The administration of succinic acid (50 mg/kg) as a bioenergetic regulator corrects oxidative phosphorylation disturbances in hepatic mitochondria under diabetes in rats and reduces blood glucose and cholesterol (10). Succinate protected isolated rat liver mitochondria from peroxidative damage, proteins from cross-links, mitochondrial membranes from permeability changes (11). The known antioxidant, pineal gland hormone, melatonin, has both receptor-dependent and independent mechanisms (12). Many of the beneficial effects of melatonin administration may depend on its action on the mitochondria (13, 14). Earlier we documented some beneficial effects of melatonin under experimental diabetes in rats: correction of impaired antioxidative status in liver tissue and regulation of nitric oxide bioavailability in the aorta (15). Long-term melatonin administration to diabetic rats reduced their hyperlipidemia and hyperinsulinemia and enhanced insulin-receptor kinase and insulin receptor substrate-1 phosphorylation; this suggests the existence of a signaling pathway cross-talk between melatonin and insulin (16).
The aim of the present work was to investigate the role of a specific functional damage in rat liver mitochondria during diabetes as well as to evaluate the possibility of mitochondrial impairment corrections by the energetic substrate, succinate, and by the antioxidant, melatonin.
Chemicals
Melatonin, 5,5-dithiobis(2-nitrobenzoic acid) (Ellmans reagent), succinic acid disodium salt hexahydrate, reduced glutathione (GSH), trichloroacetic acid (TCA), 2,6-dichlorophenol-indophenol, tert-butyl hydroperoxide (tBHP), 1-chloro-2,4-dinitrobenzene (CDNB) and safranin O were from Sigma-Aldrich (St. Louis, MO, USA/Steinheim, Germany). Streptozotocin (Streptozocin) (STZ) was from Fluka Chemie AG (Buchs, Switzerland). All other reagents were of analytical grade and were purchased from Reakhim (Moscow, Russia). All the water solutions were made with water purified in the Milli-Q system.
Animal model
The investigations were performed using 60 male albino Wistar rats (150-180 g). A standard balanced diet and tap water were provided ad libitum. Lights were on daily from 08.00 to 20.00 h. Ten animals received physiological saline containing 5% ethanol intraperitoneally (i.p.) and were kept as controls. Experimental animals were injected with a single dose of STZ (45 mg/kg, i.p.), dissolved in 0.01 M citrate buffer, pH 4.5, immediately before use. Seven days later, blood glucose levels were determined in whole blood samples. The rats injected with STZ were considered diabetic if their fasting blood glucose was >200 mg/dL (Blood Glucose Sensor Electrodes, MediSense, Abbot Laboratories, Bedford, UK). The first subgroup of the hyperglycemic animals diagnosed was injected daily with physiological saline containing 5% ethanol (i.p.) (the diabetes group); the second subgroup received daily 10 mg melatonin/kg b.w. (i.p.) (diabetes+10 mg melatonin); the third subgroup received daily 50 mg succinate/kg b.w. (i.p.) (diabetes+50 mg succinate); and the fourth subgroup received daily 10 mg melatonin/kg b.w.+50 mg succinate/kg b.w (i.p.) (diabetes+10 mg melatonin+50 mg succinate). Melatonin was prepared as a 0.3% solution in physiological saline, containing 5% ethanol, and succinate was prepared as a 1.0% solution in physiological saline. Melatonin as well as succinate was injected in the morning at 08.00; thus, we evaluated the effect of exogenous melatonin administration that was used as an antioxidant. The rats were sacrificed after 30 days of melatonin and succinate (or saline) administration. Melatonin (10 mg/kg) (15, 17) and succinate (50 mg/kg) (10) were administered at doses that were applied earlier. The animals were killed by decapitation according to the rules of the European Convention for the Protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes and the study was approved by the Ethics Committee of the Institute for Pharmacology and Biochemistry of the National Academy of Sciences of Belarus.
Blood samples were drawn by an abdominal aorta puncture into tubes containing hirudin (50 µg/ml). After removing of plasma by centrifugation, the erythrocytes were washed three times with cold phosphate buffered saline (150 mM NaCl, 10 mM Na2HPO4, pH 7.4).
Isolation of rat liver mitochondria
Mitochondria were isolated by differential centrifugation from the liver as follows (18). The liver was quickly removed and was homogenized in a glass-Teflon homogenizer with ice-cold isolation medium containing 250 mM sucrose, 20 mM Tris-HCl and 1 mM EDTA, pH 7.2, at 2°C. The homogenate was centrifuged at 600 g for 10 min, and the supernatant was centrifuged at 8,500 g for 10 min. The obtained pellet was washed in buffer containing 250 mM sucrose, 20 mM Tris-HCl, pH 7.2 (at 2°C). The protein concentration was determined by the method of Lowry et al. (19). Respiration of mitochondria was measured using a laboratory-made oxygen Clark-type electrode and a hermetic polarographic cell (volume 1.25 ml) with constant gentle stirring. The incubation medium contained 50 mM sucrose, 20 mM Tris-HCl, 125 mM KCl, 2.5 mM KH2PO4, 5 mM MgSO4, 0.5 mM EDTA, pH 7.5. The experiments were performed at 25°C using 5 mM succinate or L-glutamate as substrates. The mitochondrial protein concentration in the probe was 1.0 mg/ml. The functional state of mitochondria was determined by the acceptor control ratio (ACR), equal to the ratio of the respiratory rates (V3/V2) of mitochondria in states 3 and 2, and the coefficient of phosphorylation (ADP/O). State 2 corresponded to the respiration in the presence of the substrate (glutamate, or succinate) added (V2), and the rate of mitochondrial respiration corresponding to state 3 (V3) was registered after addition of 180 µM ADP (in the presence of 210 nm ADP in the polarographic cell).
Biochemical measurements
A stable form of glycated haemoglobin (GHb) containing 1-deoxy-1(N-valyl)fructose and the activities of marker enzymes of hepatic cell membrane injury, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were assayed in blood plasma using a reagent kits from Pliva-Lachema (Brno, Czech Republic). The concentration of total and protein thiols in mitochondria was determined spectrophotometrically by the method of Ellman (20) using the molar absorption coefficient
Mitochondrial glutathione peroxidase (GPx) activity was measured by the rate of GSH oxidation according to the method of Martinez et al. (24). The reaction mixture contained 0.1 M Tris-HCl, pH 7.0, 1 mM EDTA, 12 mM sodium azide, 2 mM tBHP and 4.9 mM GSH (as cosubstrates of GPx). The reaction was started by addition of fractured mitochondria and was stopped by 0.2 ml 25% TCA after 10-min incubation at 37°C. The protein concentration in the reaction mixture was 100 µg/ml. The activity was measured as the amount of GSH oxidized in the GPx reaction using Ellmans reagent.
The activity of mitochondrial glutathione-S-transferase (GST) was determined employing the method of Habig et al. (25). The reactions were carried out in the presence of 10 µg mitochondrial protein, 1 mM CDNB (as substrate), 5 mM GSH and 100 mM potassium phosphate buffer, pH 6.5, at 25°C in the final volume of 2 ml. The conjugation of CDNB with glutathione was monitored at 340 nm, using the molar absorbtion coefficient of 9,600 M-1cm-1.
Catalase activity was measured in rat liver cell cytoplasm by the method of Aebi (26). The reaction mixture contained 4.5 µg/ml protein, 20 mM hydrogen peroxide (H2O2), 50 mM potassium phosphate buffer, pH 7.0. The H2O2 decomposition was monitored at 240 nm by the use of the molar absorption coefficient of 36 M-1cm-1 and at 25°C for 3 min.
Statistical analysis
Data for 8-10 rats in each group are presented as a mean ±S.E.M. We used the standard unpaired Student t test for the comparison of the data, p<0.05 was taken to indicate statistical significance.
Biochemical impairments and oxidative stress under diabetes
In our experiments, 30-day STZ-induced diabetes mellitus in rats resulted in elevated levels of blood glucose and glycosylated haemoglobin as well as in reduction of animal body weight (Table 1). Similarly, the activities of the markers of liver damage, ALT and AST, in rat blood plasma increased by 50% (p<0.01) and 10% (p<0.05), respectively (Table 1). Succinate, but not melatonin, administration to diabetic animals reduced hepatolysis, diminishing elevated blood plasma ALT and AST activities. Diabetes resulted in significant impairments of rat tissues and organs: the kidney weight / body weight ratio increased in diabetic rats (Table 1). In our experiments, the melatonin injection to diabetic animals, but not succinate or melatonin plus succinate, recovered this parameter to the control value (p<0.01 in comparison with the diabetic animals) but did not affect body weight (Table 1). The level of lipid peroxidation products (TBARS) increased in the heart (by 20%, p<0.05) but not in the liver tissues of diabetic rats. The melatonin (but not succinate) administration prevented elevation of TBARS levels in heart tissue (Table 2). The melatonin or succinate (or both) administration to diabetic rats reduced the hepatic TBARS levels (p<0.05 in comparison with diabetes) (Table 2). The level of reduced glutathione decreased in red blood cells (by 25%, p<0.05) (Table 2). The liver mitochondrial protein thiol group content and mitochondrial total thiol group content did not change as a result of diabetes (Table 2). The level of PSSG increased in mitochondria of diabetic rats (by 50%, p<0.05). We evaluated the activities of antioxidative and glutathione metabolizing enzymes: liver mitochondrial GPx activity (did not change), liver mitochondrial GST activity (the main detoxifying enzyme) (decreased by 12%, p<0.05) and cytoplasmic catalase activity (decreased by 45%, p<0.001). Thus, the activities of the enzymatic antioxidative defense system diminished in the liver of diabetic rats (Table 2). Melatonin administration to diabetic animals enhanced the depressed activity of catalase (by 35%, p<0.05) in the cytoplasm of liver cells and mitochondrial GST (by 20%, p<0.05) in comparison with diabetes, respectively (Table 2).
| Table 1. Blood glucose, glycated haemoglobin, alanine aminotransferase (ALT) and aspartate aminotransferas (AST) activities and animal body and kidney weights in normal and STZ-treated diabetic rats. Effect of succinate and melatonin administration. |
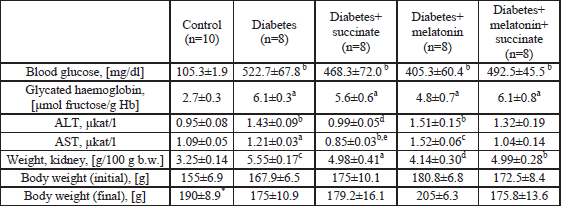 |
| a-
statistically significant in comparison with control, p<0.05; b-
statistically significant in comparison with control, p<0.01; c- statistically significant in comparison with control, p<0.001; d- statistically significant in comparison with diabetes, p<0.01; e- statistically significant in comparison with diabetes, p<0.001; *- statistically significant in comparison with initial body weight; n- the number of animals |
| Table 2. The total (TSH) and protein (PSH) thiol groups, mixed glutathione-protein disulfides (PSSG), lipid peroxidation product levels and antioxidative enzyme activities in rat liver cell mitochondria under diabetes. Effects of succinate and melatonin administration. |
 |
| a-
statistically significant in comparison with control, p<0.05; b-
statistically significant in comparison with control, p<0.01; c- statistically significant in comparison with control, p<0.001; d- statistically significant in comparison with diabetes, p<0.05; e- statistically significant in comparison with diabetes, p<0.01; n- the number of animals. |
Mitochondrial impairments under diabetes
30-day diabetes was accompanied by a considerable impairment of mitochondrial respiratory activity. In the case of succinate as a respiratory substrate, the ADP-stimulated respiration rate V3 markedly decreased (by 25%, p<0.05) (Fig. 1A) and the acceptor control ratio (ACR) V3/V2 was also diminished (by 25%, p<0.01) (Fig. 1B). We observed a drop in the respiration rate V2 (by 20%, p<0.05) and the ADP-stimulated respiration rate V3 (by 35%, p<0.05) with glutamate as substrate (Fig. 1A). In this case, the ACR also decreased (by 20%, p<0.05). Surprisingly, the phosphorylation coefficient ADP/O did not change during diabetic liver damage (Fig. 1B).
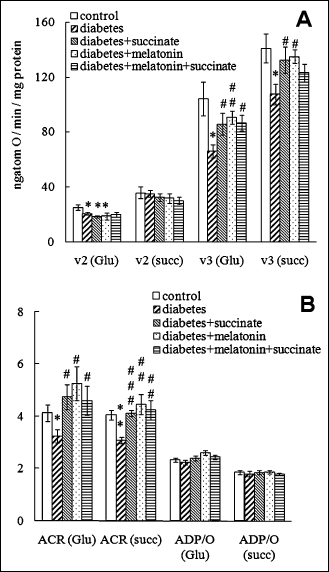 |
Fig. 1. Parameters of respiratory activity of liver mitochondria in normal and streptozotocin-treated diabetic rats. Effect of succinate and melatonin administration: substrate-dependent respiration rate V2 and ADP-stimulated respiration rate V3 (A) as well as acceptor control ratio (V3/V2) and coefficient of phosphorylation ADP/O (B). Glutamate (Glu) and succinate (succ) were used as respiratory substrates. Data for 8-10 rats in each group are presented as a mean ±S.E.M. The standard unpaired Student t test was used for the comparison of the data. * - statistically significant in comparison with control, p<0.05; ** - statistically significant in comparison with control, p<0.01; # - statistically significant in comparison with diabetes, p<0.05; ## - statistically significant in comparison with diabetes, p<0.01; ###- statistically significant in comparison with diabetes, p<0.001. |
The activity of the key enzyme of the Krebs cycle, KGDH, rose (by 60%, p<0.05) (Fig. 2); probably, this reflected the activation of protein degradation and amino acid turnover during diabetes. Despite the substantial damage in respiratory activity, the activity of SDH, respiratory complex II, did not change (Fig. 2).
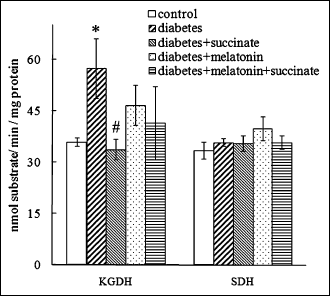 |
Fig. 2. Activities of t test was used for the comparison of data.* - statistically significant in comparison with control, p<0.05; ** - statistically significant in comparison with control, p<0.01; # - statistically significant in comparison with diabetes, p<0.05; ## - statistically significant in comparison with diabetes, p<0.01; ###- statistically significant in comparison with diabetes, p<0.001. |
Succinate administration during diabetes in rats exerted a protective effect on the liver mitochondrial function. We observed an enhancement of the reduced ADP-stimulated oxygen consumption rate V3 (by 25%, p<0.05 in comparison with diabetic animals) (Fig. 1A), when we used succinate as a respiratory substrate. The ACR considerably increased to the control values (p<0.01 in comparison with diabetic animals) (Fig. 1B). Similarly, succinate administration to diabetic animals reversed the ADP-stimulated oxygen consumption rate V3 (increased by 30%, p<0.05 in comparison with diabetic animals) and completely reversed the ARC value when mitochondria oxidized glutamate as a substrate (Fig. 1A, 1B). Melatonin administration during diabetes exerted a marked protective effect on the liver mitochondria function, reversing the decreased respiration rate V3 to the control values both for succinate-dependent respiration (p<0.05 in comparison with diabetic animals) and for glutamate-dependent respiration (p<0.01). Similarly, the melatonin treatment of diabetic rats reversed the effect of diabetes on the ACR value both for succinate-dependent respiration and for glutamate-dependent respiration (Fig. 1A, 1B). The combined administration of melatonin and succinate to diabetic animals also reversed the V3 rate value, and the ACR value for both oxidizing substrates (Fig. 1A, 1B) and only succinate administration prevented a significant KGDH activation during diabetes (Fig. 2).
There is interest in determining whether antioxidants will reduce mitochondria oxidative damage and prevent diabetic complications (6). Excess ROS accumulation stimulates various serine/threonine kinases, including IKKß, JNK and PKCs, and inflammatory signaling, resulting in reduced insulin signaling cascade (insulin resistance) (7). The protective effect of antioxidants (
Numerous studies have evaluated increased oxidative stress in mitochondria, as well as alterations in mitochondrial metabolism and function in different tissues under diabetes (9, 27). However, the results reported are controversial. Thus, it was reported that 9 weeks after STZ-induction of diabetes in rats, respiratory function of liver mitochondria declined as assessed by the mitochondrial membrane potential and respiratory ratios, succinate dehydrogenase and cytochrome C oxidase activities: all of them were significantly augmented (27). However, a decline in state 3 respiration in heart mitochondria was shown only in diabetic animals exhibiting a marked reduction in body weight (28). The data argue against hyperglycemia being a direct cause of the decline in state 3 oxygen consumption observed in cardiac mitochondria of type 1 diabetic rats (28). Moreira et al. (29) similarly concluded that 4-week and 9-week STZ-induced diabetes in rats was not accompanied by brain mitochondria dysfunction (4-week diabetic rats showed even higher mitochondrial respiratory chain enzymatic activities, compared to control rats), suggesting that oxidative stress associated with type 1 diabetes is not directly related to aberrant mitochondrial physiology. Recently it was shown that glycated LDL, which accumulates in patients with diabetes, increased ROS production by mitochondria and attenuated the activities of key enzymes in a mitochondrial electron-transport chain (30).
In the current experiments, we observed a considerable impairment of rat liver mitochondria function during diabetes, concomitant with liver damage. Significant diminishing of the ADP-depended oxygen consumption rate V3 and impairment of the coupling oxidation and phosphorylation processes in liver mitochonria (the ACR values decreased) without changes in the efficacy of oxygen consumption (the ADP/O did not change during diabetes) were shown. 30-day diabetes resulted in an increase of animal growth retardation, absolute and relative kidney weight (Table 1), and in typical signs of elevated oxidative stress. In rat liver mitochondria, diabetes was accompanied by marked impairments of metabolism: we observed a significant activation of KGDH, a key enzyme of the Krebs cycle, without changes in SDH activity. At the same time we did not observe alterations in the mitochondrial thiol group levels, or in the mitochondrial membrane potential (data not shown) as a result of diabetes. The reduction of the oxygen consumption rate V3 with glutamate or succinate as substrates, with KGDH or SDH activity diminution being unobserved, could be explained either by impaired substrate oxidation in the electron-transport chain or by reduced H+ translocation through the inner mitochondrial membrane.
We observed some beneficial effects of the melatonin and succinate administration on the complications of diabetes. In our experiments, the melatonin administration had no effect on diabetic growth retardation but partially prevented diabetic increase of kidney weight. Other authors demonstrated that melatonin supplementation to hamsters, rats, and mice reduced their body weight and the mass of white adipose tissue (31). Succinate-treated diabetic animals showed decreased ALT and AST blood plasma activities in comparison with diabetic group values. Melatonin treatment prevented GST inactivation and succinate treatment prevented KGDH activation under diabetes. The more important thing is that melatonin, as well as succinate administration to diabetic animals improved the mitochondrial physiology, i.e. increased the respiration rate of isolated rat liver mitochondria and prevented a loss of respiratory control, increasing the acceptor control ratio value. Generally, the simultaneous administration of melatonin and succinate to diabetic rats did not show any additional beneficial effect in comparison with the melatonin or succinate injections.
Succinate and its mitochondrial metabolites may participate in triggering of insulin release by pancreatic islets (32). By acting as ligands for appropriate receptors, succinate and a-ketoglutarate have unexpected signaling functions beyond their traditional roles in the regulation of energy homeostasis and cellular metabolism (33). Mitochondrial oxidation of succinate in isolated diabetic rat hepatocytes and succinate carbon incorporation into proteins were markedly lowered, probably due to impairment in the Krebs cycle activity (34). At the same time we observed a significant activation of
Melatonin exhibits a variety of biological activities, including antioxidative protection of cells and anti-inflammatory functions (35, 36). The highest intracellular concentration of melatonin seems to be in mitochondria (37), which suggests its involvement in regulation of mitochondrial function. Based on the observations that melatonin treatment does not measurably influence food intake and only has a limited impact on the physical activity, the weight loss promoting effect of melatonin must be attributed to alterations in energy metabolism, i.e. increased energy expenditure induced by melatonin (31). Uncoupling action of melatonin on oxidative phosphorylation in isolated mitochondria has been observed by Lopez et al. (38). We demonstrated this mild uncoupling effect of melatonin in experimental rat model (39).
Melatonin influences insulin secretion, with this effect being mediated by specific high-affinity, pertussis-toxin-sensitive G-protein coupled receptors MT (1) as well MT (2) in pancreatic islets (40). At the same time the reduced insulin levels in type 1 diabetes (streptozotocin-induced rat model of diabetes) associated with higher melatonin concentrations in the blood and elevated insulin levels observed in type 2 diabetes are associated with reduced melatonin levels (40). The results suggest that a melatonin-insulin antagonism may exist (41). It was shown earlier that melatonin inhibits glucose-induced insulin secretion in isolated rat islets without interfering with glucose metabolism (42). Peschke et al. pointed out the fact that melatonin protects the ß-cells against functional overcharge and, consequently, hinders the development of type 2 diabetes (43). The high concentrations of blood plasma melatonin found in type 1 diabetes are supposed to be beneficial for preventing diabetic lesions. At the same time the possible effect of melatonin on insulin secretion by pancreatic islets should be verified. Melatonin directly inhibits the mitochondrial permeability transition pore in a dose-dependent manner (IC50=0.8 µM) in rat liver mitoplasts; this inhibition may contribute to melatonins anti-apoptotic effects during transient brain ischemia (44). The combination of an antioxidant, melatonin, and a PARP inhibitor, nicotinamide, caused an essential reversal of biochemical alterations in diabetic neuropathy (45). The results of our study suggest that the antioxidant melatonin and the energetic substrate succinate, being signaling molecules, may be useful for the pharmacotherapy of diabetic complications.
In summary, the data support an important role of mitochondria dysfunction in the development of liver injury during diabetes as well as the possibility of corrections of mitochondrial disorders by melatonin and succinate. The mechanism of mitochondrial dysfunction might be impairment of cellular and mitochondrial redox-balance, changes of mitochondrial metabolism, damage of the mitochondrial membrane and components of the electron-transport chain. Succinate, an energetic mitochondrial substrate, and melatonin, a powerful antioxidant, effectively prevented the diabetic lesions of rat liver mitochondria and should be considered as effectors regulating mitochondria function. The effects of melatonin might be due to both its radical scavenging properties, its signaling effects and its interaction with complexes of the respiratory chain.
Conflict of interests: None declared.
- Rosen P, Nawroth PP, King G, Moller W, Tritschler HJ, Packer L. The role of oxidative stress in the onset and progression of diabetes and its complications: a summary of a Congress Series sponsored by UNESCO-MCBN, the American Diabetes Association and the German Diabetes Society. Diabetes Metab Res Rev 2001; 17: 189-212.
- Baynes JW, Thorpe SR. Role of oxidative stress in diabetes complications: a new perspective on an old paradigm. Diabetes 1999; 48: 1-9.
- Schaffer SW, Azuma J, Mozaffari M. Role of antioxidant activity of taurine in diabetes. Can J Physiol Pharmacol 2009; 87: 91-99.
- Goldstein BJ, Mahadev K, Wu X. Insulin action is facilitated by insulin-stimulated reactive oxygen species with multiple potential signaling targets. Diabetes 2005; 54: 311-321.
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes 2005; 54: 1615-1625.
- Green K, Brand MD, Murphy P. Prevention of mitochondrial oxidative damage as a therapeutic strategy in diabetes. Diabetes 2004; 53: 110-118.
- Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res 2008; 102: 401-414.
- Moreira PI, Cardoso SM, Pereira CM, Santos MS, Oliveira CR. Mitochondria as a therapeutic target in Alzheimers disease and diabetes. CNS Neurol Disord Drug Targets 2009; 8: 492-511.
- Dey A, Swaminathan K. Hyperglycemia-induced mitochondrial alterations in liver. Life Sci 2010; 87: 197-214.
- Vengerovskii AI, Khazanov VA, Eskina KA, Vasilyev KY. Effects of silymarin (hepatoprotector) and succinic acid (bioenergy regulator) on metabolic disorders in experimental diabetes mellitus. Bull Exp Biol Med 2007; 144: 53-56.
- Tretter L, Szabados G, Ando A, Horvath I. Effect of succinate on mitochondrial lipid peroxidation. 2. The protective effect of succinate against functional and structural changes induced by lipid peroxidation. J Bioenerg Biomembr 1987; 19: 31-44.
- Reiter RJ, Paredes SD, Manchester LC, Tan DX. Reducing oxidative/nitrosative stress: a newly discovered genre for melatonin. Crit Rev Biochem Mol Biol 2009; 44: 175-200.
- Jou MJ, Peng TI, Hsu LF, et al. Visualization of melatonins multiple mitochondrial level of protection against mitochondrial Ca2+-mediated permeability transition and beyond in rat brain astrocytes. J Pineal Res 2010; 48: 20-38.
- Paradies G, Petrosillo G, Paradies V, Reiter RJ, Ruggiero FM. Melatonin, cardiolipin and mitochondrial bioenergetics in health and disease. J Pineal Res 2010; 48: 297-310.
- Sudnikovich EJ, Maksimchik YZ, Zabrodskaya SV, et al. Melatonin attenuates metabolic disorders due to streptozotocin-induced diabetes in rats. Eur J Pharmacol 2007; 569: 180-187.
- Nishida S. Metabolic effects of melatonin on oxidative stress and diabetes mellitus. Endocrine 2005; 27: 131-136.
- Drobnik J, Slotwinska D, Olczak S, et al. Pharmacological doses of melatonin reduce the glycosaminoglycan level within the infarcted heart scar. J Physiol Pharmacol 2011; 62: 29-35.
- Johnson D, Lardy HA. Isolation of liver or kidney mitochondria. Meth Enzymol 1967; 10: 94-101.
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 1951; 193: 265-275.
- Ellman G. Tissue sulfhydryl groups. Arch Biochem Biophys 1959; 82: 70-77.
- Rossi R, Cardaioli E, Scaloni A, Ammmiooni G, Di Simplicio P. Thiol groups in proteins as endogenous reductants to determine glutathione-protein mixed disulphides in biological systems. Biochim Biophys Acta 1993;1164: 289-298.
- Stocks J, Dormandy TL. The autoxidation of human red cell lipids induced by hydrogen peroxide. Br J Haematol 1971; 20: 95-111.
- Nulton-Persson AC, Szweda LI. Modulation of mitochondrial function by hydrogen peroxide. J Biol Chem 2001; 276: 23357-23361.
- Martinez JI, Launay JM, Dreux C. A sensitive fluorimetric microassay for the determination of glutathione peroxidase activity. Application to human blood platelets. Anal Biochem 1979; 98: 154-159.
- Habig WH, Pabst MJ, Jacoby WB. Glutathione-S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem 1974; 249: 7130-7139.
- Aebi H. Catalase in vitro. In Oxygen Radicals in Biological Systems, L Packer (ed). Orlando, Academic Press, 1984, pp. 121-126.
- Ferreira FM, Palmeira CM, Seica R, Moreno AJ, Santos MS. Diabetes and mitochondrial bioenergetics alterations with age. J Biochem Mol Toxicol 2003; 17: 214-222.
- Lashin O, Romani A. Mitochondria respiration and susceptibility to ischemia-reperfusion injury in diabetic hearts. Arch Biochem Biophys 2003; 420: 298-304.
- Moreira PI, Santos MS, Moreno AJ, Proenca T, Seica R, Oliveira CR. Effect of sreptozotocin-induced diabetes on rat brain mitochondria. J Neuroendocrinol 2004; 16: 32-38.
- Sangle GV, Chowdhury SK, Xie X, Stelmack GL, Halayko AJ, Shen GX. Impairment of mitochondrial respiratory chain activity in aortic endothelial cells induced by glycated LDL. Free Radic Biol Med 2010; 48: 781-790.
- Tan DX, Manchester LC, Fuentes-Broto L, Paredes SD, Reiter RJ. Significance and application of melatonin in the regulation of brown adipose tissue metabolism: relation to human obesity. Obes Rev 2011; 12: 167-188.
- Fahien LA, MacDonald MJ. The succinate mechanism of insulin release. Diabetes 2002; 51: 2669-2676.
- He W, Miao FG, Linn DC, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004; 429: 188-193.
- Memon RA, Bessman SP, Mohan C. Impaired mitochondrial metabolism and reduced amphibolic Krebs cycle activity in diabetic rat hepatocytes. Biochem Mol Biol Int 1995; 37: 1079-1089.
- Carrillo-Vico A, Guerrero JM, Lardone PJ, Reiter RJ. A review of the multiple actions of melatonin on the immune system. Endocrine 2005; 27: 189-200.
- Gallano A, Tan DX, Reiter RJ. Melatonin as a natural ally against oxidative stress: a physicochemical examination. J Pineal Res 2011; 51: 1-16.
- Reiter RJ, Tan DX, Mayo JC, Sainz RM, Leon J, Czarnocki Z. Melatonin as an antioxidant: biochemical mechanisms and pathophysiological implications in humans. Acta Biochim Pol 2003; 50: 1129-1146.
- Lopez A, Garcia JA, Escames G, et al. Melatonin protects the mitochondria from oxidative damage reducing oxygen consumption, membrane potential, and superoxide anion production. J Pineal Res 2009; 46: 188-198.
- Maksimchik YZ, Dremza IK, Lapshina EA, et al. Rat liver mitochondria impairments under acute carbon tetrachloride-induced intoxication. Effects of melatonin. Biochemistry (Moscow) Suppl Series A: Membrane and Cell Biology 2010; 4: 187-195.
- Peschke E, Wolgast S, Bazwinsky I, Ponicke K, Muhlbauer E. Increased melatonin synthesis in pineal glands of rats in streptozotocin induced type 1 diabetes. J Pineal Res 2008; 45: 439-448.
- Peschke E, Hofmann K, Bahr I, et al. The insulin-melatonin antagonism: studies in the LEW.1AR1-iddm rat (an animal model of human type 1 diabetes mellitus). Diabetologia 2011; 54: 1831-1840.
- Picinato MC, Haber EP, Cipolla-Neto J, Curi R, de Oliveira Carvalho CR, Carpinelli AR. Melatonin inhibits insulin secretion and decreases PKA levels without interfering with glucose metabolism in rat pancreatic islets. Pineal Res 2002; 33: 156-160.
- Peschke E, Hofman K, Ponicke K, Wedekind D, Muhlbauer E. Catecholamines are the key for explaining the biological relevance of insulin-melatonin antagonism in type 1 and type 2 diabetes. J Pineal Res 2011; doi: 10.1111/j.1600-079X.2011.00951.x. Epub ahead of print.
- Andrabi SA, Sayeed I, Siemen D, Wolf G, Horn TF. Direct inhibition of the mitochondrial permeability transition pore: a possible mechanism responsible for anti-apoptotic effects of melatonin. FASEB J 2004; 18: 869-871.
- Negi G, Kumar A, Kaundal RK, Gulati A, Sharma SS. Functional and biochemical evidence indicating beneficial effect of melatonin and nicotinamide alone and in combination in experimental diabetic neuropathy. Neuropharmacology 2010; 58: 585-592.