THE EFFECT OF REACTIVE OXYGEN SPECIES
ON THE SYNTHESIS OF PROSTANOIDS FROM ARACHIDONIC ACID
2Department of Biochemistry and Human Nutrition, Pomeranian Medical University, Szczecin, Poland
INTRODUCTION
Aerobic metabolism constituted an advantage in the evolutionary race. Aerobic organisms were able to obtain much more energy from the combustion of organic compounds with oxygen than organisms that obtained their energy from anaerobic processes. However, this progress came at a huge price, i.e. generation of reactive oxygen species (ROS) that damage and destroy cell structures. Most important reactive oxygen species include superoxide anion radical (O2![]() -), hydrogen peroxide (H2O2) and hydroxyl radical (OH
-), hydrogen peroxide (H2O2) and hydroxyl radical (OH![]() ). Due to constant mitochondrial production of ROS and their continuous neutralization by antioxidative enzymes, cellular levels of ROS remain essentially at the same level. Concentrations of O2
). Due to constant mitochondrial production of ROS and their continuous neutralization by antioxidative enzymes, cellular levels of ROS remain essentially at the same level. Concentrations of O2![]() - and H2O2 in hepatocytes have their respective values of 10 pM (10-11 M) and 10 nM; however, the latter may reach up to 25 µM in lens epithelial cells (1, 2). Cellular concentrations of OH
- and H2O2 in hepatocytes have their respective values of 10 pM (10-11 M) and 10 nM; however, the latter may reach up to 25 µM in lens epithelial cells (1, 2). Cellular concentrations of OH![]() can not be determined by direct methods due to high reactivity of the radical.
can not be determined by direct methods due to high reactivity of the radical.
Many xenobiotics increase the ROS levels after entering the cells. Examples include compounds or cations of metals, such as iron, lead, mercury, chromium, vanadium, cadmium, as well as non-metals, e.g. fluoride anions (3-5). High concentrations of ROS have many adverse consequences. ROS damage cell structures, leading to pathological processes (3).
Recently it was shown that ROS, when present at low levels, are involved in intracellular signal transduction. Activation of the receptor of certain interleukins results in increased ROS levels in its vicinity. Moreover, multiple signaling pathways are activated and different cellular responses are induced upon treatment of cells with ROS or ROS-generating xenobiotics. One of these pathways is the prostanoid synthesis pathway.
Prostanoids are a group of tissue hormones, derivatives of polyunsaturated fatty acids with chains consisting of 20 carbon atoms, synthesized mainly from arachidonic acid, but also from other C20 fatty acids, e.g. eicosapentaenoic acid (EPA) (6-8). The first enzyme, and one that also restricts the rate of the entire pathway, is phospholipase A2 (PLA2), releasing arachidonic acid from sn-2 positions within phospholipids (9). About 20 isoforms of this enzyme were identified and classified into three categories: Ca2+-dependent cytoplasmic PLA2 (cPLA2), also present within the cytoplasm Ca2+-independent PLA2 (iPLA2) and Ca2+-dependent secreted PLA2 (sPLA2). The IVA isoform of cPLA2, activated by Ca2+ influx and phosphorylation, specifically cleaves arachidonic acid from phospholipids molecules and plays the main role in the release of this fatty acid in prostanoid synthesis (10). Isoforms of sPLA2 that play an important role in the prostanoid synthesis include isoforms V, X, and, in chronic inflammation, IIA (10). Isoforms of iPLA2 cleave fatty acids from phospholipid position sn-2 in a non-specific manner and thus they do not play an important role in prostanoid synthesis (10).
REACTIVE OXYGEN SPECIES IN SIGNALING PATHWAYS VERSUS THE ACTIVITY AND EXPRESSION OF PHOSPHOLIPASE A2
ROS play an important role in activation and expression of cPLA2 via the interleukin 1b (IL-1β) or thromboxane A2 (TxA2) receptors. Addition of antioxidants or inhibition of NADPH oxidase in response to receptor activation within smooth muscle cells inhibits the expression and activation of this phospholipase in response to the tested ligands (Fig. 1) (11-13). Binding the ligands at IL-1β or TxA2 receptors leads to an increase in ROS levels due to the activity of NADPH oxidase, and thus to activation of p38 (via protein kinase C alpha (PKC-α)), extracellular signal-regulated kinase (Erk) and c-Jun N-terminal kinase (JNK) mitogen-activated protein kinase (MAPK) cascades, which activated cPLA2 by phosphorylation (Fig. 2) (11, 12). Following activation of MAPK cascades, the signal is transmitted via JNK and p38 MAPK onto CREB-binding protein (CBP/p300) (a transcription factor responsible for cPLA2 expression) and transcription factor activator protein 1 (AP-1) (11, 13). Also activated is nuclear factor κB (NF-κB), a transcription factor that plays an important role in inflammatory and antioxidative responses in cells (11).
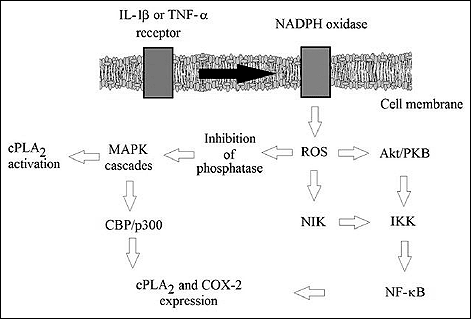 |
Fig. 1. The mechanism of activation and expression of COX-2 and cPLA2 by external ligands accd. to. (11-13, 28, 48-50). |
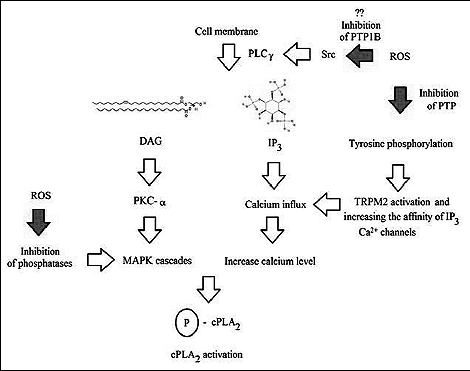 |
Fig. 2. The mechanism of activation of cPLA2 by ROS for review see references: (11, 21, 22, 24, 28, 46-48, 50, 52 - 54, 56, 57, 59, 61-65, 68, 73). |
THE EFFECT OF OXIDATIVE STRESS ON THE RELEASE OF ARACHIDONIC ACID BY ACTIVATION AND EXPRESSION OF PHOSPHOLIPASE A2
Besides the involvement of ROS in the transduction of signals generated at membrane receptors, direct effect of H2O2 or O2![]() - on the increase in the amounts of arachidonic acid by cPLA2 was also documented (14-17). Incubation of cells with 200 µM H2O2 for 30 min. leads to a distinct increase in the amounts of arachidonic acid released by cPLA2 (14-16). ROS activate PKC-a which in turn activates p38 and Erk MAPK (14-17). In astrocytes iPLA2 may be involved in the process along with cPLA2 (16). However, experiments in macrophages demonstrated that the mechanism of action of both enzymes is different, as iPLA2 is involved mainly in the release of arachidonic acid. This isoform is less sensitive to H2O2, as distinct activation of this enzyme was observed not upon incubation at 100 µM H2O2, but only at as much as 500 µM (18). Most probably, the activity of the enzyme responsible for the process depends not only on the cell type, but also on the intensity of oxidative stress. Incubation of astrocytes with H2O2 for 45 min caused an increase in the activity of Erk and JNK MAPK already at ROS levels of 100 µM; at concentration of 300 µM, the activity of the tested kinases began to drop significantly (14). However, experiments in rat pheochromocytoma PC12 cells showed that the treatment of cells with H2O2 at the level of 100 µM for 4 hours reduced expression of cPLA2 by means of the same signaling pathways as those responsible for the enhancement in enzyme expression and activation, i.e. Erk, p38 MAPK, but not JNK MAPK (19). This might be due to the reduced strength of signal transduction by cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) as a result of chronic oxidative stress (via protein kinase D1 (PKD1) and kinases involved in MAPK cascades) (19, 20). One should also consider the possible lack of NF-κB activation. As shown by the results of the studies in lens epithelial cells, identical cellular exposure to ROS (100 µM H2O2 for 4 hours), caused a reduction in the activity of this transcription factor compared with the activity after one hour of incubation by inhibiting the activity of proteasomes important for NF-κB activation (21, 22).
- on the increase in the amounts of arachidonic acid by cPLA2 was also documented (14-17). Incubation of cells with 200 µM H2O2 for 30 min. leads to a distinct increase in the amounts of arachidonic acid released by cPLA2 (14-16). ROS activate PKC-a which in turn activates p38 and Erk MAPK (14-17). In astrocytes iPLA2 may be involved in the process along with cPLA2 (16). However, experiments in macrophages demonstrated that the mechanism of action of both enzymes is different, as iPLA2 is involved mainly in the release of arachidonic acid. This isoform is less sensitive to H2O2, as distinct activation of this enzyme was observed not upon incubation at 100 µM H2O2, but only at as much as 500 µM (18). Most probably, the activity of the enzyme responsible for the process depends not only on the cell type, but also on the intensity of oxidative stress. Incubation of astrocytes with H2O2 for 45 min caused an increase in the activity of Erk and JNK MAPK already at ROS levels of 100 µM; at concentration of 300 µM, the activity of the tested kinases began to drop significantly (14). However, experiments in rat pheochromocytoma PC12 cells showed that the treatment of cells with H2O2 at the level of 100 µM for 4 hours reduced expression of cPLA2 by means of the same signaling pathways as those responsible for the enhancement in enzyme expression and activation, i.e. Erk, p38 MAPK, but not JNK MAPK (19). This might be due to the reduced strength of signal transduction by cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) as a result of chronic oxidative stress (via protein kinase D1 (PKD1) and kinases involved in MAPK cascades) (19, 20). One should also consider the possible lack of NF-κB activation. As shown by the results of the studies in lens epithelial cells, identical cellular exposure to ROS (100 µM H2O2 for 4 hours), caused a reduction in the activity of this transcription factor compared with the activity after one hour of incubation by inhibiting the activity of proteasomes important for NF-κB activation (21, 22).
THE EFFECT OF REACTIVE OXYGEN SPECIES ON MITOGEN-ACTIVATED PROTEIN KINASE CASCADES ACTIVATION
The main mechanism of ROS-mediated activation of MAPK cascades followed by cPLA2 activation or COX-2 expression may involve direct inhibition of phosphothyrosine phosphatases and serine/threonine phosphatases (Table 1). These phosphatases contain cysteine moieties that are susceptible to oxidation by ROS within their active centers. It appears that H2O2, used in a vast majority of cited experiments, can not directly impact the activity of phosphatases due to a very low course of these reactions (23). Phosphatases are inactivated directly by the OH![]() radical, generated spontaneously or in Fenton's reaction from H2O2, generating sulfenic acid -SOH moieties at cysteine residues (23).
radical, generated spontaneously or in Fenton's reaction from H2O2, generating sulfenic acid -SOH moieties at cysteine residues (23).

Experiments involving transduction of cells with genes of appropriate phosphatases showed that correlation of phosphatases with MAPK cascades is very complex, as many different phosphatases are involved in individual pathways. In T lymphocytes, SH2 domain-containing protein tyrosine phosphatase 1 (SHP-1) is responsible for inhibition of Erk and JNK MAPK, while hematopoietic phosphotyrosine phosphatase (HePTP) is responsible for inhibition of Erk MAPK, but also p38 MAPK (24). The results obtained in other experiments suggest that MAPK phosphatase 1 (MKP-1) and MAPK phosphatase 7 (MKP-7) are responsible for inhibition of JNK and p38 MAPK cascades (25). However, such experiments in enhanced expression of particular enzymes generate very simplified models: they prove that particular phosphatase is involved in inhibition of particular MAPK cascade, but do not show whether these enzymes are important for the transduction of signals in cells treated with ROS. Experiments in inhibition of individual phosphatases showed that serine/threonine protein phosphatase 1 (PP1) and protein phosphatase 2A (PP2A) play an important role in regulation of cyclooxygenase 2 (COX-2) expression, e.g. by means of ROS generated by the activity of NADPH oxidase or directly by H2O2 (HUVEC cell model), while SH2 domain-containing protein tyrosine phosphatase 2 (SHP-2) has no significant effect on the expression of COX-2 (26). One might suspect that PP2A plays an important role in inhibition of MAPK cascades and ROS-mediated activation of MAPK cascades in non-cancer cells (26, 27). It appears that inhibition of protein phosphatase 2 (PP2) leads to activation of non-receptor tyrosine kinase family Src (Src), which in turn leads to phosphorylation of epidermal growth factor receptor (EGFR) and then to the transduction of this signal into MAPK cascades (28). In lung cancer cell line A549, important part in inhibition of p38 and JNK MAPKs was played by MKP-1 (29). The importance of a particular phosphatase in inhibition of MAPK cascades is complicated, as the several different phosphatases, non-specifically inactivated by ROS, are responsible for the process. Inactivation of one phosphatase may involve some degree of compensation of the inhibition by other enzymes.
ROS also reduce the protein phosphatase 5 (PP5) expression levels, which translates into increased activation of MAPK cascades (30). In addition, ROS reduce the protein phosphatase 2A A subunit (PP2A-A) expression levels, leading to reduced activity of PP2A and PP5; however, the mechanism of this effect remains unclear (30).
Besides phosphatase-controlled activation of JNK and p38 MAPKs, another mechanism of activating these cascades by means of ROS is also possible. The cascades are sensitive to oxidative stress, e.g. at apoptosis signal-regulating kinase 1 (ASK-1) MAPK kinase kinase (MAPKKK), which is inhibited by thioredoxine - a protein inactivated by ROS (28, 31). Another mechanism of JNK MAPK activation is inhibition of phosphatases responsible for dephosphorylation, i.e. direct inhibition of JNK MAPK (31).
THE EFFECT OF REACTIVE OXYGEN SPECIES ON CALCIUM ION LEVELS
Besides phosphorylation of cPLA2, ROS are also involved in activation of this enzyme by increasing the calcium influx, as observed in many types of cells (32-35). Increase in the Ca2+ levels, occurring via activation of cation channels, was observed after incubation of cells with H2O2 at levels from 10 µM in platelets to 100 µM in human aortic endothelial cells, or even up to 300 µM for pulmonary arterial smooth muscle cells (32, 33, 36, 37).
The effect of ROS on calcium influx is a complex process that occurs along multiple routes. It appears that at least in astrocytes, the key route involves activation of PLCγ1, leading to the release of diacylglycerols (DAG) and inositol triphosphate (IP3) and activation of IP3-sensitive Ca2+ channels (36). The mechanism of activation of PLCγ1 by ROS remains unclear. Probably, ROS may inactivate SHP-1, which in turn leads to phosphorylation and activation of Src, and thus to activation of phospholipase C-gamma1 (PLCγ1). However, the precise mechanism requires further studies (38).
By mediation of DAG, released by PLCγ, ROS activate protein kinase C (PKC), which translates into the activity and expression of cPLA2 (38). In addition, ROS increase the affinity of IP3 to their specific receptors, probably by their oxidation or by inhibition of protein tyrosine phosphatases (PTPs), i.e. the enzymes that dephosphorylate these receptors (36, 39). One of these mechanisms may be inhibition of protein tyrosine phosphatase 1B (PTP1B), which leads to increased phosphorylation of amino acid residues within calcium channels, and thus to increased influx of Ca2+ into cytoplasm (40, 41). This process occurs along multiple routes; individual signaling pathways may be additive and not affect one another. ROS may also impact the Ca2+ levels by affecting actin polymerization that takes place in platelet cells (33). This mechanism may be the result of PTP inactivation, which in turn leads to phosphorylation of proteins responsible for polymerization of actin cytoskeleton (33). Direct oxidation of sensitive cysteine residues within actin, disturbing its polymerization, is also possible (33). Another mechanism responsible for the increase in Ca2+ levels is oxidation of thiol moieties within ryanodine receptors, leading to their activation in myocardial cells and increasing the cytoplasmic calcium ion levels (42).
Yet another route consists in activation of transient potential receptor melastatin-2 (TRPM2) by ROS (43). However, this mechanism remains unclear, as it appears that as much as three processes of different importance in different experiments are responsible for the process: the release of adenosine diphosphate ribose (ADPR) from mitochondria, direct oxidation of cysteine residues in TRPM2 and the increase in the number of phosphorylated thyrosine residues within the receptor as a result of protein tyrosine phosphatase L1 (PTPL1) inhibition by ROS (43, 44).
THE MECHANISM OF REACTIVE OXYGEN SPECIES-MEDIATED ACTIVATION OR INHIBITION OF NUCLEAR FACTOR-κB
NF-κB is a family of five transcription factors: RelA/p65, RelB, cRel, p50 and p52, occurring as homo- or heterodimers (45). NF-κB is found in cytoplasm in its inactive form as a complex with inhibitor of NF-κB α subunit (IκBα), inhibitor of NF-κB β subunit (IκBβ) and inhibitor of NF-κB ε subunit (IκBε), which are in turn regulated by IκB kinase (IKK), being a complex of three proteins: IκB kinase a subunit (IKKα), IκB kinase β subunit (IKKβ) and IκB kinase γ subunit/NF-κB essential modulator (IKKγ/NEMO) (46). Phosphorylation of Ser32 and Ser36 residues of IκBα by IKK triggers a signal for ubiquitination and degradation of IκBα and thus to activation of NF-κB (45, 47). Another route of activation is in place for p52 NF-κB, which is present within the cytoplasm as a precursor, p100. When phosphorylated by IKK, p100 undergoes proteolysis into the active form (45).
Activation of NF-κB by IKK occurs in association with ROS produced by NADPH oxidase in response to activation of receptors, i.e. the IL-1β or TNF-α receptors (Fig. 3) (48, 49). protein kinase B (Akt/PKB) and NF-κB-inducing kinase (NIK) are involved in this process (48, 50). ROS activate phosphoinositide 3-kinase (PI3K) and inactivate (by oxidizing cysteine residues within the active center of tumor suppressor phosphatase and tensin homolog (PTEN)) the phosphatase responsible for the hydrolysis of phosphates at phosphoinositol carbon atom 3, leading to an increase in the levels of Akt/PKB activator (51, 52). In addition, by phosphorylation by casein kinase II, PTEN enters the proteolytic degradation pathway (51). Besides affecting the metabolism of inositol, ROS affect activation of Akt/PKB by other pathways as well (53). Akt/PKB is inhibited by PP2A, which in turn may be inactivated by ROS (54). However, it appears that at lower levels, ROS oxidize the disulphide bridges in Akt/PKB, leading to association of Akt/PKB with PP2A and thus short-term activation of Akt/PKB (54). Next, Akt/PKB activate IKKα and IKKβ, which in turn stimulate p38 MAPK in the transduction of signals originating e.g. from IL-1β receptors (53). The process of Akt/PKB-mediated activation is more complex, as IKK may be present in the form of a complex with mammalian target of rapamycin (mTOR), and the kinase may thus also activate IKK, as demonstrated in the studies on the transduction of signals from the receptors to TNF (55). Besides the effect exerted by ROS on Akt/PKB, NIK, which is a serine/threonine kinase of IKKα playing a key role in activation of NF-κB, is also activated as a result of IL-1β, TNF-α, or lipopolysaccharide (LPS) receptor stimulation (50, 52). As shown in in vitro studies, NIK is activated at low (1–10 µM) concentrations of H2O2, while being inactivated at concentrations within the range of 100 µM–1 mM (50). The process may be of key importance for the switching of NF-κB activation pathway into an IKK-independent one by means of high ROS levels. It appears that the mechanism of activation of NIK by means of ROS consists in direct inactivation of phosphatases responsible for inhibition of the activity of this kinase and in oxidation of cysteine residues; however the mechanism has not been studied well yet (50, 52).
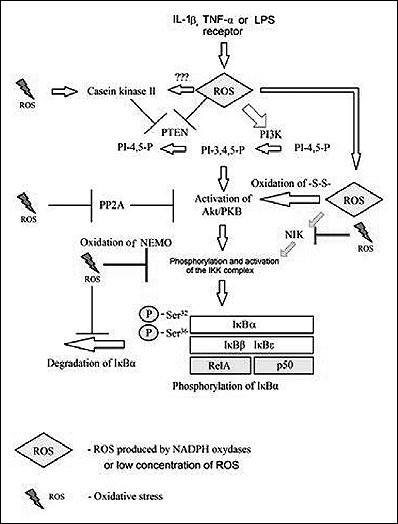 |
Fig. 3. The mechanism of IKK-dependent ROS-mediated activation of NF-κB (for review see references: 21, 22, 24, 28, 46, 48, 50, 52 - 54, 56, 68, 73). |
MAPK play an important role in the expression of e.g. COX-2 or cPLA2. Activation of JNK and Erk MAPK occurs via Src and further via EGFR onto these cascades (28) In MAPK cascades, the IKK complex may be activated by mitogen-activated protein kinase kinase 1 (MEKK1) (56). Activation of p38 and JNK MAPK cascades may also occur by means of ASK-1 (28, 31). In addition, IKK may activate p38 MAPK (48). Activated MAPK cascades affect the transcription of NF-κB-dependent genes by activation of CBP/p300 (11, 48). In addition, p38 and Erk MAPK activate mitogen and stress activated protein kinase 1 (MSK-1), which phosphorylates Ser276 within RelA/p65 subunit (28). Combined with Src-mediated phosphorylation of Ser536 within RelA/p65 subunit of NF-κB, the process translates into translocation of this transcription factor from the cytoplasm into the nucleus (Fig. 4) (28). Phosphorylation of Ser276 may be achieved not only via MSK-1, but also via protein kinase A (PKA); however, this may take place at lower levels of ROS in the transduction of signals from the TNF-α receptor (28, 57).
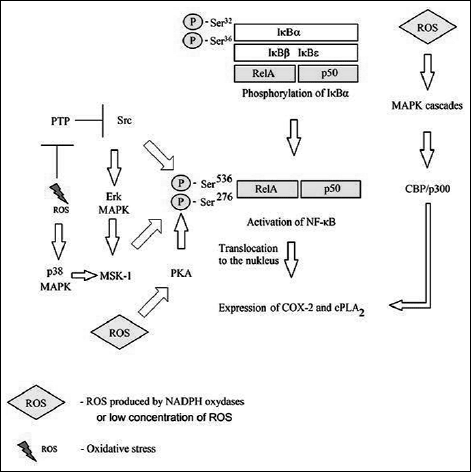 |
Fig. 4. The mechanism of activation of NF-κB ROS-mediated after degradation of IκBκ for review see references: (11, 28, 57, 48). |
Oxidative stress may also activate IKK along another pathway. Inhibition of phosphatases activates Src, which in turn activates Abl and protein kinase C delta (PKC-d), which then activate protein kinase D (PKD) (58, 59). Next, PKD directly activates IKK (58, 59). The mechanism is important for the survival of cells upon oxidative stress (58, 59).
However, activation of NF-κB in H2O2-treated cells, may occur independently of the IKK complex, in contrast to ROS involved in receptor signaling (Fig. 5) (47, 60, 61). At high concentrations (250 µM), exogenous H2O2 activates spleen tyrosine kinase (Syk) and non-receptor tyrosine kinase c-Src (c-Src), which play an important role in phosphorylation of IκBα at Tyr42, which means that phosphorylation occurs at different residues that in the case of IKK (47, 61-65). IKK-independent activation of NF-κB is characterized by the lack of enhanced degradation of IκBα following activation of this transcription factor (47, 61-65). Casein kinase II is also involved in this process, e.g. by phosphorylating domain rich in proline, glutamic acid, serine and threonine (PEST) within IκBα, as these sequences are responsible for degradation of this inhibitory protein (47, 66). Phosphorylation of Tyr42 is negatively controlled by PTP1B - a phosphatase frequently inactivated by oxidative stress (62). High ROS levels may simultaneously activate Syk and c-Src phosphatases to phosphorylate Tyr42 and inactivate PTP1B (66).
However, it appears that the process depends on the cell type, as IKK-independent activation of NF-κB in HeLa cells or T lymphocytes occurs in response to TNF-α (67). This mechanism, with an important role played by c-Src, is also important during reoxygenation following hypoxia (65, 68). By inhibition of PTP, ROS enhance phosphorylation at Tyr416 within c-Src, which activates this kinase and transmits the signal onto NF-κB (68). The pathway of Src-mediated activation of NF-κB is more complex, as Src may also deactivate PP2A, which is a phosphatase that dephoshorylates IKKb, translating into activation of IKK and disturbance of IκBα resynthesis (69).
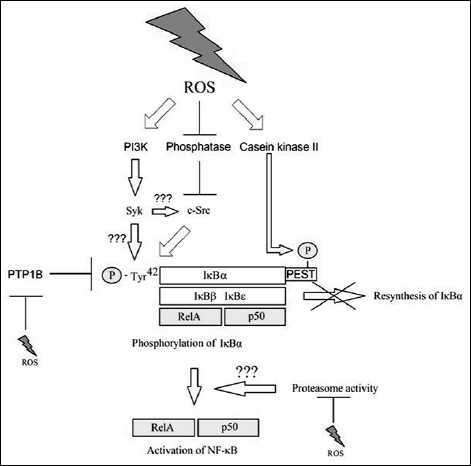 |
Fig. 5. The mechanism of IKK-independent ROS-mediated activation of NF-κB (for review see references: 47, 61-66). |
In addition, ROS disturb activation of NF-κB via IKK by means of oxidation of IKKγ/NEMO, which leads to polymerization of this IKK regulator subunit within the cytoplasm (46, 60). The activity of proteasomes is required for activation of NF-κB by oxidation stress, while degradation of IκBα is not a required condition (21, 47, 61, 63). Chronic oxidative stress leads to inhibition in proteasome activity and thus on disturbed activation of NF-κB in both mechanisms (22). The process is confirmed by the results of incubation of lens epithelial cells with H2O2 at 100 µM for 4 hours (22). However, the role of proteasomes in the IκBα degradation-independent mechanism remains unclear (22). It appears that ROS-mediated oxidation of amino acid residues within proteasomes is important for their inhibition, as is the disturbed production of ATP, translating into disturbed activity of ATP-dependent proteasomes (70, 71). Nonetheless, it appears that this effect depends on the type of cells, as e.g. it is not important in macrophages, where increased oxidation stress does not disturb activation of NF-κB (71).
Besides c-Src, other kinases are also involved in IKK-independent activation of NF-κB. Syk, which phosphorylates IκBα, is a kinase expressed in immune cells (61). However, it appears that IKK-independent activation of NF-κB in these cells may occur even regardless of signal transduction, e.g. from the TNF-α receptor (64). It appears that Syk is activated by PI3K; however, the mechanism has not been fully understood yet (61). Syk activates JNK, p38 and Erk MAPK cascades and leads to phosphorylation and degradation of IκBα. However, at higher ROS levels, an IκBα-independent mechanism of NF-κB activation takes place (61, 64). However, at higher levels of ROS in other cells, e.g. in lung epithelial cells, inhibition of NF-κB may occur due to lack of Syk expression and inhibition of the IKK complex activity (46, 61). Also important may be the increase in oxidative stress, as short exposition to H2O2 leads to poor IKK activation that does not significantly affect NF-κB activation or IκBα degradation (72). Another enzyme important in IKK-independent NF-κB activation mediated by ROS within leukocytes is SH2-containing inositol 5'-phosphatase 1 (SHIP-1) (73).
H2O2 activates NF-κB along an IKK-dependent or IKK-independent route, although the latter mechanism occurs much later (61, 63). Upon activation of NF-κB via the specific membrane receptor, activation occurs via the IKK complex. With no doubt, this may be due to various proteins being present in the vicinity of the receptor and to association with other signaling pathways originating from the same receptor. Upon H2O2 treatment of cells, entire cytoplasm undergoes oxidative stress, which may be manifested by activation of pathways different than those activated in the vicinity of the receptor (47). Also important is the intensity of the oxidation stress, as different ROS-susceptible proteins have different sensitivity to these signaling molecules. An example of such protein is NIK, which is activated at low ROS levels and inactivated at high ROS levels, which might explain the differences in NF-κB activation (50). At high ROS levels, IKKγ/NEMO and NIK are inactivated, which translates into inhibition of IKK-dependent activation of NF-κB (46, 50). In addition, inactivation if PTP1B and activation of Syk and Src occurs, leading to IKK-independent activation of NF-κB (61, 65, 66, 68).
COX PATHWAY
The released arachidonic acid may enter the prostaglandin and thromboxane synthetic pathway (COX pathway) or the leukotriene and lipoxin synthetic pathway (LOX pathway) (Fig. 6) (74-76). It may also be oxidized by other enzymes, e.g. by cytochrome P450 (CYP) into 20-hydroxyeicosatetraenoic acid (20-HETE) or into cis-epoxyeicosatrienoic acids (EETs) (77). In the cyclooxygenase pathway, arachidonic acid is oxidized by cyclooxygenase 1 (COX-1) or cyclooxygenase 2 (COX-2) into prostaglandin G2 (PGG2) and prostaglandin H2 (PGH2). PGH2 may in turn be transformed into thromboxanes or other prostaglandins. It is of significant importance in inflammatory reactions and has been associated with numerous diseases. PGH2 may also be oxidized by ROS to form levuglandins: levuglandin E2 (LGE2) and levuglandin D2 (LGD2) (78). The synthesis of prostaglandins is achieved in most cells by means of constitutive COX-1. COX-2 is an inducible enzyme, which may occur in macrophages and monocytes, where it plays an important role in inflammatory processes. Prostaglandin E2 (PGE2) was shown to play an important role in cancer diseases. Chronic inflammation leads to immune escape of cancer cells, promotes cell division and participates in angiogenesis (79, 80). Other diseases in which the prostaglandin synthetic route plays an important role include neurodegenerative diseases or atherosclerosis (81-83). ROS oxidize proinflammatory lipids cause inflammation develops, leading to atherosclerosis.
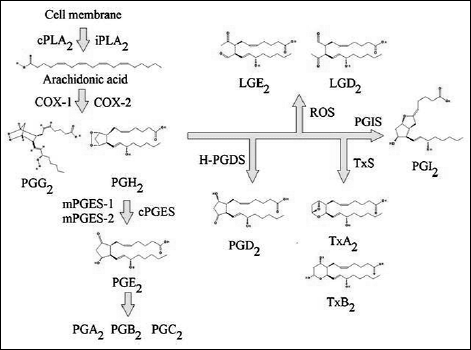 |
Fig. 6. The synthesis of prostanoids from arachidonic acid (for review see references: 11, 21, 22, 24, 28, 46-48, 50, 52 - 54, 56, 57, 59, 61-65, 68, 73). |
THE EFFECT OF REACTIVE OXYGEN SPECIES ON COX-2 EXPRESSION
The treatment of cells with LPS, IL-1β or TNF-α results in NADPH oxidase activation and increased cytoplasmic ROS levels followed by COX-2 expression (Fig. 7) (26, 84-86). MAPK cascades play an important role in this signaling pathway (85, 86). They activate CBP/p300, while p38 and JNK MAPK activate AP-1, which is a transcription factor required for COX-2 expression. These processes are also involved in the ROS-mediated induction of cPLA2 transcription. Thus, expression of cPLA2, COX-2 and membrane-bound prostaglandin E synthase 1 (mPGES-1) is activated simultaneously (11, 13). However, in the transduction of signals originating at LPS or IL-1β receptors, inhibition of NADPH oxidase only partially reduces COX-2 expression: ROS play an important, albeit not crucial role in the pathways originating at these receptors (84). ROS generated by NADPH oxidase play a crucial role in the transduction of signals originating at the TNF-α receptor (84).
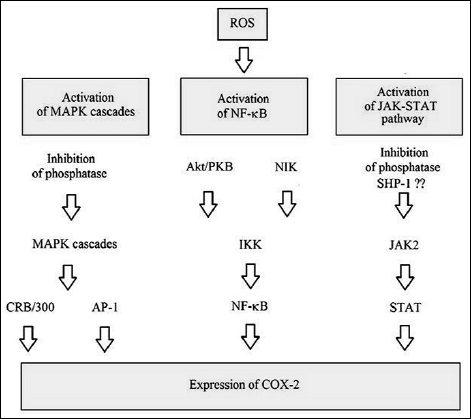 |
Fig. 7. The mechanism of ROS-mediated expression of COX-2 (for review see references: 1, 26, 50-52, 84-86, 90, 92, 93). |
Chronic oxidative stress may disturb COX-2 expression in response to LPS or IL-1β (87). In human chondrocytes model, 3-hour preincubation in 100 µM H2O2 disturbed the cellular response to LPS and IL-1β (87). Experiments in lens epithelial cells showed that incubation with ROS, lasting several hours, disturbs activation and activity of NF-κB by inhibiting the activity of proteasomes, which might thus also explain the disturbances in COX-2 expression (22). Direct inhibition of COX-1 and COX-2 activity by H2O2 is possible, as evidenced in in vitro studies (88). Already at concentrations of 10 µM, H2O2 clearly inactivate COX-1 and COX-2; although COX-2 is more sensitive to ROS (88). Nonetheless, in vitro protein studies are very simplified models that do not include cellular antioxidation mechanisms or the replacement of damaged proteins by newly synthesized ones. Therefore, it appears that the demonstrated inhibition of cyclooxygenase activity by means of ROS is of limited importance in physiological conditions.
ROS are not only involved in the transduction of signals from the receptors; COX-2 expression is also affected by the generalized oxidative stress. Experiments involving incubation of cells with H2O2 showed that COX-2 expression is induced directly by oxidative stress (26, 89). The process involves pathways similar to this characteristic for transduction of signals from e.g. LPS receptors (26, 89). MAPK cascades are also important in the process; however, it is the Erk, not p38 MAPK cascade, and only p100 NF-κB that play an important role, for instance in primary rat neonatal cardiomyocytes (89). However, p38 MAPK is crucial for the process occurring in HUVEC cells (26). It appears that COX-2 expression is more sensitive to H2O2 than cPLA2 activation, as COX-2 expression occurred already upon 50 µM H2O2/10 min incubation of primary rat neonatal cardiomyocytes or even at 3 µM H2O2 in HUVEC (16, 26, 89). At the same time, activation of cPLA2 in astrocytes was achieved only upon incubation with 100 µM H2O2 (16). With no doubt, the sensitivity of both pathways is due to the differences in cell lines as well as in the methods of activation of both cellular responses.
ROS-mediated induction of COX-2 expression occurs not only via MAPK cascades; the process is more complex (90-92). In astrocytes, an important role in the process is played by signal transducer and activator of transcription 6 (STAT-6). In pheochromocytoma cells, an important role is played by Janus tyrosine kinase 2 (JAK2), a kinase that phosphorylates signal transducer and activator of transcription (STAT) (90-92). The mechanism of JAK-STAT pathway activation is complex. By inhibiting phosphatases, ROS cause phosphatation of receptors that bind STATs (1). The same ROS, by inhibiting phosphatases, activate JAK2 and tyrosine kinase 2 (TYK2) kinases, which then activate STAT (1). However, in astrocytes, ROS activate SHP-2, thus leading to STAT dephosphorylation and contributing to inhibition of the activity of this transcription factor (93). However, it appears that the resultant of all these processes leads to an increased expression of COX-2 in astrocytes (91-93).
THE EFFECT OF CATALASE ON THE EXPRESSION OF COX-2
Transfection of vascular smooth muscle cells with catalase gene or incubation of these cells with catalase lead to increased expression of COX-2, increased stability of the mRNA transcript for that enzyme as well as to increased PGE2 production (94, 95). However, enhanced expression of other antioxidative enzymes, Mn- and Cu/Zn-SOD, did not cause such reactions (95). Induction of COX-2 expression in vascular smooth muscle cells occurred as a result of the effects of NF-κB/IL-6 and transcription factors that were sensitive to cAMP levels (cAMP response elements) (94, 95). Overexpression of catalase leads to increased stability of COX-2 transcript, which might be due to activation of proteins that activate the AU motif within mRNA (adenosine-uridine binding factors) (95). However, experiments in macrophages and microglial cells showed that catalase increases COX-2 by NF-κB along the PI3K - Akt/PKB - IKK pathway (96, 97). Nonetheless, the mechanism of the induction of PI3K by extracellular catalase is unclear; it seems that catalase may activate unspecified receptors that activate PI3K (96, 97). It is doubtful that reduced H2O2 should lead to the expression of COX-2.
THE EFFECT OF REACTIVE OXYGEN SPECIES ON THE SYNTHESIS OF PROSTAGLANDIN E2, PROSTAGLANDIN I2, PROSTAGLANDIN D2 AND THROMBOXANES FROM PROSTAGLANDIN H2
After oxidation of arachidonic acid into PGH2 by means of COX-1 or COX-2, the prostaglandins is transformed, among others, into PGE2 by prostaglandin E synthase (PGES). The enzyme is available in three isoforms. Constitutive cytosolic prostaglandin E synthase (cPGES), associated with COX-1 activity, is regulated by phosphorylation by casein kinase II (98, 99). This kinase is activated by H2O2; ROS may increase the activity of cPGES via casein kinase II (47). Another constitutive isoform, membrane-bound prostaglandin E synthase 2 (mPGES-2), is activated by reducing agents (99). It is probable that ROS inactivate this enzyme by their oxidation properties (99). In contrast to the above synthases, membrane-bound prostaglandin E synthase 1 (mPGES-1) is inducible and is subject to strict transcriptional regulation along with COX-2 (99, 100). However, regulation of the expression of these two enzymes, i.e. mPGES-1 and COX-2, is slightly different (99, 101). Nonetheless, due to differences stated in the expression of the induction, both enzymes, COX-2 and mPGES-1, undergo synthesis in different time segments (102).
In chondrocytes, p38 MAPK is not responsible for the expression of mPGES-1 though activation of Egr-1 by NF-κB is required, while the expression of COX-2 is early growth response gene-1 (Egr-1)-independent (99). As shown by experiments conducted in glial cells, PI3K is involved in the expression of mPGES-1, but not in the expression of COX-2 (101). However, expression of both factors is regulated in a similar fashion. An important role is played by JNK MAPK, which enhance the stability of COX-2 and mPGES-1 transcripts and proteins, as well as other proteins, such as NF-κB, PKC, p42/44 and p38 MAPK (101).
Regulation of the activity of NADPH oxidase is a complex process, as PGI2 concentration and NADPH oxidase activity are strictly correlated (100). PGI2 inhibits the activity of NADPH oxidase, while O2![]() - reacts with nitric oxide (NO) to form peroxynitrite, which starts tyrosine nitration in prostaglandin I synthase (PGIS), thus inactivating the enzyme (100, 103). Concentration of NO is much higher than that of O2
- reacts with nitric oxide (NO) to form peroxynitrite, which starts tyrosine nitration in prostaglandin I synthase (PGIS), thus inactivating the enzyme (100, 103). Concentration of NO is much higher than that of O2![]() -; it appears that the limiting factor for this reaction is the activity of NADPH oxidase and particularly O2
-; it appears that the limiting factor for this reaction is the activity of NADPH oxidase and particularly O2![]() - levels.
- levels.
Biosynthesis of PGD2 is ROS-dependent (104). ROS play an important role in inducing production of this prostaglandin by regulating the hematopoietic-type PGD synthase (H-PGDS) activity (104). However, the mechanism of increasing the activity of H-PGDS is unclear, studies show that H-PGDS may be inhibited by the addition of free radical scavengers or simply require ROS to be active (104).
The effect of ROS on the synthesis of thromboxanes has not been described well. Many studies show that ROS affect the production of thromboxanes from arachidonic acid in granulocytes or activated platelets (105-107). However, it seems that the impact of ROS on the supply of substrates for thromboxane synthase (TxS) by means of cPLA2 activation is most important for the synthesis of this eicosanoid (105, 107).
FUTURE PERSPECTIVES
In the last decade, much progress has been made in understanding the effects of ROS on inflammatory reactions and prostanoid synthesis. Inflammatory reactions were associated with many disease entities, such as neurodegenerative diseases, peptic ulcers or gastrointestinal cancers (108-110).
What concerns Alzheimer's disease, the organism deposits neurotoxic amyloid-β peptide (Aβ) in the brain (111). Progress of the disease is associated with activation of NADPH oxidase, which creates ROS in cytoplasm of astrocytes and microglial cells (111). This is followed by activation of signalling pathways, which leads to inflammatory reactions, posing toxic influence on neural cells (111, 112). A multitude of experiments revealed that natural substances, such as flavonoids, polyphenols and isoflavones, common in consumed plants, not only pose an antiaggregating influence on Ab, but also present an anti-inflammatory and antioxidative activity (111-113). Effects of these actions pose influence on various links of inflammatory reactions described in this thesis, depending on the examined substance (112, 113). Nonetheless, blocking even one stage of the inflammatory reaction leads to cytoprotection of neurons in Alzheimer's disease, and as a consequence, slows the progress of this disease.
Inflammatory reactions play a crucial role in the course of many conditions relating the respiratory system, such as asthma or acute respiratory distress syndrome (ARDS) (114).
Thanks to the knowledge of induction mechanisms underlying inflammatory reactions activated by ROS, it is possible to more effectively treat and prevent diseases with these backgrounds. However, one must keep in mind that the enzymes discussed here as taking part in pathological processes are also important as far as the physiological functions within the body are concerned.
Abbreviations: 20-HETE, 20-hydroxyeicosatetraenoic acid; ADPR, adenosine diphosphate ribose; Akt/PKB, protein kinase B; AP-1, transcription factor activator protein 1; ARDS, acute respiratory distress syndrome; ASK-1, apoptosis signal-regulating kinase 1; Aβ, amyloid β peptide; c-Src, non-receptor tyrosine kinase c-Src; cAMP, cyclic adenosine monophosphate; CBP/p300, CREB-binding protein; COX-1, cyclooxygenase 1; COX-2, cyclooxygenase 2; cPGES, cytosolic prostaglandin E synthase; cPLA2, cytosolic phospholipase A2; CREB, cAMP response element-binding protein; CYP, cytochrome P450; DAG, diacylglycerol; EET, cis-epoxyeicosatrienoic acid; EGFR, epidermal growth factor receptor; Egr-1, early growth response gene-1; EPA, eicosapentaenoic acid; Erk, extracellular signal-regulated kinase; H-PGDS, hematopoietic-type PGD synthase; HePTP, hematopoietic phosphotyrosine phosphatase; IL-1β, interleukin 1β; IκBα, inhibtior of NF-κB α subunit; IκBβ, inhibitor of NF-κB β subunit; IκBε, inhibitor of NF-κB ε subunit; IKK, IκB kinase; IKKα, IκB kinase a subunit; IKKβ, IκB kinase β subunit; IKKγ/NEMO, IκB kinase γ subunit/NF-κB essential modulator; IP3, triphosphoinositol; iPLA2, calcium independent phospholipase A2; JAK2, Janus tyrosine kinase 2; JNK, c-Jun N-terminal kinase; LGD2, levuglandin D2; LGE2, levuglandin E2; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MAPKKK, MAPK kinase kinase; MEKK1, mitogen-activated protein kinase kinase kinase 1; MKP-1, MAPK phosphatase 1; MKP-7, MAPK phosphatase 7; mPGES-1, mPGES-2, membrane-bound prostaglandin E synthase 1 or 2; MSK-1, mitogen and stress activated protein kinase 1; mTOR, mammalian target of rapamycin; NADPH oxidase, nicotinamide adenine dinucleotide phosphate oxidase; NF-κB, nuclear factor kB; NIK, NF-κB-inducing kinase; NO, nitric oxide; PEST, the domain rich in proline, glutamic acid, serine and threonine; PGD2, prostaglandin D2; PGE2, prostaglandin E2; PGES, prostaglandin E synthase; PGG2, prostaglandin G2; PGH2, prostaglandin H2; PGI2, prostaglandin I2; PGIS, prostaglandin I synthase; PI-3,4,5-P, phosphatidyl-inositol-3,4,5-trisphosphate; PI3K, phosphoinositide 3-kinase; PKA, protein kinase A; PKC, protein kinase C; PKC-α, protein kinase C α; PKC-δ, protein kinase C δ; PKD, protein kinase D; PKD1, protein kinase D1; PLA2, phospholipase A2; PLC, phospholipase C; PLCγ1, phospholipase C-γ1; PP1, protein phosphatase 1; PP2, protein phosphatase 2; PP2A, protein phosphatase 2A; PP2A-A, protein phosphatase 2A A subunit; PP5, protein phosphatase 5; PTEN, tumor suppressor phosphatase and tensin homolog; PTP, protein tyrosine phosphatase; PTP1B, protein tyrosine phosphatase 1B; PTPL1, protein tyrosine phosphatase L1; ROS, reactive oxygen species; SHIP-1, SH2-containing inositol 5'-phosphatase 1; SHP-1, SH2 domain-containing protein tyrosine phosphatase 1; SHP-2, SH2 domain-containing protein tyrosine phosphatase 2; sPLA2, secretory phospholipase A2; Src, non-receptor tyrosine kinase family Src; STAT, signal transducer and activator of transcription; STAT-6, signal transducer and activator of transcription 6; Syk, spleen tyrosine kinase; TNF-α, tumor necrosis factor α; TRPM2, transient potential receptor melastatin-2; TxA2, thromboxane A2; TxS, thromboxane synthase; TYK2, tyrosine kinase 2;
Conflict of interests: None declared.
REFERENCES
- Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol 1998; 275: C1640-C1652.
- Bartosz G. Strategia ataku. In: Druga Twarz Tlenu. Warszawa, PWN, 2003, pp. 81-82.
- Patrick L. Lead toxicity part II: the role of free radical damage and the use of antioxidants in the pathology and treatment of lead toxicity. Altern Med Rev 2006; 11: 114-127.
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006; 160: 1-40.
- Barbier O, Arreola-Mendoza L, Del Razo LM. Molecular mechanisms of fluoride toxicity. Chem Biol Interact 2010; 188: 319-333.
- Groeger AL, Cipollina C, Cole MP, et al. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol 2010; 6: 433-341.
- Vecchio AJ, Simmons DM, Malkowski MG. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J Biol Chem 2010; 285: 22152-22163.
- Norambuena F, Mackenzie S, Bell JG, Callol A, Estevez A, Duncan N. Prostaglandin (F and E, 2- and 3-series) production and cyclooxygenase (COX-2) gene expression of wild and cultured broodstock of Senegalese sole (Solea senegalensis). Gen Comp Endocrinol 2012; 177: 256-262.
- Ivanov AI, Romanovsky AA. Prostaglandin E2 as a mediator of fever: synthesis and catabolism. Front Biosci 2004; 9: 1977-1993.
- Boyanovsky BB, Webb NR. Biology of secretory phospholipase A2. Cardiovasc Drugs Ther 2009; 23: 61-72.
- Lee CW, Lin CC, Lee IT, Lee HC, Yang CM. Activation and induction of cytosolic phospholipase A2 by TNF-α mediated through Nox2, MAPKs, NF-κB, and p300 in human tracheal smooth muscle cells. J Cell Physiol 2011; 226: 2103-2114.
- Chakraborti S, Roy S, Mandal A, et al. Role of PKCα-p(38)MAPK-G(i)α axis in NADPH oxidase derived O(2)(·-)-mediated activation of cPLA(2) under U46619 stimulation in pulmonary artery smooth muscle cells. Arch Biochem Biophys 2012; 523: 169-180.
- Chi PL, Chen YW, Hsiao LD, Chen YL, Yang CM. Heme oxygenase 1 attenuates interleukin-1β-induced cytosolic phospholipase A2 expression via a decrease in NADPH oxidase/reactive oxygen species/activator protein 1 activation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 2012; 64: 2114-25.
- Tournier C, Thomas G, Pierre J, Jacquemin C, Pierre M, Saunier B. Mediation by arachidonic acid metabolites of the H2O2-induced stimulation of mitogen-activated protein kinases (extracellular-signal-regulated kinase and c-Jun NH2-terminal kinase). Eur J Biochem 1997; 244: 587-595.
- Yasuda Y, Yoshinaga N, Murayama T, Nomura Y. Inhibition of hydrogen peroxide-induced apoptosis but not arachidonic acid release in GH3 cell by EGF. Brain Res 1999; 850: 197-206.
- Xu J, Yu S, Sun AY, Sun GY. Oxidant-mediated AA release from astrocytes involves cPLA(2) and iPLA(2). Free Radic Biol Med 2003; 34: 1531-1543.
- van Rossum GS, Drummen GP, Verkleij AJ, Post JA, Boonstra J. Activation of cytosolic phospholipase A2 in Her14 fibroblasts by hydrogen peroxide: a p42/44(MAPK)-dependent and phosphorylation-independent mechanism. Biochim Biophys Acta 2004; 1636: 183-195.
- Martinez J, Moreno JJ. Role of Ca2+-independent phospholipase A2 on arachidonic acid release induced by reactive oxygen species. Arch Biochem Biophys 2001; 392: 257-262.
- Akiyama N, Shimma N, Takashiro Y, et al. Decrease in cytosolic phospholipase A2alpha mRNA levels by reactive oxygen species via MAP kinase pathways in PC12 cells: effects of dopaminergic neurotoxins. Cell Signal 2005; 17: 597-604.
- Ozgen N, Guo J, Gertsberg Z, Danilo P, Rosen MR, Steinberg SF. Reactive oxygen species decrease cAMP response element binding protein expression in cardiomyocytes via a protein kinase D1-dependent mechanism that does not require Ser133 phosphorylation. Mol Pharmacol 2009; 76: 896-902.
- Dudek EJ, Shang F, Taylor A. H(2)O(2)-mediated oxidative stress activates NF-kappa B in lens epithelial cells. Free Radic Biol Med 2001; 31: 651-658.
- Wu M, Bian Q, Liu Y, et al. Sustained oxidative stress inhibits NF-kappaB activation partially via inactivating the proteasome. Free Radic Biol Med 2009; 46: 62-69.
- Meng FG, Zhang ZY. Redox regulation of protein tyrosine phosphatase activity by hydroxyl radical. Biochim Biophys Acta 2013; 1834: 464-469.
- Lee K, Esselman WJ. Inhibition of PTPs by H(2)O(2) regulates the activation of distinct MAPK pathways. Free Radic Biol Med 2002; 33: 1121-1132.
- Hou N, Torii S, Saito N, Hosaka M, Takeuchi T. Reactive oxygen species-mediated pancreatic beta-cell death is regulated by interactions between stress-activated protein kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated protein kinase phosphatases. Endocrinology 2008; 149: 1654-1665.
- Eligini S, Arenaz I, Barbieri SS, et al. Cyclooxygenase-2 mediates hydrogen peroxide-induced wound repair in human endothelial cells. Free Radic Biol Med 2009; 46: 1428-1436.
- Foley TD, Armstrong JJ, Kupchak BR. Identification and H2O2 sensitivity of the major constitutive MAPK phosphatase from rat brain. Biochem Biophys Res Commun 2004; 315: 568-574.
- Kefaloyianni E, Gaitanaki C, Beis I. ERK1/2 and p38-MAPK signalling pathways, through MSK1, are involved in NF-kappaB transactivation during oxidative stress in skeletal myoblasts. Cell Signal 2006; 18: 2238-2251.
- Turpeinen T, Nieminen R, Moilanen E, Korhonen R. Mitogen-activated protein kinase phosphatase-1 negatively regulates the expression of interleukin-6, interleukin-8, and cyclooxygenase-2 in A549 human lung epithelial cells. J Pharmacol Exp Ther 2010; 333: 310-318.
- Chen L, Liu L, Yin J, Luo Y, Huang S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int J Biochem Cell Biol 2009; 41: 1284-1295.
- Han D, Ybanez MD, Ahmadi S, Yeh K, Kaplowitz N. Redox regulation of tumor necrosis factor signaling. Antioxid Redox Signal 2009; 11: 2245-2263.
- Hu Q, Corda S, Zweier JL, Capogrossi MC, Ziegelstein RC. Hydrogen peroxide induces intracellular calcium oscillations in human aortic endothelial cells. Circulation 1998; 97: 268-275.
- Redondo PC, Salido GM, Pariente JA, Rosado JA. Dual effect of hydrogen peroxide on store-mediated calcium entry in human platelets. Biochem Pharmacol 2004; 67: 1065-1076.
- Giambelluca MS, Gende OA. Hydrogen peroxide activates calcium influx in human neutrophils. Mol Cell Biochem 2008; 309: 151-156.
- Zhou X, Wen K, Yuan D, Ai L, He P. Calcium influx-dependent differential actions of superoxide and hydrogen peroxide on microvessel permeability. Am J Physiol Heart Circ Physiol 2009; 296: H1096-H1107.
- Hong JH, Moon SJ, Byun HM, et al. Critical role of phospholipase Cgamma1 in the generation of H2O2-evoked [Ca2+]i oscillations in cultured rat cortical astrocytes. J Biol Chem 2006; 281: 13057-13067.
- Lin MJ, Yang XR, Cao YN, Sham JS. Hydrogen peroxide-induced Ca2+ mobilization in pulmonary arterial smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007; 292: L1598-L1608.
- Tao Q, Spring SC, Terman BI. Comparison of the signaling mechanisms by which VEGF, H2O2, and phosphatase inhibitors activate endothelial cell ERK1/2 MAP-kinase. Microvasc Res 2005; 69: 36-44.
- Hu Q, Yu ZX, Ferrans VJ, Takeda K, Irani K, Ziegelstein RC. Critical role of NADPH oxidase-derived reactive oxygen species in generating Ca2+ oscillations in human aortic endothelial cells stimulated by histamine. J Biol Chem 2002; 277: 32546-32551.
- Hsu S, Schmid A, Sternfeld L, et al. Tyrosine phosphatase PTP1B modulates store-operated calcium influx. Cell Signal 2003; 15: 1149-1156.
- Bogeski I, Bozem M, Sternfeld L, Hofer HW, Schulz I. Inhibition of protein tyrosine phosphatase 1B by reactive oxygen species leads to maintenance of Ca2+ influx following store depletion in HEK 293 cells. Cell Calcium 2006; 40: 1-10.
- Meissner G. Molecular regulation of cardiac ryanodine receptor ion channel. Cell Calcium 2004; 35: 621-628.
- Takahashi N, Kozai D, Kobayashi R, Ebert M, Mori Y. Roles of TRPM2 in oxidative stress. Cell Calcium 2011; 50: 279-287.
- Zhang W, Tong Q, Conrad K, Wozney J, Cheung JY, Miller BA. Regulation of TRP channel TRPM2 by the tyrosine phosphatase PTPL1. Am J Physiol Cell Physiol 2007; 292: C1746-C1758.
- Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res 2011; 21: 103-115.
- Herscovitch M, Comb W, Ennis T, et al. Intermolecular disulfide bond formation in the NEMO dimer requires Cys54 and Cys347. Biochem Biophys Res Commun 2008; 367: 103-108.
- Schoonbroodt S, Ferreira V, Best-Belpomme M, et al. Crucial role of the amino-terminal tyrosine residue 42 and the carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B activation by an oxidative stress. J Immunol 2000; 164: 4292-4300.
- Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS Jr, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol Cell Biol 2000; 20: 1626-1638.
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 2006; 72: 1493-1505.
- Li Q, Engelhardt JF. Interleukin-1βeta induction of NFkappaB is partially regulated by H2O2-mediated activation of NFkappaB-inducing kinase. J Biol Chem 2006; 281: 1495-1505.
- Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J Biol Chem 2002; 277: 20336-20342.
- Kim JH, Na HJ, Kim CK, et al. The non-provitamin A carotenoid, lutein, inhibits NF-kappaB-dependent gene expression through redox-based regulation of the phosphatidylinositol 3-kinase/PTEN/Akt and NF-kappaB-inducing kinase pathways: role of H2O2 in NF-kappaB activation. Free Radic Biol Med 2008; 45: 885-896.
- Madrid LV, Mayo MW, Reuther JY, Baldwin AS Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-kappa B through utilization of the Ikappa B kinase and activation of the mitogen-activated protein kinase p38. J Biol Chem 2001; 276: 18934-18940.
- Murata H, Ihara Y, Nakamura H, Yodoi J, Sumikawa K, Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem 2003; 278: 50226-50233.
- Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP, Baldwin AS. Akt-dependent regulation of NF-{kappa}B is controlled by mTOR and Raptor in association with IKK. Genes Dev 2008; 22: 1490-1500.
- Lee FS, Hagler J, Chen ZJ, Maniatis T. Activation of the IkappaB alpha kinase complex by MEKK1, a kinase of the JNK pathway. Cell 1997; 88: 213-222.
- Jamaluddin M, Wang S, Boldogh I, Tian B, Brasier AR. TNF-αlpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal 2007; 19: 1419-1433.
- Storz P, Toker A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J 2003; 22: 109-120.
- Storz P, Doppler H, Toker A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol Cell Biol 2004; 24: 2614-2626.
- Korn SH, Wouters EF, Vos N, Janssen-Heininger YM. Cytokine-induced activation of nuclear factor-kappa B is inhibited by hydrogen peroxide through oxidative inactivation of IkappaB kinase. J Biol Chem 2001; 276: 35693-35700.
- Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J Biol Chem 2003; 278: 24233-24241.
- Singh S, Darnay BG, Aggarwal BB. Site-specific tyrosine phosphorylation of I kappa B alpha negatively regulates its inducible phosphorylation and degradation. J Biol Chem 1996; 271: 31049-31054.
- Canty TG Jr, Boyle EM Jr, Farr A, Morgan EN, Verrier ED, Pohlman TH. Oxidative stress induces NF-κB nuclear translocation without degradation of IκBα. Circulation 1999; 100 (Suppl. 19): II361-II364.
- Takada Y, Aggarwal BB. TNF activates Syk protein tyrosine kinase leading to TNF-induced MAPK activation, NF-κB activation, and apoptosis. J Immunol 2004; 173: 1066-1077.
- Lluis JM, Buricchi F, Chiarugi P, Morales A, Fernandez-Checa JC. Dual role of mitochondrial reactive oxygen species in hypoxia signaling: activation of nuclear factor-(kappa)B via c-SRC and oxidant-dependent cell death. Cancer Res 2007; 67: 7368-7377.
- Schwarz EM, Van Antwerp D, Verma IM. Constitutive phosphorylation of IκBα by casein kinase II occurs preferentially at serine 293: requirement for degradation of free I Ba. Mol Cell Biol 1996; 16: 3554-3559.
- Fan C, Li Q, Ross D, Engelhardt JF. Tyrosine phosphorylation of I kappa B alpha activates NF kappa B through a redox-regulated and c-Src-dependent mechanism following hypoxia/reoxygenation. J Biol Chem 2003; 278: 2072-2080.
- Li Q, Zhang Y, Marden JJ, Banfi B, Engelhardt JF. Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury. Biochem J 2008; 411: 531-541.
- Barisic S, Schmidt C, Walczak H, Kulms D. Tyrosine phosphatase inhibition triggers sustained canonical serine-dependent NFkB activation via Src-dependent blockade of PP2A. Biochem Pharmacol 2010; 80: 439-447.
- Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJ, Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J 1998; 335: 637-642.
- Zmijewski JW, Zhao X, Xu Z, Abraham E. Exposure to hydrogen peroxide diminishes NF-κB activation, IκB-α degradation, and proteasome activity in neutrophils. Am J Physiol Cell Physiol 2007; 293: C255-C266.
- Jaspers I, Zhang W, Fraser A, Samet JM, Reed W. Hydrogen peroxide has opposing effects on IKK activity and IκBα breakdown in airway epithelial cells. Am J Respir Cell Mol Biol 2001; 24: 769-777.
- Gloire G, Charlier E, Rahmouni S, et al. Restoration of SHIP-1 activity in human leukemic cells modifies NF-κB activation pathway and cellular survival upon oxidative stress. Oncogene 2006; 25: 5485-5494.
- Straus DS, Glass CK. Cyclopentenone prostaglandins: new insights on biological activities and cellular targets. Med Res Rev 2001; 21: 185-210.
- Herlong JL, Scott TR. Positioning prostanoids of the D and J series in the immunopathogenic scheme. Immunol Lett 2006; 102: 121-131.
- Murakami M. Lipid mediators in life science. Exp Anim 2011; 60: 7-20.
- Arnold C, Konkel A, Fischer R, Schunck WH. Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids. Pharmacol Rep 2010; 62: 536-547.
- Boutaud O, Li J, Zagol I, Shipp EA, et al. Levuglandinyl adducts of proteins are formed via a prostaglandin H2 synthase-dependent pathway after platelet activation. J Biol Chem 2003; 278: 16926-16928.
- Chang SH, Liu CH, Conway R, et al. Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc Natl Acad Sci USA 2004; 101: 591-596.
- Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E2 (PGE2) suppresses natural killer cell function primarily through the PGE2 receptor EP4. Cancer Immunol Immunother 2011; 60: 1577-1586.
- Linton MF, Fazio S. Cyclooxygenase-2 and inflammation in atherosclerosis. Curr Opin Pharmacol 2004; 4: 116-123.
- O'Banion MK. Prostaglandin E2 synthases in neurologic homeostasis and disease. Prostaglandins Other Lipid Mediat 2010; 91: 113-117.
- Shi J, Wang Q, Johansson JU, et al. Inflammatory prostaglandin E2 signaling in a mouse model of Alzheimer disease. Ann Neurol 2012; 72: 788-798.
- Feng L, Xia Y, Garcia GE, Hwang D, Wilson CB. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipopolysaccharide. J Clin Invest 1995; 95: 1669-1675.
- Wang T, Qin L, Liu B, et al. Role of reactive oxygen species in LPS-induced production of prostaglandin E2 in microglia. J Neurochem 2004; 88: 939-947.
- Peng T, Lu X, Feng Q. NADH oxidase signaling induces cyclooxygenase-2 expression during lipopolysaccharide stimulation in cardiomyocytes. FASEB J 2005; 19: 293-295.
- Mathy-Hartert M, Martin G, Devel P, et al. Reactive oxygen species downregulate the expression of pro-inflammatory genes by human chondrocytes. Inflamm Res 2003; 52: 111-118.
- Fujimoto Y, Uno E, Sakuma S. Effects of reactive oxygen and nitrogen species on cyclooxygenase-1 and -2 activities. Prostaglandins Leukot Essent Fatty Acids 2004; 71: 335-340.
- Adderley SR, Fitzgerald DJ. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J Biol Chem 1999; 274: 5038-5046.
- Tang XQ, Yu HM, Zhi JL, et al. Inducible nitric oxide synthase and cyclooxgenase-2 mediate protection of hydrogen peroxide preconditioning against apoptosis induced by oxidative stress in PC12 cells. Life Sci 2006; 79: 870-876.
- Park SJ, Lee JH, Kim HY, et al. Astrocytes, but not microglia, rapidly sense H2O2 via STAT6 phosphorylation, resulting in cyclooxygenase-2 expression and prostaglandin release. J Immunol 2012; 188: 5132-5141.
- Yu H, Liu Z, Zhou H, et al. JAK-STAT pathway modulates the roles of iNOS and COX-2 in the cytoprotection of early phase of hydrogen peroxide preconditioning against apoptosis induced by oxidative stress. Neurosci Lett 2012; 529: 166-171.
- Park SJ, Kim HY, Kim H, et al. Oxidative stress induces lipid-raft-mediated activation of Src homology 2 domain-containing protein-tyrosine phosphatase 2 in astrocytes. Free Radic Biol Med 2009; 46: 1694-1702.
- Chen G, Kamal M, Hannon R, Warner TD. Regulation of cyclo-oxygenase gene expression in rat smooth muscle cells by catalase. Biochem Pharmacol 1998; 55: 1621-1631.
- Fang X, Moore AS, Nwankwo JO, et al. Induction of cyclooxygenase-2 by overexpression of the human catalase gene in cerebral microvascular endothelial cells. J Neurochem 2000; 75: 614-623.
- Jang BC, Kim DH, Park JW, et al. Induction of cyclooxygenase-2 in macrophages by catalase: role of NF-kappaB and PI3K signaling pathways. Biochem Biophys Res Commun 2004; 316: 398-406.
- Jang BC, Paik JH, Kim SP, et al. Catalase induced expression of inflammatory mediators via activation of NF-kappaB, PI3K/AKT, p70S6K, and JNKs in BV2 microglia. Cell Signal 2005; 17: 625-633.
- Kobayashi T, Nakatani Y, Tanioka T, et al. Regulation of cytosolic prostaglandin E synthase by phosphorylation. Biochem J 2004; 381: 59-69.
- Park JY, Pillinger MH, Abramson SB. Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol 2006; 119: 229-240.
- Barbieri SS, Amadio P, Gianellini S, Zacchi E, Weksler BB, Tremoli E. Tobacco smoke regulates the expression and activity of microsomal prostaglandin E synthase-1: role of prostacyclin and NADPH-oxidase. FASEB J 2011; 25: 3731-3740.
- de Oliveira AC, Candelario-Jalil E, Bhatia HS, Lieb K, Hüll M, Fiebich BL. Regulation of prostaglandin E2 synthase expression in activated primary rat microglia: evidence for uncoupled regulation of mPGES-1 and COX-2. Glia 2008; 56: 844-855.
- Xiao L, Ornatowska M, Zhao G, et al. Lipopolysaccharide-induced expression of microsomal prostaglandin E synthase-1 mediates late-phase PGE2 production in bone marrow derived macrophages. PLoS One. 2012; 7: e50244.
- Zou MH, Klein T, Pasquet JP, Ullrich V. Interleukin 1beta decreases prostacyclin synthase activity in rat mesangial cells via endogenous peroxynitrite formation. Biochem J 1998; 336: 507-512.
- Zhao G, Yu R, Deng J, et al. Pivotal role of reactive oxygen species in differential regulation of lipopolysaccharide-induced prostaglandins production in macrophages. Mol Pharmacol 2013; 83: 167-178.
- Segal ML, Fertel RH, Kraut EH, Sagone AL. The role of reactive oxygen species in thromboxane b2 generation by polymorphonuclear leukocytes. J Lab Clin Med 1983; 102: 788-794.
- Basili S, Pignatelli P, Tanzilli G, et al. Anoxia-reoxygenation enhances platelet thromboxane A2 production via reactive oxygen species-generated NOX2: effect in patients undergoing elective percutaneous coronary intervention. Arterioscler Thromb Vasc Biol 2011; 31: 1766-1771.
- Tang WH, Stitham J, Gleim S, et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J Clin Invest 2011; 121: 4462-4476.
- Konturek PC, Kania J, Burnat G, Hahn EG, Konturek SJ. Prostaglandins as mediators of COX-2 derived carcinogenesis in gastrointestinal tract. J Physiol Pharmacol 2005; 56(Suppl. 5): 57-73.
- Cha B, Lim JW, Kim KH, Kim H. 15-deoxy-D12,14-prostaglandin J2 suppresses RANTES expression by inhibiting NADPH oxidase activation in Helicobacter pylori-infected gastric epithelial cells. J Physiol Pharmacol 2011; 62: 167-174.
- Kwiecien S, Konturek PC, Sliwowski Z, et al. Interaction between selective cyclooxygenase inhibitors and capsaicin-sensitive afferent sensory nerves in pathogenesis of stress-induced gastric lesions. Role of oxidative stress. J Physiol Pharmacol 2012; 63: 143-151.
- Sun GY, Horrocks LA, Farooqui AA. The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem 2007; 103: 1-16.
- Weinreb O, Mandel S, Amit T, Youdim MB. Neurological mechanisms of green tea polyphenols in Alzheimer's and Parkinson's diseases. J Nutr Biochem 2004; 15: 506-516.
- Zhao B. Natural antioxidants protect neurons in Alzheimer's disease and Parkinson's disease. Neurochem Res 2009; 34: 630-638.
- Lee IT, Yang CM. Role of NADPH oxidase/ROS in pro-inflammatory mediators-induced airway and pulmonary diseases. Biochem Pharmacol 2012; 84: 581-590.
A c c e p t e d : August 14, 2013