THE EXPRESSION OF THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM
IN THE SKIN AND ITS EFFECTS ON SKIN PHYSIOLOGY AND PATHOPHYSIOLOGY
2Department of Esthetic Medicine, Medical University of Bialystok, Bialystok, Poland;
INTRODUCTION
The renin-angiotensin-aldosterone system (RAAS) is mainly recognized as a regulator of blood pressure and salt-water balance. It was originally considered to be a systemic, endocrine system; however, subsequently, it was discovered that in addition to systemic RAAS (Fig. 1), there is a similarly composed local tissue RAAS. The components of local RAAS can be expressed de novo and affect tissue biology through paracrine signaling synergistically and independently to systemic RAAS (1). Local RAAS has been documented in many different tissues (2), including in the skin (3-7). However, its function in the skin, as shown in this article, goes far beyond the regulation of local microcirculation.
The characteristics and regulatory significance of long-known components of local RAAS and its therapeutic targets have been reviewed extensively (8-12) and will not be the subject of this paper. The goal of this review is to summarize current aspects of the expression and function of local RAAS in the skin in relation to physiological and certain pathological conditions.
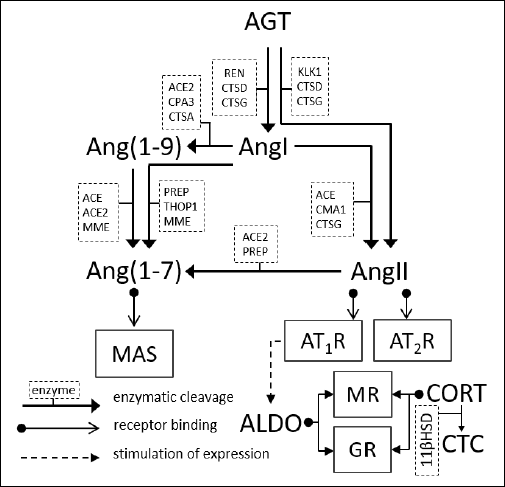 |
Fig. 1. Components of the renin-angiotensin-aldosterone system. 11βHSD, hydroxysteroid 11-beta dehydrogenase; ACE, angiotensin-converting type 1 enzyme; ACE2, angiotensin-converting type 2 enzyme; ALDO, aldosterone; Ang(1–7), angiotensin (1–7), Ang(1–9), angiotensin (1–9); AngI, angiotensin I; AngII, angiotensin II; AngIII, angiotensin III; AT1R, AT1 receptor; AT2R, AT2 receptor; CMA1, chymase 1; CORT, cortisol; CPA3, carboxypeptidase A3; CTC, cortisone; CTSA, cathepsin A; CTSD, cathepsin D; CTSG, cathepsin G; GCs, glucocorticoids; GR, glucocorticoid receptor; KLK1, kallikrein-1; MAS, specific G protein-coupled receptor MAS; MME, membrane metalloendopeptidase; MR, mineralocorticoid receptor; PREP, prolyl-endopeptidase; REN, renin; THOP1, thiometoligopeptidase 1 |
RENIN-ANGIOTENSIN-ALDOSTERONE SYSYTEM EXPRESSION
AND PHYSIOLOGY IN THE SKIN
Transcriptomic analysis has shown that the components of local RAAS in the skin are widely expressed on the mRNA level (2), and many sources also confirm their expression on the protein level (Table 1).
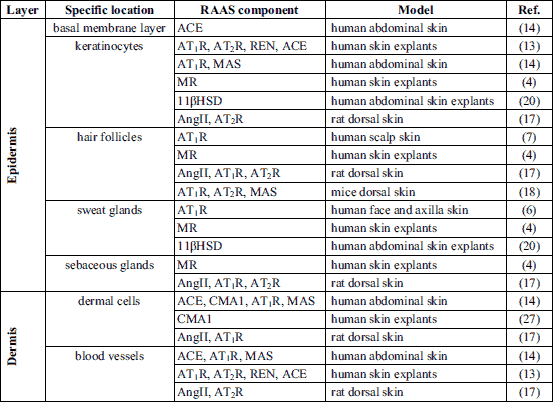
Epidermal expression of angiotensinogen, renin and angiotensin-converting enzymes (ACE) has been reported in the human skin (13,14). Keratinocytes are abundant in AT1 receptors (AT1R) (6, 13) and G protein-coupled receptors MAS (MAS) (14); however, data indicating the presence of AT2 receptors (AT2R) in this layer are not consistent (6, 13). It has been shown that cultured human keratinocytes express angiotensin II (AngII) (13), which is responsible for the up-regulation of their proliferation independently of AT1R and AT2R (15). The presence of the mineralocorticoid receptor (MR) has also been confirmed in the human epidermis (4). MR expression is associated with down-regulation of keratinocyte proliferation (16).
In hair follicles, AT1R has been labeled in the keratinizing zone of anagen hair follicles localized in the squamous and granular layers of the intermolecular epidermis (7). AT2R has not been detected in human hair follicles (7); however, it has been observed in rodents (17, 18). Data about the presence of MAS in the hair follicles come only from the murine model (18). Expression of angiotensinogen, renin and ACE has been documented in human hair follicles (13). Hair follicles are abundant in MR, as has been confirmed in both human and rat models (4, 19). The role of angiotensin receptors and MR in the regulation of hair follicle physiology is unknown.
Sweat glands express AT1R (6,13), MR, and hydroxysteroid 11-beta dehydrogenase (11βHSD) (4, 20). Expression of the RAAS receptors in sweat glands suggests that they are involved in the regulation of the sweating process. AngII administered intradermally decreases sweat rate and reduces cutaneous blood flow during post-exercise rest in healthy volunteers (21). This AngII-mediated decrease in sweating was linked to an increase in reactive oxygen species (ROS) production, since co-administration of ascorbate (non-selective antioxidant) abolished the AngII effect on sweating, simultaneously causing no changes in the cutaneous blood flow (21). MR also participates in sweating process regulation. It has been shown that aldosterone is responsible for the modulation of sweat ionic composition during exercise and heat acclimatization (22). This observation matches the expected Na+-reabsorbing function of epithelial Na+ channels (ENaC) which are highly expressed in human eccrine sweat glands (23) and are regulated by aldosterone (24). What is more, it has been proven that exercise and heat accumulation amplifies eccrine sweat gland responsiveness to aldosterone (25).
Sebaceous glands express AT1R, AT2R, and MR (4, 13), but their role in these structures has not been fully described. There is only one report showing that topical treatment with MR antagonist spironolactone cream in patients with moderately severe facial acne did not reduce sebum production (26).
In the dermis, RAAS components are expressed in dermal cells. It has been shown that fibroblasts express AT1R, AT2R, MAS, angiotensinogen, renin, ACE, and AngII (13, 14). The expression of chymase has been observed in mast cells (27). Cultured melanocytes express AT1R, angiotensinogen, renin, and ACE (13). It has been shown that melanogenesis is increased trough AT1R signaling in human melanocyte cultures (28).
In the hypodermis, RAAS components are expressed (excluding blood vessels, skin appendages, and fibroblasts) in the subcutaneous fat. Human subcutaneous adipose tissue expresses angiotensinogen, renin, ACE, angiotensin-converting enzyme 2 (ACE2), chymase, AT1R, AT2R and MAS on the mRNA level (29). There are no data available on RAAS expression in the hypodermis on the protein level. It has been shown that the expression pattern of RAAS components in subcutaneous fat is significantly different than in visceral fat, which may be explained by the different roles of these tissues (29). Unfortunately, there is a lack of data describing RAAS function in subcutaneous fat.
RENIN-ANGIOTENSIN-ALDOSTERONE SYSYTEM
IN THE SKIN MICROCIRCULATION
RAAS components are widely expressed in dermal blood vessels (13, 14, 17) (Table 1). It has been presented that dermal blood vessels are able to express the receptors for AngII and angiotensin (1-7) (Ang(1-7)) (13, 14). The MRs are widely expressed in the cardiovascular system in which they are responsible mainly for the deleterious effects like pathological remodeling and fibrosis (30, 31). However, their presence has not been confirmed in the skin vasculature. The role of RAAS in the skin microcirculation is summarized in Table 2.
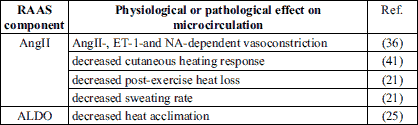
Role of angiotensin II in the skin microcirculation
The role of AngII in the regulation of cutaneous microcirculation is complex and depends on many variables, e.g. age or dose (32).
It has been shown that intradermal perfusion of high AngII doses has a greater vasoconstriction response in skin of older than younger individuals. The vasoconstrictive effect of AngII in older individuals is increased by AT2R antagonists, while it is attenuated with an adrenoreceptor blockade. However, young skin exhibits a vasodilation in response to lower AngII doses that is blunted by AT2R blockers (32). The authors of this study explain this phenomenon by the lower AT1R/AT2R ratio in young individuals (33) which, in response to the lower AngII concentration, favors a reduction in vascular resistance. A similar AT2R-dependent vasodilatory effect in response to low AngII doses has been observed in animal models (34, 35), however only in females. Further studies have shown that the effect is estrogen-dependent (35).
What is more, the mechanism of AngII-dependent vasoconstriction in the human skin also involves endothelin-1 pathways. It has been shown that a selective ETA receptor (ETA) blockade blunts AngII-dependent and noradrenaline-dependent vasoconstriction in healthy volunteers (36). In turn, administration of an AT1R blocker (ARB) valsartan not only reduces the endothelin-1-induced vasoconstriction in human skin but is able to switch it into vasodilatation (37).
The involvement of adrenergic and endothelin pathways in AngII-mediated vascular response in skin microcirculation could be explained by the heterodimerization of the AT1R with beta-adrenergic (38) and endothelin-1 receptors (39). It is well known that heterodimerization of receptors alters their ligand-binding properties and in some cases this appears to be essential for activation of downstream signaling (40).
It is known that AngII is also involved in the thermal physiological response to local heating. Intradermal administration of AngII decreases the vasodilatory reaction to local heating in the skin of the human forearm. This described effect is mediated through AT1R since the subcutaneous injection of ARB-losartan increases the baseline flow in both control and AngII groups (Table 3), while co-administration of PD123319 (selective AT2R blocker) does not affect local heating response (41). Despite the vasodilatory effect of intradermal losartan administration (41), it has been shown that the chronical oral losartan treatment does not affect the skin microvascular blood flow in young healthy women on a low sodium diet (42). Moreover, the mechanism of AngII-dependent vascular response in skin microcirculation could be ROS-dependent, since intradermal administration of ascorbate restores the AngII-blunted vasodilatory effect to local heating in the skin of healthy volunteers (41). Recently, it has been stated that the peroxisome proliferator-activated receptor gamma activation is involved in the vasodilatiation by mechanism dependent on AT2R and MAS receptor expression and improvement of NO availability (43).
Renin-angiotensin-aldosterone system and Reynaud phenomenon
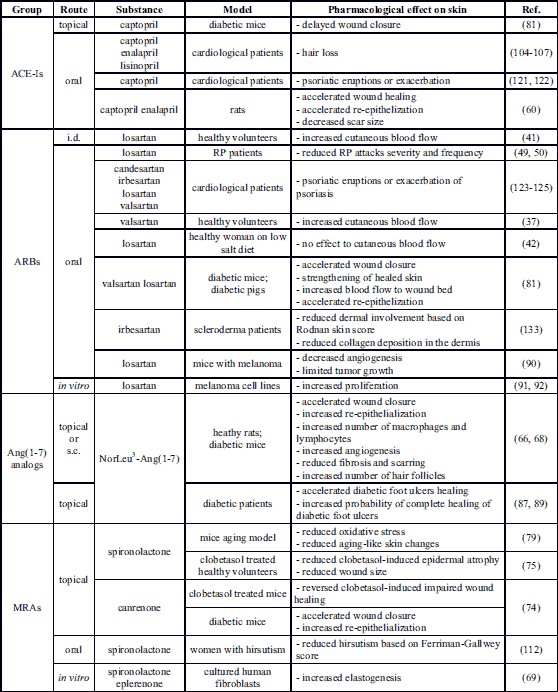
Reynaud phenomenon (RP) is manifested by episodic ischemia in digits caused by overstated cold-induced vasoconstriction in cutaneous arterioles due to increased sensitization of the vasculature (44). Due to their vasodilator effect, both ACE inhibitors (ACE-Is) and ARBs are widely tested as a potent therapy for RP.
Case studies and noncomparative studies show no consistent efficiency of ACE-I therapy in the reduction of the frequency and severity of RP episodes (45, 46). However, the recent review by Cochrane shows that enalapril (ACE-I) slightly increases the frequency of attacks and captopril (ACE-I) does not change the frequency or the duration of attacks (47). A double-blinded trial has also shown that quinapril does not reduce the frequency or severity of RP episodes (48).
Short-term losartan treatment reduces the severity and frequency of RP attacks (49, 50) and this effect is more pronounced in patients with primary RP. Nevertheless, losartan shows an additional clinical benefit in patients with secondary RP associated with scleroderma by reducing N-terminal peptide and vascular cell adhesion molecule expression. However, losartan does not influence the response to the mild cold water stress (49).
To sum up, there is no strong evidence to consider RAAS modulators as an effective first-line therapy; however, they may be recommended if initial therapies are not tolerated or ineffective. Losartan may be preferred over ACE-I, especially in patients with secondary RP associated with systemic scleroderma (51).
RENIN-ANGIOTENSIN-ALDOSTERONE SYSYTEM
IN THE WOUND HEALING PROCESS
The wound healing process consists sequentially of inflammatory, proliferative and remodeling phases and results in scar formation. It has been shown that angiotensins are the key regulator of this process, and this issue has recently been described in detail (52) (Fig. 2). The role of aldosterone is less known.
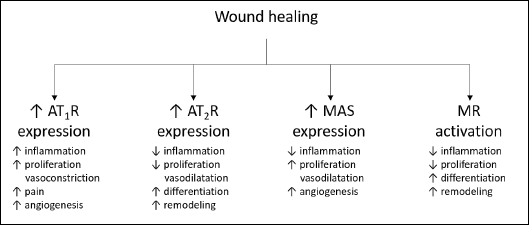 |
Fig. 2. Role of renin-angiotensin-aldosterone system receptors in the wound healing process. AT1R, AT1 receptor; AT2R, AT2 receptor; MAS, specific G protein-coupled receptor MAS; MR, mineralocorticoid receptor |
Angiotensins and angiotensin receptors in the wound healing process
Injury induces dynamic changes in the RAAS expression pattern which highlights the role of the local system in the wound healing process. The expression of each component depends on the current wound healing phase. Using a mice model, it has been shown that, after full-thickness cutaneous wounding, AngII expression is increased during the first 7 days (inflammatory phase) to reach its peak on day 7 (proliferative phase), then it gradually decreases (53). ACE expression in the human keratinocytes increases a short time after wounding (13). What is more, the expression of ACE and chymase is elevated in 1.5 – 3.5-month-old human post-burn scars (14).
A short time after wounding, AT1R, AT2R, and MAS are up-regulated. About 24 hours after injury, the expression of receptors decreases to a near-baseline level and remains constant until the proliferative phase in both human and animal models (3, 18, 53, 54). During the proliferative phase, AT1R, AT2R, and MAS are once again up-regulated, which heightens the expression of AT1R during the early stages of the proliferative phase and increases expression of AT2R and MAS during later stages. Expression of AT2R is predominant during the remodeling phase and is noticeably stronger than that of AT1R in the area of scarring (53, 54). In old human scars (> 12 months), the expression of AT1R is highly increased (14).
AngII plays a key role in the wound healing process in humans by activating both AT1R and AT2R (52). Besides its vasoconstrictive action, AT1R shows pro-inflammatory, pro-oxidative and pro-proliferative potential while AT2R shows an opposite activity (55, 56).
The role of the AngII receptors in the wound healing process has been clearly determined in knock-out animals. It has been shown that an AT1R–/– mice model presents a delayed healing pattern, while AT2R–/– mice display attenuated healing only at the first stage and accelerated during later stages of wound healing. Regardless of the shortened healing time, AT2R–/– mice show impaired quality of wound repair and worse skin quality. Healed skin in AT2R–/– mice is more fragile and fractures under lower tension than in wild type mice (57-59). These data indicate that AT1R is involved in the regulation of proliferation during wound healing, while AT2R regulates the differentiation process. Thus, the functional balance between the expression of AT1R and AT2R in the wounding area is crucial to maintain the optimal healing process.
Results from studies evaluating the effects on skin of pharmacological RAAS modulators confirm the role of RAAS in the wound healing process. Oral administration of ACE-Is (captopril or enalapril) shortens wound closure time, reduces time to complete epithelization and decreases scar size. Furthermore, ACE-I intake in rats results in an enhanced granulation tissue formation process (60). This phenomenon can be explained by the fact that ACE inhibition in the skin does not exert a direct influence on the AngII concentration in the wounded area but increases the local bradykinin level by inhibition of its breakdown which prolongs the duration of the inflammatory phase (61, 62). What is more, during wound healing, local AngI-AngII conversion is mainly catalyzed not by ACE, but by one of the predominant mast cell proteins - chymase (63). Following tissue injury, chymase is secreted into the extracellular matrix (ECM) and activated (27). However, overactivity of chymase during the wound healing process may result in the overgrowth of granulation tissue at the site of wounding, an overabundance of dermal collagen and production of keloid scars (63).
Due to its high pro-proliferative activity (64-66) and anti-inflammatory potential (67), Ang(1-7) has been widely tested as a wound healing supporting factor. Ang(1-7) administered topically increases the thickness of the epidermal layer, accelerates re-epithelization of wounds and angiogenesis, increases ECM production and normalizes tissue architecture in animal models (64, 65, 68). Ang(1-7) synthetic analog NorLeu3-Ang(1-7), which is an MAS agonist, when administered subcutaneously or topically presents even more pronounced dermal repair acceleration, reduces scarring at the site of the wounding and increases the number of new skin appendages and hair follicles (68).
Aldosterone and mineralocorticoid receptors in the wound healing process
Data describing the role of aldosterone and MR in wound healing are limited. Over the last decade, only a few studies have revealed their importance in this process.
MR is responsible for the regulation of epidermal differentiation and for the remodeling phase of the wound healing process (16, 69). The antiproliferative potential of MR has been presented in knock-out animals. In transgenic mice with an overexpression of human MR in keratinocytes (19), highly atrophic epidermis with an abnormally high degree of keratinocyte apoptosis is developed. Correspondingly, the epidermal thickness in MR–/– knock-out mice embryos increases compared to the MR+/– group (70). What is more, in the adult epidermal-specific MR knock-out mice (MREKO) model (16), the epidermal thickness is also increased. Despite increased keratinocyte proliferation and augmented susceptibility to epidermal damage, there is no difference in wound closure time between MREKO and wild type mice. However, MREKO mice present impaired quality of wound repair stemming from defective keratinocyte migration and reduced collagen deposition (16).
Interestingly, regardless of the general trend describing MR as a receptor with pro-inflammatory potential (71, 72), in MREKO mice, MR plays an anti-inflammatory role in the skin through the nuclear factor kappa-light-chain-enhancer of the activated B cells (NF-κB) inhibition mechanism (16). It has been stated that the anti-inflammatory potential of MR is keratinocyte-specific (16).
On the cellular level, aldosterone enhanced elastogenesis improves ECM remodeling and increases collagen type I expression in cultures of human fibroblasts derived from dermal scars and keloids explants (69). Interestingly, the addition of MR antagonist spironolactone or eplerenone to the aldosterone-containing medium results in further amplification of elastogenesis. The mechanism of this process has an MR-independent nongenomic character and is related to the insulin-like growth factor I receptor (IGF1R) (69). However, patients with primary aldosteronism, despite their high aldosterone serum levels, show no changes in dermal collagen deposition (73).
Some of the activities attributed to glucocorticoids are linked to MR antiproliferative activity. The main side effects of topically administered glucocorticoids, e.g. epidermal atrophy or impaired wound healing, have for years been associated primarily with glucocorticoid receptor (GR) activation. However, the most recent studies indicate that this effect also results from the MR-dependent mechanism. It has been shown that MR knock-out mice are characterized by increased keratinocyte proliferation and show resistance to glucocorticoid-prompted epidermal thinning (16). Moreover, the co-administration of canrenone (MR antagonist) to clobetasol (synthetic GR agonist) reverses the delayed wound re-epithelialization and restores keratinocyte outgrowth in both mice in vivo and human ex vivo models (74). Furthermore, a SPIREPI clinical trial showed that spironolactone reduced skin atrophy in clobetasol treated patients (75).
RENIN-ANGIOTENSIN-ALDOSTERONE
SYSYTEM AND SKIN AGING
The aging of the skin contributes to physiological dysfunctions such as mitochondrial disorder, genomic instability, excessive ECM breakdown and disturbances in intracellular communication. Aging is a combined effect of both intrinsic and extrinsic factors contributing to increased generation of ROS (76). The most explained extrinsic factor for skin aging is ultraviolet radiation (UV) exposure, also referred to as photoaging (77).
Recently, it has been shown that the expression of skin RAAS components can be altered in photoaging experimental models. Repetitive UV irradiation of a hairless mouse resulted in increased expression of AngII, ACE, and AT1R in the epidermis as well as amplified expression of AT2R in the upper dermis. Repetitive topical administration of ACE-I, enalapril, reduced expression of AngII in the epidermis, decreased transepidermal water loss and reduced wrinkle grade. Despite the improvement in epidermal barrier function, epidermal thickness decreases in enalapril-treated mice, which indicates the antiproliferative potential of ACE-Is (78).
UV irradiation of metabolic syndrome mice (aging-like model) results in increased MR downstream genes expression, which suggests augmented MR activation, whereas MR expression does not change. Topical application of spironolactone decreases UV-induced aging-like histological skin changes to nearly normal condition and reduces expression of MR downstream genes. Interestingly, chronic exposure of healthy mice to UV results in similar skin damage, however not related to MR signaling up-regulation (79).
There are limited data describing changes in RAAS expression in the physiological aging process; however, it has been noticed that the pharmacological response to RAAS stimulation in the skin is age-dependent. The vasodilatory effect of low AngII doses is less pronounced in the skin of older volunteers, while high AngII doses result in greater vasoconstriction in this group. These results are explained by reduced AT2R expression with age since AT2R selective inhibition has no effect on the older adult group, while it significantly reduces cutaneous perfusion in the younger group (32).
Moreover, RAAS may also be involved in age-related increase in the skin sympathetic nerve activity. It has been shown that attenuation of AngII-dependent vasoconstriction after adrenoreceptor blockade is more pronounced in the older adult group (32). Similarly, local administration of losartan to older volunteers results in a 50% reduction in vasoconstrictive response to the whole body cooling, whereas there is no such reaction in the younger group (80). An analogous observation has been made in an animal wound healing model. Topical valsartan administration results in more pronounced acceleration of wound healing in old mice than in young ones (81). The observed differences suggest that the pharmacological response to RAAS modulators is shifted with age from the protective AT2R pathway to the more pathogenic AT1R pathway (32).
RENIN-ANGIOTENSIN-ALDOSTERONE
SYSYTEM IN DIABETIC SKIN
About 30% of diabetic patients suffer from variable skin complications associated with local inflammation, increased vascular permeability or impaired wound healing. Since all of these symptoms can be modulated by local RAAS, a potential relationship between RAAS and diabetes skin complications could be assumed.
Changes in renin-angiotensin-aldosterone system expression and histological changes in diabetic skin
There are no data describing RAAS components expression in the skin of patients with diabetes; however, it is known that the activity of systemic RAAS is augmented in diabetic patients (82). The available data come mainly from experimental models. The concentration of AngII in the skin of streptozotocin (STZ)-induced diabetic rats is almost 30% higher than in normoglycemic animals (17). Skin AT1R expression is increased, while the expression of AT2R is decreased. The AT1R/AT2R ratio disturbances in the skin of diabetic patients may be partly explained by insulin deficiency since insulin and insulin-like growth factors (IGF) have been reported to be responsible for the AT2R expression up-regulation (83). The unopposed overactivation of AT1R may lead to the development of local inflammatory phenotypes in the skin, over-pronounced vasoconstriction, and decreased local microcirculation resulting in tissue malnutrition and impaired wound healing (84, 85). Moreover, diabetes leads to differing RAAS components expression pattern in the skin structures. The expression of AT1R is observed both in the inner and outer hair root sheaths in STZ diabetic rats, while in healthy animals it is expressed only in the membrane cells of the inner hair root sheath (17).
Histological analysis has shown that the skin of STZ-induced rats displays an atrophic character and presents a decreased number of fibroblasts. Moreover, the collagen fibers in ECM are fractured and degenerated (17). This abnormal ECM microstructure is at least partially caused by augmented AngII level. It has been shown that AngII stimulation of fibroblasts derived from the STZ-induced diabetic rat skin leads to enhanced expression of collagen type I and III, as well as, tissue growth factor b and inhibition of metalloproteinase-I. These changes are mediated via an AT1R dependent mechanism since the administration of losartan inhibits AngII-induced ECM degradation, whereas PD123319 is not effective (86).
Recently, it has been stated that MR mRNA expression is increased in the skin of STZ diabetic mice (74). However, more specific data on MR levels in diabetes are not available.
Effect of pharmacological renin-angiotensin-aldosterone system modulators on diabetic skin
There are several studies describing the beneficial effects of RAAS-modulating drugs on diabetic skin. In diabetic murine and porcine models, valsartan accelerates wound closure and strengthens the healed skin when administered topically from the 7th day after wounding. The epidermal layer in valsartan-treated groups is thicker and the dermis is characterized by more organized collagen fiber structure. Interestingly, valsartan applied during the early stages of wound healing resulted in delayed wound closure, according to the authors of the cited study probably due to the disruption of the inflammatory phase and impaired transition to the proliferative and remodeling phases (81). In contrast, topical administration of captopril applied 7 days after wounding delayed the wound closure time in diabetic mice. Co-addition of 1% valsartan to the captopril formulation did not shorten this parameter (81). The discrepancies in wound healing activity between valsartan and captopril may be related both to the fact that administration of ARB results selectively in AT1R blockade, while ACE-Is administration results in reduced activation of both AT1R and AT2R, as well as to the fact that ACE-I increases local bradykinin level by inhibition of its breakdown (61).
Clinical studies have confirmed the effectiveness of NorLeu3-Ang(1-7) as a well-tolerated topical agent for the treatment of chronic nonhealing diabetic wounds. NorLeu3-Ang(1-7) shortens the wound closure time, reaching an over 3 fold higher probability of complete healing by week 24 compared to placebos (87, 88).
MR blockade also shows a beneficial effect in the treatment of diabetic wounds. It has been shown that topical application of canrenone improves wound closure time, augments re-epithelization and increases keratinocyte proliferation in STZ diabetic mice (74).
RENIN-ANGIOTENSIN-ALDOSTERONE
SYSYTEM IN SKIN CANCERS
It has been widely documented that, during carcinogenesis and malignancy, the normal physiological actions of RAAS in inflammation, proliferation, and angiogenesis might promote tumor growth during malignancy (89). Most of the data regarding the link between RAAS and skin cancers are related to melanoma.
The expression of RAAS components, at both the mRNA and protein levels, has been confirmed in melanoma tissues and tumor-associated vessels. The immunohistochemical analysis of human explanted melanoma tissue has shown the presence of AngII in tumors and surrounding stromal cells, while AT1R has been found mainly in the tumor-associated vessels (90). Moreover, in vitro studies have shown that both AT1R and AT2R are expressed at the mRNA level in most melanoma cell lines (91, 92).
It has been shown that the blockade of AT1R by losartan results in decreased angiogenesis and limited tumor growth in mice with melanoma (90), indicating the involvement of AngII-dependent pathways in melanoma. Moreover, it has been shown that AngII stimulation of human melanoma cell cultures increases proliferation, amplifies expression of F-actin, and enhances Na+/H+ exchanger isoform 1 (NHE1) activity; however, it decreases cell migration. Interestingly, losartan treatment, independently of the presence of AngII, increases proliferation, and reduces NHE1 and migratory activity, but increases cell adhesion and invasion. AT2R blockade with PD123319 increases adhesion and invasion but has no effect on proliferation and migration (91). The mechanism explaining these results was not proposed by the authors of this study. However, corresponding observations were described in another in vitro study (92) in which losartan treatment or AT1R gene knockdown also promoted proliferation in human melanoma cell lines in serum-free conditions, while in serum-replete conditions losartan or gene knockout had no effect on the proliferation. AT2R blockade with PD123319 or EMA401 (highly selective antagonist) inhibited melanoma growth and angiogenesis in serum-free condition while in serum-replete conditions the inhibitors had no effect on proliferation. Thus, unlike usual AT1R presented growth suppressor-activity, AT2R acted as an oncogene. According to the authors, the observed differences depending on serum presence might be caused by the fact that bovine serum contains an abundance of growth-promoting as well as inhibitory factors that could mask the effect of the studied factors (92).
ACE overexpression in macrophages leads to increased resistance to melanoma, due to increased inflammatory response, in ACE10/10 transgenic mice. Captopril treatment abolishes the protective character of the ACE10/10 phenotype and leads to more rapid tumor growth (93).
The involvement of RAAS in non-melanoma skin cancers is less described. The immunohistochemical study of human basal cell carcinoma (BCC) has shown weakly positive AT1R staining in the cells at the center of tumor nests and the tumor cells surrounding the keratinizing cysts (7). In squamous cell carcinoma (SCC), the expression of both AT1R and AT2R has been observed on cells within the tumor nests and the stroma (94). ACE staining has been observed in the cytoplasm and the nuclei of both BCC and SCC, while ACE2 staining has not been observed in any tumor cells (94, 95).
However, recent clinical studies on relationship between ARB- and ACE-I intake and the melanoma and non-melanoma skin cancer risk are inconsistent (96-101).
RENIN-ANGIOTENSIN-ALDOSTERONE SYSYTEM
AND HAIR LOSS PROCESS
Although hair follicles express almost a full set of RAAS-associated receptors (Table 1), the role of this system in the regulation of hair growth has not yet been described.
The data describing serum ACE activity in patients with alopecia areata are not consistent; however, both of the studies performed to date have exploited relatively small groups (102, 103). Moreover, one study has shown that in patients with mild or moderate alopecia areata, ACE tissue activity is decreased in the epidermis, as well as in follicular epithelium and endothelium (103).
The role of RAAS in the hair loss process should be considered as highly possible due to reports linking to the intake of ACE-I to hair loss (104-107). Interestingly, discontinuation of ACE-I treatment (105) or switching patients from ACE-I to ARB (104) has resulted in hair regrowth. However, the mechanism of ACE-I-induced alopecia is unknown.
There are some studies indicating the potential role of aldosterone and MR in hair loss. Postnatal MR overexpression in keratinocytes in mice has resulted in delayed alopecia, hair follicle dystrophy, and abnormalities of the hair cycle, without alternation of the interfollicular epidermis (19). Moreover, clinical studies have shown that both male and female patients with androgenic alopecia have higher aldosterone serum levels (108-110). The mechanism underlying this process may be associated with the skin microinflammation found in those patients (111). Despite the potential role of aldosterone, there is only one study evaluating the influence of MR blockade on hair. This showed that spironolactone treatment reduced hair shaft size and weight resulting in softer, finer hair and a slower growth rate in patients with hirsutism (112). In this study, the action of spironolactone was mainly associated with its antiandrogenic activity. Nevertheless, the potential role of MR blockade cannot be excluded (112).
RENIN-ANGIOTENSIN-ALDOSTERONE
SYSYTEM AND PSORIASIS
Psoriasis is a genetically determined autoimmunological disease manifesting in the skin, triggered through mechanisms involving genes, and interacting with environmental factors (113).
It has been reported that ACE activity in both serum and skin is elevated in patients with psoriasis, whereas tissue activity is almost doubled in lesional skin compared to uninvolved skin (114, 115). ACE tissue activity in other inflammatory skin conditions such as lichen planus or seborrheic dermatitis is not statistically different from control groups, which suggests that ACE may be associated specifically with the psoriasis pathomechanism (114). Moreover, effective psoriasis treatment (local therapy, photochemotherapy or cytostatic therapy) results in a reduction of both ACE serum and skin activity to the base level (114).
Epidemiological studies (116-119) have shown that that ACE I/D polymorphism may be associated with the risk of psoriasis in some populations and the psoriasis odds ratio is higher in genotype I/I patients (119). AT1R-A1166C/A polymorphism is also associated with an increased risk and severity of psoriasis. The presence of the AT1R-A1166C allele increases the risk of psoriasis by almost 5-fold (120).
Interestingly, both ACE-Is (121, 122) and ARBs (123-125) are reported as factors triggering psoriasis eruption or flare-up of the existing disease. For ACE-I, it has been suggested that the mechanism underlying these effects is related to inhibition of bradykinin degradation and alternation of the kallikrein-kinin system leading to augmented local kinin concentrations and induction of inflammatory lesions (61, 126). The mechanism of psoriasis related to ARBs is unknown.
The aldosterone serum level in patients with psoriasis is elevated compared with patients with other skin diseases (127). Moreover, the pathohistological similarities between primary hyperaldosteronism and psoriasis-like epidermal hyperplasia and high immune infiltrates may suggest the involvement of aldosterone in the pathomechanism of psoriasis (73). Nevertheless, there is no direct evidence confirming this association.
RENIN-ANGIOTENSIN-ALDOSTERONE
SYSYTEM IN SCLERODERMA
Scleroderma is a chronic disease of unknown etiology causing excessive collagen deposition in the skin and internal organs, as well as widespread microvascular damage in small-and medium-sized vessels (128).
There are few studies indicating the role of RAAS in the pathogenesis of this disease. The expression of angiotensinogen has been found in fibroblasts from patients with diffuse cutaneous scleroderma who have high levels of serum AngII, but not in fibroblasts from healthy donors (129). However, the data regarding AngII serum level are conflicting. One study has shown that patients with scleroderma have elevated AngII levels (129), while a second study has shown that scleroderma patients present reduced serum levels of AngII, along with Ang(1-7), and ACE (130). Despite contrary AngII level results, it has been shown that scleroderma patients present significantly higher serum levels of AT1R agonistic autoantibodies (anti-AT1R) than is the case in a healthy population, as well as patients with rheumatoid arthritis, primary Sjogren syndrome, primary RP, and morphea (131). Moreover, the severity of scleroderma has been positively correlated with the anti-AT1R level (131). The activity of the anti-AT1R level has been evaluated in further studies, which showed that the addition of IgG (containing anti-AT1R and anti-ETA) obtained from systemic scleroderma patients (SSc-IgG) to human microvascular endothelial cell cultures results in increased IL-8 expression and decreased wound repair. Moreover, SSc-IgG increased type I collagen expression and increased migration of fibroblasts obtained from healthy donors (132). All of the anti-AT1R-induced parameters were reduced by AT1R blockade with valsartan (ARB) (132). These results confirmed that anti-AT1R, similarly to Ang II (84), presents pro-inflammatory and profibrotic potential trough AT1R agonism.
The provided data suggest that ARBs may show some clinical benefits in the treatment of skin-related scleroderma symptoms. There is a report showing that a 4-month irbesartan (ARB) therapy resulted in a significant improvement of dermal fibrosis in patients with systemic scleroderma expressed by a reduced collagen deposition in the dermis (133). Currently, none of the RAAS-modulating drugs are recommended in the guidelines (134, 135) to treat scleroderma skin symptoms.
Conclusions
- Fully expressed tissue RAAS is present in the human skin and it is an important player in the skin biology regulating the physiological processes such as skin microcirculation, aging, sweating and wound healing.
- RAAS activity is changed in skin diseases, e.g. diabetic skin disorders, skin cancers, alopecia, psoriasis or scleroderma.
- Orally-taken ACE-Is can affect the skin causing side effects such as alopecia and psoriasis exacerbation.
- Some RAAS-modulating drugs, as well as AngII, Ang(1-7), and its analog NorLeu3-Ang(1-7), when applied topically can provide some beneficial effects on skin wound healing.
- The addition of a mineralocorticoid receptor antagonist to glucocorticoid may decrease the side effects of glucocorticoid during wound healing treatment. Moreover, it seems that this combination may also be an effective treatment for inflammatory skin diseases, e.g. atopic dermatitis or psoriasis.
- Orally-taken ARBs may reduce skin-related symptoms in patients with scleroderma.
Acknowledgments: This study was supported by projects N/ST/MN/17/003/2226 and N/ST/MN/18/001/2226 of the Medical University of Bialystok.
Conflict of interests: None declared.
REFERENCES
- Paul M, Poyan Mehr A, Kreutz R. Physiology of local renin-angiotensin systems. Physiol Rev 2006; 86: 747-803.
- Nehme A, Cerutti C, Dhaouadi N, et al. Atlas of tissue renin-angiotensin-aldosterone system in human: a transcriptomic meta-analysis. Sci Rep 2015; 5: 10035. doi: 10.1038/srep10035
- Kimura B, Sumners C, Phillips MI. Changes in skin angiotensin II receptors in rats during wound healing. Biochem Biophys Res Commun 1992; 187: 1083-1090.
- Kenouch S, Lombes M, Delahaye F, Eugene E, Bonvalet JP, Farman N. Human skin as target for aldosterone: coexpression of mineralocorticoid receptors and 11 beta-hydroxysteroid dehydrogenase. J Clin Endocrinol Metab 1994; 79: 1334-1341.
- Steckelings UM, Czarnetzki BM. The renin-angiotensin-system in the skin. Evidence for its presence and possible functional implications. Exp Dermatol 1995; 4: 329-334.
- Takeda H, Kondo S. Immunohistochemical study of angiotensin receptors in normal human sweat glands and eccrine poroma. Br J Dermatol 2001; 144: 1189-1192.
- Takeda H, Katagata Y, Kondo S. Immunohistochemical study of angiotensin receptors in human anagen hair follicles and basal cell carcinoma. Br J Dermatol 2002; 147: 276-280.
- Bader M. Tissue renin-angiotensin-aldosterone systems: targets for pharmacological therapy. Annu Rev Pharmacol Toxicol 2010; 50: 439-465.
- Farman N, Maubec E, Poeggeler B, Klatte JE, Jaisser F, Paus R. The mineralocorticoid receptor as a novel player in skin biology: beyond the renal horizon? Exp Dermatol 2010; 19: 100-107.
- Romero CA, Orias M, Weir MR. Novel RAAS agonists and antagonists: clinical applications and controversies. Nat Rev Endocrinol 2015; 11: 242-252.
- Mirabito Colafella KM, Bovee DM, Danser AH. The renin angiotensin aldosterone system and its therapeutic targets. Exp Eye Res 2019; May 23: S0014-4835(19)30157. doi: 10.1016/j.exer.2019.05.020.
- Nehme A, Zouein FA, Zayeri ZD, Zibara K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J Cardiovasc Dev Dis 2019; 6: E14. doi: 10.3390/jcdd6020014
- Steckelings UM, Wollschlager T, Peters J, Henz BM, Hermes B, Artuc M. Human skin: source of and target organ for angiotensin II. Exp Dermatol 2004; 13: 148-154.
- Akershoek JJ, Vlig M, Brouwer K, et al. The presence of tissue renin-angiotensin system components in human burn wounds and scars. Burn Open 2018; 2: 114-121.
- Steckelings UM, Artuc M, Paul M, Stoll M, Henz BM. Angiotensin II stimulates proliferation of primary human keratinocytes via a non-AT1, non-AT2 angiotensin receptor. Biochem Biophys Res Commun 1996; 229: 329-333.
- Boix J, Sevilla LM, Saez Z, Carceller E, Perez P. Epidermal mineralocorticoid receptor plays beneficial and adverse effects in skin and mediates glucocorticoid responses. J Invest Dermatol 2016; 136: 2417-2426.
- Hao S yun, Ren M, Yang C, et al. Activation of skin renin-angiotensin system in diabetic rats. Endocrine 2011; 39: 242-250.
- Jadhav SS, Sharma N, Meeks CJ, et al. Effects of combined radiation and burn injury on the renin-angiotensin system. Wound Repair Regen 2013; 21: 131-140.
- Sainte Marie Y, Toulon A, Paus R, et al. Targeted skin overexpression of the mineralocorticoid receptor in mice causes epidermal atrophy, premature skin barrier formation, eye abnormalities, and alopecia. Am J Pathol 2007; 171: 846-860.
- Tomlinson JW, Walker EA, Bujalska IJ, et al. 11b-Hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev 2004; 25: 831-866.
- Fujii N, Meade RD, Paull G, et al. Can intradermal administration of angiotensin II influence human heat loss responses during whole body heat stress? J Appl Physiol 2015; 118: 1145-1153.
- Conn JW. Aldosteronism in man. JAMA 1963; 183: 775-781.
- Hanukoglu I, Boggula VR, Vaknine H, Sharma S, Kleyman T, Hanukoglu A. Expression of epithelial sodium channel (ENaC) and CFTR in the human epidermis and epidermal appendages. Histochem Cell Biol 2017; 147: 733-748.
- Lee IH, Campbell CR, Cook DI, Dinudom A. Regulation of epithelial Na+ channels by aldosterone: role of Sgk1. Clin Exp Pharmacol Physiol 2008; 35: 235-241.
- Kirby CR, Convertino VA. Plasma aldosterone and sweat sodium concentrations after exercise and heat acclimation. J Appl Physiol 1986; 61: 967-9 70.
- Walton S, Cunliffe WJ, Lookingbill P, Keczkes K. Lack of effect of topical spironolactone on sebum excretion. Br J Dermatol 1986; 114: 261-264.
- Riekki R, Harvima IT, Jukkola A, Risteli J, Oikarinen A. The production of collagen and the activity of mast-cell chymase increase in human skin after irradiation therapy. Exp Dermatol 2004; 13: 364-371.
- Liu LH, Fan X, Li HT, An XX, Yang RY. Angiotensin II promotes melanogenesis via angiotensin II type 1 receptors in human melanocytes. Mol Med Rep 2015; 12: 651-656.
- Zhang Y, Somers KR, Becari C, et al. Comparative expression of renin-angiotensin pathway proteins in visceral versus subcutaneous fat. Front Physiol 2018; 9: 1370. doi: 10.3389/fphys.2018.01370
- Bafford R, Sui XX, Park M, et al. Mineralocorticoid receptor expression in human venous smooth muscle cells: a potential role for aldosterone signaling in vein graft arterialization. Am J Physiol Circ Physiol 2011; 301: H41-H47.
- Sztechman D, Czarzasta K, Cudnoch-Jedrzejewska A, Szczepanska-Sadowska E, Zera T. Aldosterone and mineralocorticoid receptors in regulation of the cardiovascular system and pathological remodelling of the heart and arteries. J Physiol Pharmacol 2018; 69: 829-845.
- Lang JA, Krajek AC. Age-related differences in the cutaneous vascular response to exogenous angiotensin II. Am J Physiol Circ Physiol 2019; 316: H516-H521.
- Wang M, Zhang J, Jiang LQ, et al. Proinflammatory profile within the grossly normal aged human aortic wall. Hypertension 2007; 50: 219-227.
- Sampson AK, Moritz KM, Jones ES, Flower RL, Widdop RE, Denton KM. Enhanced angiotensin II type 2 receptor mechanisms mediate decreases in arterial pressure attributable to chronic low-dose angiotensin II in female rats. Hypertension 2008; 52: 666-671.
- Sampson AK, Hilliard LM, Moritz KM, et al. The arterial depressor response to chronic low-dose angiotensin II infusion in female rats is estrogen dependent. Am J Physiol Integr Comp Physiol 2012; 302: R159-R165.
- Wenzel RR, Ruthemann J, Bruck H, Schafers RF, Michel MC, Philipp T. Endothelin-A receptor antagonist inhibits angiotensin II and noradrenaline in man. Br J Clin Pharmacol 2001; 52: 151-157.
- Mitchell A, Rushentsova U, Siffert W, Philipp T, Wenzel RR. The angiotensin II receptor antagonist valsartan inhibits endothelin 1-induced vasoconstriction in the skin microcirculation in humans in vivo: influence of the G-protein b3 subunit (GNB3) C825T polymorphism. Clin Pharmacol Ther 2006; 79: 274-281.
- Barki-Harrington L, Luttrell LM, Rockman HA. Dual inhibition of b-adrenergic and angiotensin II receptors by a single antagonist. Circulation 2003; 108: 1611-1618.
- Zeng C, Hopfer U, Asico LD, Eisner GM, Felder RA, Jose PA. Altered AT1 receptor regulation of ETB receptors in renal proximal tubule cells of spontaneously hypertensive rats. Hypertension 2005; 46: 926-931.
- Hydar A, Haribabu B, Natarajan K, Berk BC. Crosstalk coregulation mechanisms of G protein-coupled receptors and receptor tyrosine kinases. Transmembrane Signal Protoc 2006; 332: 51-78.
- Stewart JM, Taneja I, Raghunath N, Clarke D, Medow MS. Intradermal angiotensin II administration attenuates the local cutaneous vasodilator heating response. Am J Physiol Heart Circ Physiol 2008; 295: H327-H334.
- Cavka A, Cosic A, Grizelj I, et al. Effects of AT1 receptor blockade on plasma thromboxane A2 (TXA2) level and skin microcirculation in young healthy women on low salt diet. Kidney Blood Press Res 2013; 37: 432-442.
- Kvandova M, Barancik M, Balis P, Puzserova A, Majzunova M, Dovinova I. The peroxisome proliferator-activated receptor gamma agonist pioglitazone improves nitric oxide availability, renin-angiotensin system and aberrant redox regulation in the kidney of pre-hypertensive rats. J Physiol Pharmacol 2018; 69: 231-243.
- Fardoun MM, Nassif J, Issa K, Baydoun E, Eid AH. Raynaud’s phenomenon: a brief review of the underlying mechanisms. Front Pharmacol 2016; 7: 438. doi: 10.3389/fphar.2016.00438
- Wood HM, Ernst ME. Renin-angiotensin system mediators and Raynaud’s phenomenon. Ann Pharmacother 2006; 40: 1998-2002.
- Henness S, Wigley FM. Current drug therapy for scleroderma and secondary Raynaud’s phenomenon: evidence-based review. Curr Opin Rheumatol 2007; 19: 611-618.
- Stewart M, Morling JR. Oral vasodilators for primary Raynaud’s phenomenon. Cochrane Database Syst Rev 2012; doi: 10.1002/14651858.CD006687.pub3.
- Gliddon AE, Dore CJ, Black CM, et al. Prevention of vascular damage in scleroderma and autoimmune Raynaud’s phenomenon: a multicenter, randomized, double-blind, placebo-controlled trial of the angiotensin-converting enzyme inhibitor quinapril. Arthritis Rheum 2007; 56: 3837-3846.
- Dziadzio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week, randomized, parallel-group, controlled trial. Arthritis Rheum 1999; 42: 2646-2655.
- Pancera P, Sansone S, Secchi S, Covi G, Lechi A. The effects of thromboxane A2 inhibition (Picotamide) and angiotensin II receptor blockade (Losartan) in primary Raynaud’s phenomenon. J Intern Med 1997; 242: 373-376.
- Shinn BW. The description and treatment of Raynaud’s disease/phenomenon. US Pharmacist 2008; 33: 36-48.
- Bernasconi R, Nystrom A. Balance and circumstance: the renin angiotensin system in wound healing and fibrosis. Cell Signal 2018; 51: 34-46.
- Wu HJ, Liu HW, Cheng B, et al. The change of angiotensin II production and its receptor expression during wound healing: possible role of angiotensin II in wound healing [in Chinese]. Zhonghua Zheng Xing Wai Ke Za Zhi 2011; 27: 124-128.
- Steckelings UM, Henz BM, Wiehstutz S, Unger T, Artuc M. Differential expression of angiotensin receptors in human cutaneous wound healing. Br J Dermatol 2005; 153: 887-893.
- Vajapey R, Rini D, Walston J, Abadir P. The impact of age-related dysregulation of the angiotensin system on mitochondrial redox balance. Front Physiol 2014; 5: 439. doi: 10.3389/fphys.2014.00439
- Kaschina E, Unger T. Angiotensin AT1/AT2 receptors: regulation, signalling and function. Blood Press 2003; 12: 70-88.
- Yahata Y, Shirakata Y, Tokumaru S, et al. A novel function of angiotensin II in skin wound healing. Induction of fibroblast and keratinocyte migration by angiotensin II via heparin-binding epidermal growth factor (EGF)-like growth factor-mediated EGF receptor transactivation. J Biol Chem 2006; 281: 13209-13216.
- Mizoue S, Iwai M, Ide A, et al. Role of angiotensin II receptor subtypes in conjunctival wound healing. Curr Eye Res 2006; 31: 129-136.
- Faghih M, Hosseini SM, Smith B, , et al. Knockout of angiotensin AT 2 receptors accelerates healing but impairs quality. Aging (Albany NY) 2015; 7: 1185-1197.
- Torgal SS. Evaluation of wound healing activity of angiotensin converting enzyme inhibitors in Wistar rats. Recent Res Sci Technol 2010; 2: 76-80.
- Scholzen TE, Stander S, Riemann H, Brzoska T, Luger TA. Modulation of cutaneous inflammation by angiotensin-converting enzyme. J Immunol 2003; 170: 3866-3873.
- Parenti A, Morbidelli L, Ledda F, Granger HJ, Ziche M. The bradykinin/B1 receptor promotes angiogenesis by up-regulation of endogenous FGF-2 in endothelium via the nitric oxide synthase pathway. FASEB J 2001; 15: 1487-1489.
- Wang R, Chen J, Zhang Z, Cen Y. Role of chymase in the local renin-angiotensin system in keloids: inhibition of chymase may be an effective therapeutic approach to treat keloids. Drug Des Devel Ther 2015; 9: 4979-4988.
- Rodgers K, Xiong S, Felix J, et al. Development of angiotensin (1-7) as an agent to accelerate dermal repair. Wound Repair Regen 2001; 9: 238-247.
- Rodgers K, Xiong S, DiZerega G. Accelerated recovery from irradiation injury by angiotensin peptides. Cancer Chemother Pharmacol 2002; 49: 403-411.
- Rodgers KE, Roda N, Felix JC, Espinoza T, Maldonado S, DiZerega G. Histological evaluation of the effects of angiotensin peptides on wound repair in diabetic mice. Exp Dermatol 2003; 12: 784-790.
- Rodrigues Prestes TR, Rocha NP, Miranda AS, Teixeira AL, Simoes-e-Silva AC. The anti-inflammatory potential of ACE2/angiotensin-(1-7)/Mas receptor axis: evidence from basic and clinical research. Curr Drug Targets 2017; 18: 1301-1313.
- Rodgers KE, Espinoza T, Felix J, Roda N, Maldonado S, DiZerega G. Acceleration of healing, reduction of fibrotic scar, and normalization of tissue architecture by an angiotensin analogue, NorLeu3-A(1-7). Plast Reconstr Surg 2003; 111: 1195-1206.
- Mitts TF, Bunda S, Wang Y, Hinek A. Aldosterone and mineralocorticoid receptor antagonists modulate elastin and collagen deposition in human skin. J Invest Dermatol 2010; 130: 2396-2406.
- Boix J, Carceller E, Sevilla LM, Marcos-Garces V, Perez P. The mineralocorticoid receptor plays a transient role in mouse skin development. Exp Dermatol 2016; 25: 69-71.
- Munoz-Durango N, Vecchiola A, Gonzalez-Gomez LM, et al. Modulation of immunity and inflammation by the mineralocorticoid receptor and aldosterone. Biomed Res Int 2015; 2015: 652738. doi: 10.1155/2015/652738
- Gilbert KC, Brown NJ. Aldosterone and inflammation. Curr Opin Endocrinol Diabetes Obes 2010; 17: 199-204.
- Boix J, Bigas J, Sevilla LM, et al. Primary aldosteronism patients show skin alterations and abnormal activation of glucocorticoid receptor in keratinocytes. Sci Rep 2017; 7: 15806. doi: 10.1038/s41598-017-16216-5
- Nguyen VT, Farman N, Maubec E, et al. Re-epithelialization of pathological cutaneous wounds is improved by local mineralocorticoid receptor antagonism. J Invest Dermatol 2016; 136: 2080-2089.
- Maubec E, Laouenan C, Deschamps L, et al. Topical mineralocorticoid receptor blockade limits glucocorticoid-induced epidermal atrophy in human skin. J Invest Dermatol 2015; 135: 1781-1789.
- Farage MA, Miller KW, Elsner P, Maibach HI. Characteristics of the aging skin. Adv Wound Care (New Rochelle) 2013; 2: 5-10.
- Kammeyer A, Luiten RM. Oxidation events and skin aging. Ageing Res Rev 2015; 21: 16-29.
- Matsuura-Hachiya Y, Arai KY, Ozeki R, Kikuta A, Nishiyama T. Angiotensin-converting enzyme inhibitor (enalapril maleate) accelerates recovery of mouse skin from UVB-induced wrinkles. Biochem Biophys Res Commun 2013; 442: 38-43.
- Nagase T, Akase T, Sanada H, et al. Aging-like skin changes in metabolic syndrome model mice are mediated by mineralocorticoid receptor signaling. Aging Cell 2013; 12: 50-57.
- Lang JA, Kolb KE. Angiotensin II type I receptor blockade attenuates reflex cutaneous vasoconstriction in aged but not young skin. Am J Physiol Circ Physiol 2015; 308: H1215-H1220.
- Abadir P, Hosseini S, Faghih M, et al. Topical reformulation of valsartan for treatment of chronic diabetic wounds. J Invest Dermatol 2018; 138: 434-443.
- Hollenberg NK, Stevanovic R, Agarwal A, et al. Plasma aldosterone concentration in the patient with diabetes mellitus Rapid Communication. Kidney Int 2004; 65: 1435-1439.
- Kambayashi Y, Nagata K, Ichiki T, Inagami T. Insulin and insulin-like growth factors induce expression of angiotensin type-2 receptor in vascular-smooth-muscle cells. Eur J Biochem 1996; 239: 558-565.
- Stawski L, Han R, Bujor AM, Trojanowska M. Angiotensin II induces skin fibrosis: a novel mouse model of dermal fibrosis. Arthritis Res Ther 2012; 14: R194. doi: 10.1186/ar4028
- Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol 2003; 35: 881-900.
- Ren M, Hao S, Yang C, et al. Angiotensin II regulates collagen metabolism through modulating tissue inhibitor of metalloproteinase-1 in diabetic skin tissues. Diabetes Vasc Dis Res 2013; 10: 426-435.
- Rodgers KE, Bolton LL, Verco S, DiZerega GS. NorLeu(3)-angiotensin (1-7) [DSC127] as a therapy for the healing of diabetic foot ulcers. Adv Wound Care 2015; 4: 339-345.
- Rodgers K, Verco S, Bolton L, Dizerega G. Accelerated healing of diabetic wounds by NorLeu(3)-angiotensin (1-7). Expert Opin Investig Drugs 2011; 20: 1575-1581.
- George AJ, Thomas WG, Hannan RD. The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer 2010; 10: 745-759.
- Otake AH, Mattar AL, Freitas HC, et al. Inhibition of angiotensin II receptor 1 limits tumor-associated angiogenesis and attenuates growth of murine melanoma. Cancer Chemother Pharmacol 2010; 66: 79-87.
- Olschewski DN, Hofschroer V, Nielsen N, Seidler DG, Schwab A, Stock C. The angiotensin II type 1 receptor antagonist losartan affects NHE1-dependent melanoma cell behavior. Cell Physiol Biochem 2018; 45: 2560-2576.
- Renziehausen A, Wang H, Rao B, et al. The renin angiotensin system (RAS) mediates bifunctional growth regulation in melanoma and is a novel target for therapeutic intervention. Oncogene 2019; 38: 2320-2336.
- Shen XZ, Li P, Weiss D, et al. Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am J Pathol 2007; 170: 2122-2134.
- Ram RS, Brasch HD, Dunne JC, Davis PF, Tan ST, Itinteang T. Cancer stem cells in moderately differentiated lip squamous cell carcinoma express components of the renin-angiotensin system. Front Surg 2017; 4: 30. doi: 10.3389/fsurg.2017.00030
- Grzegrzolka J, Swiatko K, Pula B, et al. ACE and ACE2 expression in normal and malignant skin lesions. Folia Histochem Cytobiol 2013; 51: 232-238.
- Moscarelli L, Zanazzi M, Mancini G, et al. Keratinocyte cancer prevention with ACE inhibitors, angiotensin receptor blockers or their combination in renal transplant recipients. Clin Nephrol 2010; 73: 439-445.
- Yoon C, Yang H-S, Jeon I, Chang Y, Park SM. Use of angiotensin-converting-enzyme inhibitors or angiotensin-receptor blockers and cancer risk: a meta-analysis of observational studies. Can Med Assoc J 2011; 183: E1073-E1084.
- Xiong MY, Rizzo AE, Cohen TS, et al. Predictors of squamous cell carcinoma in high-risk patients in the VATTC trial. J Invest Dermatol 2013; 133: 1521-1532.
- Schmidt SAJ, Schmidt M, Mehnert F, Lemeshow S, Sorensen HT. Use of antihypertensive drugs and risk of skin cancer. J Eur Acad Dermatology Venereol 2015; 29: 1545-1554.
- Nardone B, Majewski S, Kim AS, et al. Melanoma and non-melanoma skin cancer associated with angiotensin-converting-enzyme inhibitors, angiotensin-receptor blockers and thiazides: a matched cohort study. Drug Saf 2017; 40: 249-255.
- Gandini S, Palli D, Spadola G, et al. Anti-hypertensive drugs and skin cancer risk: a review of the literature and meta-analysis. Crit Rev Oncol Hematol 2018; 122: 1-9. doi: 10.1016/j.critrevonc.2017.12.003
- Namazi MR, Ashraf A, Handjani F, Eftekhar E, Kalafi A. Angiotensin converting enzyme activity in alopecia areata. Enzyme Res 2014; 2014: 694148. doi: 10.1155/2014/694148
- Fahim S, Montazer F, Tohidinik H, et al. Serum and tissue angiotensin-converting enzyme in patients with alopecia areata. Indian J Dermatol Venereol Leprol 2019; 85: 295-299.
- Kataria V, Wang H, Wald JW, Phan YL. Lisinopril-induced alopecia: a case report. J Pharm Pract 2017; 30: 562-566.
- Ahmad S. Enalapril and reversible alopecia. Arch Intern Med 1991; 151: 404.
- Motel PJ. Captopril and alopecia: a case report and review of known cutaneous reactions in captopril use. J Am Acad Dermatol 1990; 23: 124-125.
- Leaker B, Whitworth JA. Alopecia associated with captopril treatment. Aust NZJ Med 1984; 14: 866.
- Arias-Santiago S, Gutierrez-Salmeron MT, Castellote-Caballero L, Naranjo-Sintes R. Elevated aldosterone levels in patients with androgenetic alopecia. Br J Dermatol 2009; 161: 1196-1198.
- Arias-Santiago S, Gutierrez-Salmeron MT, Buendia-Eisman A, Giron-Prieto MS, Naranjo-Sintes R. Hypertension and aldosterone levels in women with early-onset androgenetic alopecia. Br J Dermatol 2010; 162: 786-789.
- Arias-Santiago S, Gutierrez-Salmeron MT, Castellote-Caballero L, Buendia-Eisman A, Naranjo-Sintes R. Androgenetic alopecia and cardiovascular risk factors in men and women: a comparative study. J Am Acad Dermatol 2010; 63: 420-429.
- Hirsso P, Rajala U, Hiltunen L, Jokelainen J, Keinanen-Kiukaanniemi S, Nayha S. Obesity and low-grade inflammation among young Finnish men with early-onset alopecia. Dermatology 2007; 214: 125-129.
- Brown J, Farquhar C, Lee O, Toomath R, Jepson RG. Spironolactone versus placebo or in combination with steroids for hirsutism and/or acne. Cochrane Database Syst Rev 2009; doi: 10.1002/14651858.CD000194.pub2
- Boehncke WH, Schoen MP. Psoriasis. Lancet 2015; 386: 983-994.
- Huskic J, Mulabegovic N, Alendar F, et al. Serum and tissue angiotensin converting enzyme in patients with psoriasis. Coll Antropol 2008; 32: 1215-1219.
- Rahmati-Roudsari M, Saeedi M, Eshghi G. Serum angiotensin converting enzyme in patients with psoriasis. Iran J Dermatology 2009; 12: 127-130.
- Liu T, Han Y, Lu L. Angiotensin-converting enzyme gene polymorphisms and the risk of psoriasis: a meta-analysis. Clin Exp Dermatol 2013; 38: 352-359.
- Song GG, Bae SC, Kim JH, Lee YH. The angiotensin-converting enzyme insertion/deletion polymorphism and susceptibility to rheumatoid arthritis, vitiligo and psoriasis: a meta-analysis. J Renin-Angiotensin-Aldosterone Syst 2015; 16: 195-202.
- Yang K, Zhang F, Li F, et al. Angiotensin-converting enzyme insertion/deletion polymorphism and susceptibility to psoriasis in a Chinese population. J Renin-Angiotensin-Aldosterone Syst 2014; 15: 39-43.
- Munir S, Rahman SB, Rehman S, et al. The angiotensin-converting enzyme gene insertion polymorphism: a higher risk for psoriasis in male patients. Br J Dermatol 2016; 175: 824-826.
- Mohammadi Y, Vaisi-Raygani A, Shakiba E, et al. Angiotensin II type 1 receptor A 1166 C (rs5186) gene polymorphism increased risk and severity of psoriasis, contribution to oxidative stress, antioxidant statues, lipid peroxidation and correlation with vascular adhesion protein 1, preliminary. J Eur Acad Dermatol Venereol 2016; 30: 1395-1397.
- Wolf R, Tamir A, Brenner S. Psoriasis related to angiotensin-converting enzyme inhibitors. Dermatologica 1990; 181: 51-53.
- Gilleaudeau P, Vallat VP, Carter DM, Gottlieb AB. Angiotensin-converting enzyme inhibitors as possible exacerbating drugs in psoriasis. J Am Acad Dermatol 1993; 28: 490-492.
- Saraceno R, Citarella L, Spallone G, Chimenti S. A biological approach in a patient with psoriasis and bullous pemphigoid associated with losartan therapy. Clin Exp Dermatol 2008; 33: 154-155.
- Marquart-Elbaz C, Grosshans E, Lipsker D, Lipsker D. Sartans, angiotensin II receptor antagonists, can induce psoriasis. Br J Dermatol 2002; 147: 617-618.
- Azzouz B, Abou Taam M, Morel A, Trenque T. Psoriasis during angiotensin receptor blocker exposure: an underestimated adverse drug reaction. Expert Opin Drug Saf 2018; 17: 853-857.
- Ozkur M, Erbagci Z, Nacak M, Tuncel AA, Alasehirli B, Aynacioglu AS. Association of insertion/deletion polymorphism of the angiotensin-converting enzyme gene with psoriasis. Br J Dermatol 2004; 151: 792-795.
- van de Kerkhof PC. Plasma aldosterone and cortisol levels in psoriasis and atopic dermatitis. Br J Dermatol 1982; 106: 423-427.
- Ferreli C, Gasparini G, Parodi A, Cozzani E, Rongioletti F, Atzori L. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol 2017; 53: 306-336.
- Kawaguchi Y, Takagi K, Hara M, et al. Angiotensin II in the lesional skin of systemic sclerosis patients contributes to tissue fibrosis via angiotensin II type 1 receptors. Arthritis Rheum 2004; 50: 216-226.
- Pignone A, Del Rosso A, Brosnihan KB, et al. Reduced circulating levels of angiotensin-(1-7) in systemic sclerosis: A new pathway in the dysregulation of endothelial-dependent vascular tone control. Ann Rheum Dis 2007; 66: 1305-1310.
- Riemekasten G, Philippe A, Nather M, et al. Involvement of functional autoantibodies against vascular receptors in systemic sclerosis. Ann Rheum Dis 2011; 70: 530-536.
- Kill A, Tabeling C, Undeutsch R, et al. Autoantibodies to angiotensin and endothelin receptors in systemic sclerosis induce cellular and systemic events associated with disease pathogenesis. Arthritis Res Ther 2014; 16: R29. doi: 10.1186/ar4457
- Abou-Raya A, Abou-Raya S, Helmii M. Effects of angiotensin II receptor blockade in systemic sclerosis: randomized controlled trial. Ann Rheum Dis 2014; 72: A61.
- Kowal-Bielecka O, Fransen J, Avouac J, et al. Update of EULAR recommendations for the treatment of systemic sclerosis. Ann Rheum Dis 2017; 76: 1327-1339.
- Fernandez-Codina A, Walker KM, Pope JE. Treatment algorithms for systemic sclerosis according to experts. Arthritis Rheumatol 2018; 70: 1820-1828.
A c c e p t e d : June 17, 2019