induced eNOS expression.
It would be of interest to address the endoglin involvement on the ATV-induced eNOS expression in mouse models of atherosclerosis. Unfortunately, atherosclerosis models in endoglin deficient mice are not available yet. Homozygous endoglin-deficient mice are not available, as they die at day 10–10.5 of embryonic development because of alterations in their cardiovascular system formation (51). Despite the fact that 129/Ola and C57BL/6 inbred endoglin haploinsufficient mice strains are available as models of hereditary hemorrhagic telangiectasia, they do not develop atherosclerosis (51). Furthermore, apoE-knockout mice with endoglin deficiency are not available as well. Thus, our in vitro approach might represent the appropriate model nowadays, to study the link between endoglin and eNOS after statin treatment.
In conclusion, we demonstrate here for the first time that atorvastatin treatment prevents inflammation-reduced endoglin and eNOS expression in endothelial cells. In addition, ATV-induced eNOS expression strongly depends on the proper expression of endoglin in endothelial cells. Possible implications of this finding might be reflected in pathological conditions characterized by reduced levels of endoglin and eNOS as for example in hereditary hemorrhagic telangiectasia or in other endothelial dysfunctions.
Acknowledgements: We acknowledge the excellent support for flow cytometry from Dr. Pedro Lastres (Centro de Investigaciones Biologicas, CSIC, Madrid, Spain), and from Dr. Doris Vokurkova (Department of Clinical Immunology and Allergology, University Hospital Hradec Kralove) and Dr. Pavlina Haskova (Department of Biochemical Sciences, Faculty of Pharmacy in Hradec Kralove), for help with ROS measurements.
This work was supported by The Grant Agency of Charles University in Prague (300811/C and 1158413/C), Charles University in Prague (SVV/2014/260064), Ministerio de Economia y Competitividad of Spain Raras ( (SAF2010-19222 and SAF2013-42421-R to CB), and Centro de Investigacion Biomédica en Red de Enfermedades CIBERER to CB). CIBERER is an initiative of the Instituto de Salud Carlos III (ISCIII) of Spain supported by FEDER funds. The publication is co-financed by the European Social Fund and the state budget of the Czech Republic (Project No. CZ.1.07/2.3.00/30.0061).
Authors L.Zemankova and M. Varejckova contributed equally to the paper.
Conflict of interests: None declared.
REFERENCES
- ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008; 11: 79-89.
- Li C, Guo B, Ding S, et al. TNF alpha down-regulates CD105 expression in vascular endothelial cells: a comparative study with TGF beta 1. Anticancer Res 2003; 23: 1189-1196.
- Bernabeu C, Lopez-Novoa JM, Quintanilla M. The emerging role of TGF-βeta superfamily coreceptors in cancer. Biochim Biophys Acta 2009; 1792: 954-973.
- Krupinski J, Turu MM, Luque A, Badimon L, Slevin M. Increased PrPC expression correlates with endoglin (CD105) positive microvessels in advanced carotid lesions. Acta Neuropathol 2008; 116: 537-545.
- Nachtigal P, Pospisilova N, Jamborova G, et al. Endothelial expression of endoglin in normocholesterolemic and hypercholesterolemic C57BL/6J mice before and after atorvastatin treatment. Can J Physiol Pharmacol 2007; 85: 767-773.
- Nachtigal P, Vecerova L, Pospisilova N, et al. Endoglin co-expression with eNOS, SMAD2 and phosphorylated SMAD2/3 in normocholesterolemic and hypercholesterolemic mice: an immunohistochemical study. Histol Histopathol 2009; 24: 1499-1506.
- Andersson J, Sundstrom J, Gustavsson T, et al. Echogenecity of the carotid intima-media complex is related to cardiovascular risk factors, dyslipidemia, oxidative stress and inflammation: the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) study. Atherosclerosis 2009; 204: 612-618.
- Toporsian M, Gros R, Kabir MG, et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res 2005; 96: 684-692.
- Chatterjee A, Black SM, Catravas JD. Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vascul Pharmacol 2008; 49: 134-140.
- Ponnuswamy P, Schrottle A, Ostermeier E, et al. eNOS protects from atherosclerosis despite relevant superoxide production by the enzyme in apoE mice. PLoS One 2012; 7: e30193.
- Jerkic M, Rivas-Elena JV, Prieto M, et al. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J 2004; 18: 609-611.
- Santibanez JF, Letamendia A, Perez-Barriocanal F, et al. Endoglin increases eNOS expression by modulating Smad2 protein levels and Smad2-dependent TGF-βeta signaling. J Cell Physiol 2007; 210: 456-468.
- Blaha M, Cermanova M, Blaha V, et al. Elevated serum soluble endoglin (sCD105) decreased during extracorporeal elimination therapy for familial hypercholesterolemia. Atherosclerosis 2008; 197: 264-270.
- Blazquez-Medela AM, Garcia-Ortiz L, et al. Increased plasma soluble endoglin levels as an indicator of cardiovascular alterations in hypertensive and diabetic patients. BMC Med 2010; 8: 86.
- Hawinkels LJ, Kuiper P, Wiercinska E, et al. Matrix metalloproteinase-14 (MT1-MMP)-mediated endoglin shedding inhibits tumor angiogenesis. Cancer Res 2010; 70: 4141-4150.
- Valbuena-Diez AC, Blanco FJ, Oujo B, et al. Oxysterol-induced soluble endoglin release and its involvement in hypertension. Circulation 2012; 126: 2612-2624.
- Rathouska J, Vecerova L, Strasky Z, et al. Endoglin as a possible marker of atorvastatin treatment benefit in atherosclerosis. Pharmacol Res 2011; 64: 53-59.
- Dayoub JC, Ortiz F, Lopez LC, et al. Synergism between melatonin and atorvastatin against endothelial cell damage induced by lipopolysaccharide. J Pineal Res 2011; 51: 324-330.
- Feron O, Dessy C, Desager JP, Balligand JL. Hydroxy-methylglutaryl-coenzyme A reductase inhibition promotes endothelial nitric oxide synthase activation through a decrease in caveolin abundance. Circulation 2001; 103: 113-118.
- Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 1998; 273: 24266-24271.
- Aguirre L, Hijona E, Macarulla MT, et al. Several statins increase body and liver fat accumulation in a model of metabolic syndrome. J Physiol Pharmacol 2013; 64: 281-288.
- Armato J, Ruby R, Reaven G. Plasma triglyceride determination can identify increased risk of statin-induced type 2 diabetes: a hypothesis. Atherosclerosis 2015; 239: 401-404.
- Vecerova L, Strasky Z, Rathouska J, et al. Activation of TGF-βeta receptors and Smad proteins by atorvastatin is related to reduced atherogenesis in ApoE/LDLR double knockout mice. J Atheroscler Thromb 2012; 19: 115-126.
- Fernandez LA, Sanz-Rodriguez F, Zarrabeitia R, et al. Blood outgrowth endothelial cells from hereditary haemorrhagic telangiectasia patients reveal abnormalities compatible with vascular lesions. Cardiovasc Res 2005; 68: 235-248.
- Eruslanov E, Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol Biol 2010; 594: 57-72.
- Brcakova E, Fuksa L, Cermanova J, et al. Alteration of methotrexate biliary and renal elimination during extrahepatic and intrahepatic cholestasis in rats. Biol Pharm Bull 2009; 32: 1978-1985.
- Heeba G, Hassan MK, Khalifa M, Malinski T. Adverse balance of nitric oxide/peroxynitrite in the dysfunctional endothelium can be reversed by statins. J Cardiovasc Pharmacol 2007; 50: 391-398.
- Hernandez-Perera O, Perez-Sala D, Navarro-Antolin J, et al. Effects of the 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors, atorvastatin and simvastatin, on the expression of endothelin-1 and endothelial nitric oxide synthase in vascular endothelial cells. J Clin Invest 1998; 101: 2711-2719.
- Zapolska-Downar D, Siennicka A, Kaczmarczyk M, Kolodziej B, Naruszewicz M. Simvastatin modulates TNF-αlpha-induced adhesion molecules expression in human endothelial cells. Life Sci 2004; 75: 1287-1302.
- Eccles KA, Sowden H, Porter KE, Parkin SM, Homer-Vanniasinkam S, Graham AM. Simvastatin alters human endothelial cell adhesion molecule expression and inhibits leukocyte adhesion under flow. Atherosclerosis 2008; 200: 69-79.
- Nubel T, Dippold W, Kleinert H, Kaina B, Fritz G. Lovastatin inhibits Rho-regulated expression of E-selectin by TNFalpha and attenuates tumor cell adhesion. FASEB J 2004; 18: 140-142.
- Lopez-Novoa JM, Bernabeu C. The physiological role of endoglin in the cardiovascular system. Am J Physiol Heart Circ Physiol 2010; 299: H959-H974.
- Bernabeu C, Conley BA, Vary CP. Novel biochemical pathways of endoglin in vascular cell physiology. J Cell Biochem 2007; 102: 1375-1388.
- Nachtigal P, Zemankova Vecerova L, Rathouska J, Strasky Z. The role of endoglin in atherosclerosis. Atherosclerosis 2012; 224: 4-11.
- Rossi E, Sanz-Rodriguez F, Eleno N, et al. Endothelial endoglin is involved in inflammation: role in leukocyte adhesion and transmigration. Blood 2013; 121: 403-415.
- Venkatesha S, Toporsian M, Lam C, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 2006; 12: 642-649.
- Strasky Z, Vecerova L, Rathouska J, et al. Cholesterol effects on endoglin and its downstream pathways in ApoE/LDLR double knockout mice. Circ J 2011; 75: 1747-1755.
- Ikemoto T, Hojo Y, Kondo H, et al. Plasma endoglin as a marker to predict cardiovascular events in patients with chronic coronary artery diseases. Heart Vessels 2012; 27: 344-351.
- Wang BW, Chang H, Lin S, Kuan P, Shyu KG. Induction of matrix metalloproteinases-14 and -2 by cyclical mechanical stretch is mediated by tumor necrosis factor-alpha in cultured human umbilical vein endothelial cells. Cardiovasc Res 2003; 59: 460-469.
- Shyu KG, Wang BW, Chen WJ, Kuan P, Hung CR. Mechanism of the inhibitory effect of atorvastatin on endoglin expression induced by transforming growth factor-beta1 in cultured cardiac fibroblasts. Eur J Heart Fail 2010; 12: 219-226.
- Giordano A, Romano S, Monaco M, et al. Differential effect of atorvastatin and tacrolimus on proliferation of vascular smooth muscle and endothelial cells. Am J Physiol Heart Circ Physiol 2012; 302: H135-H142.
- Nachtigal P, Pospisilova N, Vecerova L, et al. Atorvastatin increases endoglin, SMAD2, phosphorylated SMAD2/3 and eNOS expression in ApoE/LDLR double knockout mice. J Atheroscler Thromb 2009; 16: 265-274.
- Paquet ME, Pece-Barbara N, Vera S, et al. Analysis of several endoglin mutants reveals no endogenous mature or secreted protein capable of interfering with normal endoglin function. Hum Mol Genet 2001; 10: 1347-1357.
- Endres M, Laufs U. Effects of statins on endothelium and signaling mechanisms. Stroke 2004; 35: 2708-2711.
- Mason RP, Corbalan JJ, Jacob RF, Dawoud H, Malinski T. Atorvastatin enhanced nitric oxide release and reduced blood pressure, nitroxidative stress and rantes levels in hypertensive rats with diabetes. J Physiol Pharmacol 2015; 66: 65-72.
- Benova T, Knezl V, Viczenczova C, Bacova BS, Radosinska J, Tribulova N. Acute anti-fibrillating and defibrillating potential of atorvastatin, melatonin, eicosapentaenoic acid and docosahexaenoic acid demonstrated in isolated heart model. J Physiol Pharmacol 2015; 66: 83-89.
- Chen XN, Xu J, Feng Z, Fan M, Han JY, Yang Z. Simvastatin combined with nifedipine enhances endothelial cell protection by inhibiting ROS generation and activating Akt phosphorylation. Acta Pharmacol Sin 2010; 31: 813-820.
- Inman GJ, Nicolas FJ, Callahan JF, et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol 2002; 62: 65-74.
- Bot PT, Hoefer IE, Sluijter JP, et al. Increased expression of the transforming growth factor-beta signaling pathway, endoglin, and early growth response-1 in stable plaques. Stroke 2009; 40: 439-447.
- DiChiara MR, Kiely JM, Gimbrone MA, Lee ME, Perrella MA, Topper JN. Inhibition of E-selectin gene expression by transforming growth factor beta in endothelial cells involves coactivator integration of Smad and nuclear factor kappaB-mediated signals. J Exp Med 2000; 192: 695-704.
- Bourdeau A, Faughnan ME, Letarte M. Endoglin-deficient mice, a unique model to study hereditary hemorrhagic telangiectasia. Trends Cardiovasc Med 2000; 10: 279-285.
A c c e p t e d : March 6, 2015