THE ANTIPROLIFERATIVE ACTION OF [D-ARG1, D-PHE5, D-TRP7,9, LEU11]
SUBSTANCE P ANALOGUE ANTAGONIST AGAINST SMALL CELL-
AND NON-SMALL-CELL LUNG CANCER CELLS COULD BE DUE TO THE PHARMACOLOGICAL PROFILE
OF ITS TACHYKININ RECEPTOR ANTAGONIST
INTRODUCTION
According to the American Cancer Society, over 1.6 million new lung cancer cases are identified worldwide annually, of which over 200,000 are in the United States (1). Lung cancer is the leading cause of cancer-due death in men and the second leading cause of cancer-due death in women. Two histological groups have been described: non-small-cell lung cancer (NSCLC) and small-cell lung cancer (SCLC). The 5-year survival rate for all stages of lung cancer is 15% (2). Furthermore, SCLC cells are characterized by their ability 1) to secrete a variety of peptides including bombesin/gastrin-releasing peptide, galanin, somatostatin, vasopressin, cholecystokinin, vasoactive intestinal peptide and neurotensin, and 2) to express receptors for these peptides (2). Consequently, the peptides secreted by these cells could exert an autocrine/paracrine action, amplifying the growth of the very lung cancer cells which also express the receptors of these peptides. In this sense, many in vitro and in vivo experiments have also reported the involvement of the substance P (SP)/neurokinin (NK)-1 receptor system in cancer progression. It is known that the number of NK-1 receptors expressed in normal human cells is lower than that expressed in human tumour cells and that the expression of NK-1 receptors is correlated with the degree of malignancy (3). The undecapeptide SP (4) (Fig. 1) belongs to the tachykinin family of peptides and is the natural ligand of the NK-1 receptor. Via this receptor, SP promotes tumour cell proliferation, metastasis and neoangiogenesis, and it induces an antiapoptotic effect (3). In fact, this system is profoundly involved in both SCLC and NSCLC. It is known that non-peptide NK-1 receptor antagonists inhibit the growth of human H-69 (SCLC) and COR-L23 (NSCLC) cell lines in a concentration-dependent manner; that both cell lines overexpress the NK-1 receptor; that the NK-1 receptor promotes the viability of H-69/COR-L23 tumour cells (in this case, the small interfering RNA gene silencing method was applied); that the specific antiproliferative action of these antagonists occurs through the NK-1 receptor, and that lung cancer cells (H-69/COR-L23) die by apoptosis in the presence of NK-1 receptor antagonists and after the application of the siRNA gene silencing method for the NK-1 receptor (5).
 |
Fig. 1. Substance P (SP) and [D-Arg1, D-Trp5,7,9, Leu11] SP analogue antagonist sequences. |
Many years ago it was also reported that SP analogue antagonists (SPAAs) (synonymous with broad-spectrum GPCR antagonist, broad-spectrum peptide antagonists, synthetic analogues of SP) also act as antiproliferative agents against SCLC in in vitro and in in vivo experiments and also against NSCLC in in vitro studies (6-13). It is also known that the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SP analogue antagonist inhibits the signal transduction and the DNA synthesis stimulated by bombesin/gastrin-releasing peptide, bradykinin, and vasopressin (14) and that SPAAs inhibit the growth of tumours in xenograft models in nude mice (15). SPAAs are obtained when D-amino acids are introduced in the SP sequence (16) (Fig. 1). However, to our knowledge the specific mechanisms underlying the antiproliferative action of SPAAs against SCLC and NSCLC cells remains unknown. Thus, by using competition assays with SP, the aim of this study was to demonstrate that the antiproliferative action of the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SP analogue antagonist against human H-69 SCLC and COR-L23 NSCLC cell lines occurs at least via the NK-1 receptor.
MATERIALS AND METHODS
Cell cultures
H-69 SCLC and COR-L23 NSCLC lung cancer cell lines (European Collection of Cell Cultures (ECC)) were cultured according to the culture conditions suggested by the ECC. Cell lines were seeded in 75 cm2 tissue culture flasks (Falcon, Heidelberg, Germany). The medium was renewed every 2 days and the cells were harvested by treatment with trypsin (0.05% and 0.02% EDTA without Ca2+ and Mg2+, Sigma-Aldrich, Madrid, Spain) when confluence had reached 100% of tumour cells after seeding. Cells were incubated at 37°C in a humidified atmosphere of 95% air/5% CO2.
Drug treatments
The [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SP analogue antagonist (SPAA) was used in this study (MW 1516.85, Sigma-Aldrich, Madrid, Spain). The antagonist was dissolved in distilled water and in order to determine the IC50, different concentrations (5 to 100 µM) were evaluated for the presence of the H-69 SCLC and COR-L23 NSCLC cell lines. SP, acetate salt (Sigma-Aldrich, Madrid, Spain), was dissolved in distilled water and different concentrations (5 to 100 nM) were also used. Each cell line was incubated with 10 nM of SP (mitogenic effect) 1 hour before the addition of the SPAA.
Proliferation assays
As previously reported (5), cell proliferation was evaluated using the tetrazolium compound 3-(4, 5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS), according to the manufacturer’s instructions (CellTiter 96 Aqueous One-Solution Cell Proliferation Assay, Promega Corp., Madison, USA). Cell numbers were quantified using a Coulter counter. The plate included blank wells (0 cells/0.1 ml), control wells (104 cells/0.1 ml), control wells with distilled water, control wells treated with SP, control wells treated with the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA, and control wells treated with SP (at nM concentration) and the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA (approximately the fifty-percent inhibition concentration (µM) of antagonist (IC50) for their first doubling times). For the proliferation assay, 20 µl of the MTS reagent was added to each well 90 min before reading the samples on a multiscanner microplate reader (TECAN Spectra classic, Barcelona, Spain) at 492 nm. Each experimental condition (blank wells, control wells, and control wells treated with the different concentrations of the SPAA and/or SP) was assayed in duplicate and all experiments were performed at least three times.
The IC50 of the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA was calculated using the linear interpolation of values providing a dose-response curve (using a polynomial model).
DAPI staining
In order to determine whether the SPAA used here induced apoptosis, DAPI staining was performed in the two cell lines studied here (5). Cells were cultured on four-chamber slides. After treatment with the SPAA (51.05 µM and 79.46 µM for H-69; 75.81 µM and 106,07 µM for COR-L23) for their first doubling times (48 hours for H-69; 24 hours for COR-L23), cells were washed twice with phosphate buffered saline (PBS) and fixed by incubation in 4% paraformaldehyde for 30 min. Following a second wash in PBS, cells were incubated in DAPI solution at a dilution of 1/1,000 for 15 min in the dark and the supporting slides were mounted with a mixture of PBS and glycerol at 70%. The cells were then observed through a fluorescence microscope. Apoptotic cells were defined by chromatin condensation and nuclear fragmentation.
Statistical analysis
Data were expressed as means ± S.D. Statistical analyses were performed with SPPS statistical software for Microsoft Windows, release 14.0 (Professional Statistic, Chicago). The homogeneity of variance was tested using the Levene test. If the variances were homogeneous, the data were analyzed using the one-way ANOVA test with Bonferroni’s correction for multiple comparisons. For data sets with non-homogeneous variances, the ANOVA test with T3 Dunnett post-hoc analysis was applied. The criterion for significance was P < 0.05 for all comparisons.
RESULTS
Antiproliferative action of [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA
The SPAA exerted growth inhibition in both the H-69 SCLC and COR-L23 NSCLC cell lines in a concentration-dependent manner (Fig. 2). The concentration required for a 50% reduction of lung cancer cells (IC50) observed with the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA was 51.05 µM for H-69, and 75.81 µM for COR-L23 respectively. Maximum inhibition was observed when the drug was present at a concentration of 79.46 µM of the SPAA for H-69 SCLC, and 106.07 µM for COR-L23 NSCLC respectively during the culture periods. No remaining living cell was observed at the maximum concentration.
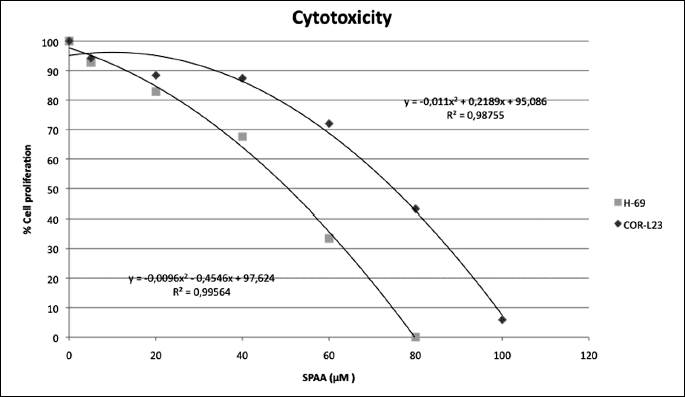
The [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA block SP-induced mitogen stimulation
Growth of the H-69 SCLC and COR-L23 NSCLC cell lines was noted after the addition of SP, and certain nanomolar concentrations of SP induced cell proliferation (Fig. 3). After the administration of 10 nM SP, the increase in the percentage in cell proliferation was from 14% for H-69 and 11% for COR-L23. In order to examine whether the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA inhibited cell proliferation via an interaction with the NK-1 receptor, this antagonist was used in competition experiments. The cellular concentrations at 50 µM (for H-69) or 60 µM (for COR-L23) of the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA and at 10 nM of SP were higher than those observed when the antagonist was administered alone (Fig. 3). Thus, [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA-induced growth inhibition was partially reversed by the administration of a nanomolar dose of SP.
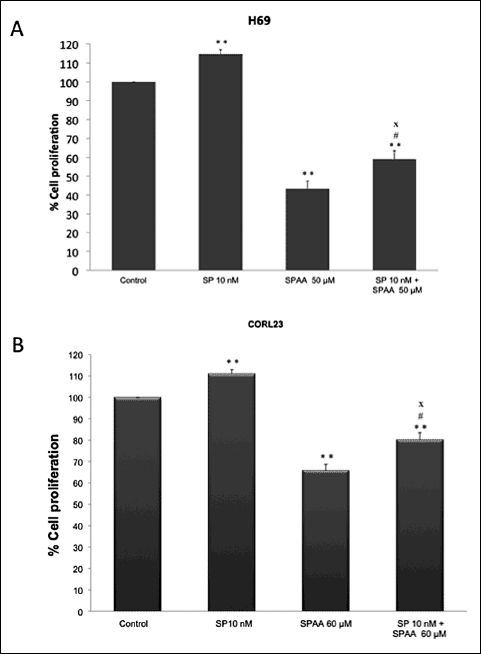 |
Fig. 3. (A) Induction of cell proliferation of the human H-69 SCLC cell line by substance (SP) at nanomolar concentration (10 nM). The [D-Arg1, D-Trp5,7,9, Leu11] SPAA was added (50 µM) in the presence (10 nM) or absence (none) of SP for their first doubling time. In both cases, proliferation of the H-69 SCLC cell line was inhibited. (B) Induction of cell proliferation of human COR-L23 NSCLC cell lines by SP at nanomolar concentration (10 nM). The [D-Arg1, D-Trp5,7,9, Leu11] SPAA was added (60 µM) in the presence (10 nM) or absence (none) of SP for their first doubling time. In both cases, COR-L23 NSCLC cell line proliferation was inhibited. Using the ANOVA test, a significant difference between each group and the control group (none-none) was found. Level of significance: * P ≤ 0.01; ** P ≤ 0.05. # indicates the value of significance of IC50-most mitogenic SP concentration versus IC50-none P < 0.05, and x indicates IC50-most mitogenic SP concentration versus none-most mitogenic SP concentration, P ≤ 0.01. Vertical bars indicate S.D. |
The [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA induces apoptosis
After the administration of the SPAA, large numbers of apoptotic cells were found in the H-69 SCLC and COR-L23 NSCLC cell lines (Fig. 4). In DAPI-stained cultures, we observed means of 35.9 ± 9.76 (S.D.) % and 34.34 ± 9.81 (S.D.) % of apoptotic cells, respectively, in H-69 SCLC and COR-L23 NSCLC cell lines after administration of IC50 SPAA (51.05 µM for H-69 and 75.81 µM for COR-L23); and 42.52 ± 5.58 (S.D.) % and 70.50 ± 5.76 (S.D.) % of apoptotic cells in the H-69 SCLC and COR-L23 NSCLC cell lines respectively, after administration of IC100 SPAA (79.46 µM for H-69 and 106.07 µM for COR-L23). By contrast, in control H-69 and COR-L23 cells (not treated with SPAA) we observed means of < 3% apoptotic cells in the two cell lines studied.
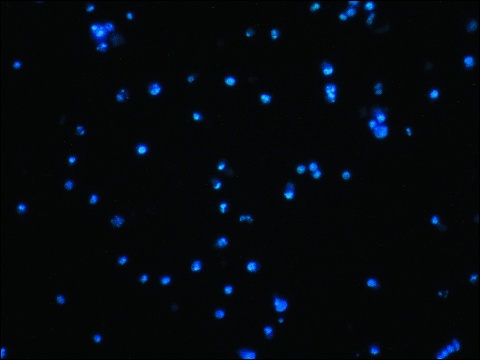 |
Fig. 4. H-69 cells treated with 79.46 µM of [D-Arg1, D-Trp5,7,9, Leu11] SPAA (48 hours). Apoptotic figures. Note chromatin condensation and nuclear fragmentation. |
DISCUSSION
Many in vivo and in vitro studies have reported that SPAAs exert an antiproliferative action (tumour cell proliferation inhibition by apoptosis) against SCLC and NSCLC cell lines (6-14, 17, 18), but the underlying mechanisms involved in this antiproliferative action remain unknown. Our results suggest that the antiproliferative action of the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA (a peptide NK-1 receptor antagonist) against human H-69 SCLC and COR-L23 NSCLC cell lines occurs at least through the NK-1 receptor, which is overexpressed in these lung cancer cell lines (5). It seems that the overexpression of this tachykinin receptor is a general feature in human tumour cells (e.g., glioblastoma, retinoblastoma, breast cancer, acute lymphoblastic leukemia, larynx, pancreatic, gastric and colon carcinomas) (3). Our results are in agreement with previous studies in which the antiproliferative action of non-peptide NK-1 receptor antagonists, via the NK-1 receptor, was also reported against the human H-69 SCLC and COR-L23 NSCLC cell lines (5). In fact, it has been reported that both lung cancer cells and samples overexpress NK-1 receptors (5). Previously, the antiproliferative action of SPAAs against lung cancer cells had not been attributed to the SP/NK-1 receptor system, but to other peptide systems (e.g., neurotensin, somatostatin, galanin, endothelin and their receptors (2)). However, our data suggest that SPAAs, although non-specific for the NK-1 receptor, exert their antiproliferative action at least via the NK-1 receptor. Our past results (3, 5) indicate that antiproliferative actions are observed with a variety of highly specific and selective, molecularly dissimilar, NK-1 receptor antagonists; importantly, that these NK-1 receptor antagonists, at the concentration tested, lack appreciable affinity for other peptidergic receptors. Thus our present results suggest that the interactions of SPAAs with the previous reported peptidergic systems (2) could be secondary or indirect since the peptides in question bind specifically to receptors other than the NK-1 receptor.
The above data suggest that via the NK-1 receptor, NK-1 receptor antagonists of distinctly different chemical types and classes (both peptide, e.g., the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA and non-peptide, e.g., the drug aprepitant, L-733,060, L-732,138) act as antiproliferative agents. This implies that the likely mechanism that SPAAs and all other studied NK-1 receptor antagonists share in mitigating lung cancer cell proliferation (3, 5) involves the SP/NK-1 receptor system. It seems that the antiproliferative action of peptide and non-peptide NK-1 receptor antagonists is related to their stereochemical features (affinity for the NK-1 receptor) and not to their chemical composition (19).
After using the siRNA gene-silencing method for the NK-1 receptor, it has been demonstrated that the number of non-transfected H-69/COR-L23 cells increases significantly in comparison with the number of H-69/COR-L23 transfected cells (5). Moreover, after the administration of siRNA TAC1R to both H-69 and COR-L23 lines of cultured cells, many apoptotic cells have been found (e.g., means of 36.2 ± 1.15 (S.D.) % of apoptotic H-69 cells versus means of 2.2 ± 0.6 (S.D.) % in siRNA-negative control H-69 cells) (5). This means that the NK-1 receptor is involved in the viability of the H-69/COR-L23 cells and it seems that the overexpression of NK-1 receptors by H-69/COR-L23 cells (and tumour cells in general) renders them highly dependent upon the mitotic signal induced by SP (3). This potent signal counteracts the death signal pathways activated in H-69/COR-L23 cells by their own suppressor gene losses, oncogene activation and genetic damage. Thus, by increasing the phenotypic expression of NK-1 receptors H-69/COR-L23 cells could neutralize the pathways leading to cell death, since the different death signals are overridden by the SP-mediated mitotic stimulus (3). Thus, after the blockade of NK-1 receptors by non-peptide NK-1 receptor antagonists/SPAAs or the application of the siRNA gene silencing method for the NK-1 receptor, H-69/COR-L23 cell dies because the balance inside the cell is favourable to apoptotic/death signals (5). To sum up, after genetic (applying the small interfering RNA gene-silencing methodology) and pharmacological (using non-peptide NK-1 receptor antagonists/SPAAs) treatments, cancer cells die by apoptosis.
Apoptosis of tumour cells is the shared outcome for these chemically distinct NK-1 receptor antagonists. It is known that the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA induces apoptosis in the H-69 SCLC and COR-L23 NSCLC cell lines (10), as confirmed here upon using the DAPI method, and that this induction is also triggered when lung cancer cells are treated with non-peptide NK-1 receptor antagonists (5). Moreover, it has also been reported that the cytotoxicity produced by the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA against normal cells is less marked than that reported for cancer cells (8). This is in accordance with the results reported for non-peptide NK-1 receptor antagonists, because the cytotoxicity exerted by these antagonists against cancer cells is greater than that produced in normal cells (5).
It is also known that in a pancreatic carcinoma xenograft nu/nu mice model the [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SPAA exerts a dual effect against this carcinoma: it inhibits both tumour cell proliferation and angiogenesis (20). It appears that this dual effect would be mediated through the NK-1 receptor, since it has been reported that pancreatic cancer cells overexpress the NK-1 receptor (21) and that endothelial cells also express this receptor (22). Again, via the NK-1 receptor non-peptide NK-1 receptor antagonists (e.g., the drug aprepitant, L-733,060) exerted an antiproliferative action against pancreatic cancer cells (19). SP is also known to be involved in neurogenic inflammation and in the growth of capillary vessels because, through the NK-1 receptor, this tachykinin promotes the proliferation of endothelial cells in a concentration-dependent manner and hence SP promotes neoangiogenesis (22). By contrast, non-peptide NK-1 receptor antagonists block endothelial cell proliferation via the NK-1 receptor (22).
In addition to the antiproliferative action (via the NK-1 receptor) exerted by SPAAs and non-peptide NK-1 receptor antagonists, other compounds (e.g., pterostilbene) also exert antiproliferative and proapoptotic actions in cancer cells. Thus, for example, it has been reported that pterostilbene induces the accumulation of autophagic vacuoles into human leukemia cells and that this phenomenon is followed by the death of tumour cells (23). Moreover, it is known that ghrelin activates the ERK 1/2 and PI-3 kinase pathways which mediate the proliferative and the antiapoptotic effects exerted by ghrelin on ovarian follicle cells and that the treatment with selective inhibitors of ERK 1/2 and PI-3 kinases decreased cell proliferation and increased apoptosis in these cells (24). This is in agreement with the results found in tumour cells, since once activated, ERK 1/2 is translocated into the nucleus of cancer cells, inducing proliferation and protecting cells from apoptosis (3). It is also known that gastrin promotes intestinal polyposis via either a direct gastrin receptor-mediated proliferative signaling or by fostering tumour microenvironment (e.g., macrophage activation) (25). Together, the above data show new targets for the treatment of cancer and hence, in the future, new strategies focused on these targets should be developed.
In summary, we demonstrate that the antiproliferative action of the peptide NK-1 receptor antagonist [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SP against the human H-69 SCLC and COR-L23 NSCLC cell lines occurs at least through the NK-1 receptor by blocking the action of SP. In addition, the results found here are in agreement with those showing that SP is the natural ligand of the NK-1 receptor and that the undecapeptide promotes the proliferation of lung cancer cells. In contrast, after binding to the NK-1 receptor, [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SP (biologically inert, yet molecularly similar to SP except for its D-amino acids) substitutes for naturally occurring biologically active SP, and thus mitigates lung cancer cell proliferation, which enables natural tumour cell apoptosis in the cell lines.
Acknowledgements: The authors wish to thank Mr. Nicholas Skinner (University of Salamanca, Spain) for supervising the English text and Professor Mark Kramer (University of Philadelphia, USA), Mr. Javier Munoz (University of Seville, Spain), Mr. Juan Manuel Praena-Fernandez and Ms. Esther Rosso for technical assistance.
Conflict of interests: USPTO Application no. 20090012086 “Use of non-peptidic NK-1 receptor antagonists for the production of apoptosis in tumor cells” (Miguel Muńoz).
REFERENCES
- Siegel R, Naishadham D, Jemal A. Cancer statistics 2012. CA Cancer J Clin 2012; 62: 10-29.
- Hodkinson PS, Mackinnon A, Sethi T. Targeting growth factors in lung cancer. Chest 2008; 133: 1209-1216.
- Munoz M, Covenas R. Involvement of substance P and the NK-1 receptor in cancer progression. Peptides 2013; 48: 1-9.
- Chang MM, Leeman SE, Nial HD. Amino-acid sequence of substance P. Nat New Biol 1971; 232: 86-87.
- Munoz M, Gonzalez-Ortega A, Rosso M, et al. The substance P/neurokinin-1 receptor system in lung cancer: Focus on the antitumor action of neurokinin-1 receptor antagonists. Peptides 2012; 38: 318-325.
- Matsumoto Y, Kawatani M, Simizu S, Tanaka M, Imoto M. Bcl-2-independent induction of apoptosis by neuropeptide receptor antagonist in human small cell lung carcinoma cells. Anticancer Res 2000; 20: 3123-3129.
- Woll PJ, Rozengurt E. A neuropeptide antagonist that inhibits the growth of small cell lung cancer in vitro. Cancer Res 1990; 50: 3968-3973.
- Everard MJ, Macaulay VM, Miller JL, Smith IE. in vitro effects of substance P analogue [D-Arg1, D-Phe5, D-Trp7,9, Leu11] substance P on human tumour and normal cell growth. Br J Cancer 1992; 65: 388-392.
- Everard MJ, Macaulay VM, Miller JL, Smith IE. [D-Arg1, D-Phe5, D-Trp7,9, Leu11] substance P inhibits the growth of human small cell lung cancer xenografts in vivo. Eur J Cancer 1993; 29: 1450-1453.
- Reeve JG, Bleehen NM. [D-Arg1, D-Phe5, D-Trp7,9, Leu11] substance P induces apoptosis in lung cancer cell lines in vitro. Biochem Biophys Res Commun 1994; 199: 1313-1319.
- Woll PJ, Rozengurt E. [D-Arg1, D-Phe5, D-Trp7,9, Leu11] substance P, a potent bombesin antagonist in murine Swiss 3T3 cells, inhibits the growth of human small cell lung cancer cells in vitro. Proc Natl Acad Sci USA 1988; 85: 1859-1863.
- Rosati R, Adil MR, Ali MA, et al. Induction of apoptosis by a short-chain neuropeptide analog in small cell lung cancer. Peptides 1998; 19: 1519-1523.
- Seckl MJ, Higgins T, Widmer F, Rozengurt E. [D-Arg1, D-Phe5, D-Trp7,9, Leu11] substance P: a novel potent inhibitor of signal transduction and growth in vitro and in vivo in small cell lung cancer cells. Cancer Res 1997; 57: 51-54.
- Seckl MJ, Higgins T, Rozengurt E. [D-Arg1, D-Phe5, D-Trp7,9, Leu11] substance P coordinately and reversibly inhibits bombesin- and vasopressin-induced signal transduction pathways in Swiss 3T3 cells. J Biol Chem 1996; 271: 29453-29460.
- Langdon S, Sethi T, Ritchie A, Muir M, Rozengurt E. Broad spectrum neuropeptide antagonists inhibit the growth of small cell lung cancer cell in vivo. Cancer Res 1992; 52: 4554-4557.
- Almeida TA, Rojo J, Nieto PM, et al. Tachykinins and tachykinin receptors: structure and activity relationships. Curr Med Chem 2004; 11: 2045-2081.
- Orosz A, Schrett J, Nagy J, Bartha L, Schon I, Nyeki O. New short-chain analogs of a substance-P antagonist inhibit proliferation of human small-cell lung cancer cells in vitro and in vivo. Int J Cancer 1995; 60: 82-87.
- MacKinnon AC, Armstrong RA, Waters CM, et al. [Arg6, D-Trp7,9, NmePhe8]- substance P (6-11) activates JNK and induces apoptosis in small cell lung cancer cells via an oxidant-dependent mechanism. Br J Cancer 1999; 80: 1026-1034.
- Munoz M, Covenas R. Involvement of substance P and the NK-1 receptor in pancreatic cancer. World J Gastroenterol 2014; 20: 2321-2334.
- Guha S, Eibl G, Kisfalvi K, et al. Broad-spectrum G protein-coupled receptor antagonist, [D-Arg1, D-Phe5, D-Trp7,9, Leu11] SP: a dual inhibitor of growth and angiogenesis in pancreatic cancer. Cancer Res 2005; 65: 2738-2745.
- Friess H, Zhu Z, Liard V, et al. Neurokinin-1 receptor expression and its potential effects on tumor growth in human pancreatic cancer. Lab Invest 2003; 83: 731-742.
- Ziche M, Morbidelli L, Pacini M, Gepetti P, Alessandri C, Maggi CA. Substance P stimulates neovascularization in vivo and proliferation of cultured endothelial cells. Microvasc Res 1990; 40: 264-278.
- Siedlecka-Kroplewska K, Jozwik A, Boguslawski W, et al. Pterostilbene induces accumulation of autophagic vacuoles followed by cell death in HL60 human leukemia cells. J Physiol Pharmacol 2013; 64: 545-556.
- Rak-Mardyla A, Gregoraszczuk EL. ERK 1/2 and PI-3 kinase pathways as a potential mechanism of ghrelin action on cell proliferation and apoptosis in the porcine ovarian follicular cells. J Physiol Pharmacol 2010; 61: 451-458.
- Han YM, Park JM, Park SH, Hahm KB, Hong SP, Kim EH. Gastrin promotes intestinal polyposis through cholecystokinin-B receptor-mediated proliferative signaling and fostering tumor microenvironment. J Physiol Pharmacol 2013; 64: 429-437.
A c c e p t e d : March 23, 2015