GASTRIN PROMOTES INTESTINAL POLYPOSIS THROUGH CHOLECYSTOKININ-B RECEPTOR-MEDIATED PROLIFERATIVE SIGNALING AND FOSTERING TUMOR MICROENVIRONMENT
INTRODUCTION
Colorectal cancer is the third most common cancer and the fourth leading cause of cancer-related death in the world, but its etiological factor is still unknown (1). One of the predisposing factors of colon cancer is inflammatory bowel disease (IBD) such as ulcerative colitis (UC) and Crohn’s disease (CD) with longstanding and extensive involvement (2). While the cause of sporadic colorectal cancer (CRC) is not clear, though the majority of CRCs have been known to develop from an adenomatous polyp (3), colitis-associated colon cancer (CAC) is fundamentally related to inflammation. Progression from adenoma to carcinoma requires a combination of genetic mutations which are either hereditary and/or occur as a result of environmental risk factors, during which gastrin has been extensively studied as a growth factor for CRC. However, few studies have investigated its role in either the adenoma-carcinoma sequence or colitis-associated carcinogenesis (4).
Gastrin is a peptide hormone produced by G cells in the gastric antrum, which is fundamentally responsible for the stimulation of acid secretion from the parietal cell, but also acts as a potent cell-growth factor that has been implicated to the maintenance of the gastric mucosa, proliferation of enterochromaffin-like cells (ECL), as well as neoplastic transformation (5). Gastrin binds to cholecystokinin-B receptor (CCK-B) receptor which mediates the proliferative action through signal transduction pathways. Since not only colorectal cancer cells but also colon epithelium has CCK-B receptor (6), high levels of gastrin had been acknowledged as risk factor for CRC. Gastrin levels were commonly increased through either Helicobacter pylori (H. pylori) infection or the administration of proton pump inhibitors (PPIs) (7), several clinical studies have concluded that hypergastrinemia can be associated with an increased risk of colorectal cancer, particularly in patients with H. pylori infection (8). At preclinical level, gastrin stimulates both in vitro and xenografted human colon cancer cells (9) and in animal models of colonic carcinogenesis, hypergastrinemia increased the incidence and growth rate of colon neoplasms (10). Though CCK-B receptors have been detected in primary colorectal tumors (11), their role in the control of cancer cell growth remains controversial and deserves further investigation. Under the hypothesis that gastrin might play a trophic role on intestinal polyposis through either proliferative action on epithelial cells or promoting tumor prone inflammatory microenvironment, we performed the experiments comprising of in vitro and in vivo intestinal polyposis model to prove the inflammatory and proliferative effects of gastrin via CCK-B receptor-mediated β-catenin pathway.
MATERIALS AND METHODS
Cell culture
HCT116 cells, human colorectal cancer cell line, were cultured in Macoy’s 5A medium containing 10% (v/v) fetal bovine serum, 100 U/ml penicillin and Raw 264.7 cells, macrophage cell line, were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% (v/v) fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin and 25 mM/L HEPES. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Cell proliferation and apoptosis analysis
Cells were plated at a density of 1–2 × 104 cells/well in 96-well plates, and the cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich, St, Louis, MO) reduction assay. After incubation, cells were treated with the MTT solution (final concentration, 1 mg/ml) for 2 hours. The dark blue formazan crystals formed in intact cells were dissolved with dimethyl sulfoxide and the absorbance at 570 nm was read using a microplate reader. Results were expressed as the percentage of MTT reduction obtained in the treated cells, assuming that the absorbance of control cells was 100%.
Western blot analysis
Extracted colon tissues were washed twice with PBS and then lysed in ice-cold cell lysis buffer (Cell Signaling Technology, Denver, MA) containing 1 mM phenylmethylsulfonyl fluoride (PMSF, Sigma Aldrich). After 20 min of incubation, samples were centrifuged at 10,000 ×g for 10 min. Supernatants were then collected. Proteins in lysates were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride (PVDF) membranes, which were incubated with primary antibodies, washed, incubated with peroxidase-conjugated secondary antibodies, rewashed, and then visualized using an enhanced chemiluminescence (ECL) system (GE Healthcare, Buckinghamshire, UK). Cyclin D1, CDK4, CCKB-R, β-catenin and a-tubulin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Reverse transcription-PCR for inflammatory cytokines
Total RNA were extracted from tissues using an RNeasy mini kit (Qiagen, N.V.), PCR was performed over 30 cycles of: 94°C for 20 s, 58°C for 30 s, and 72°C for 45 s. Oligonucleotide primers were purchased from Bioneer (Seoul, Korea). The sequences of PCR primers used were as follows: forward, 5’-AAT GTA TCC GTT GTG GAT CT-3’ and reverse, 5’-TCC ACC ACC CTG TTG CTG TA-3’ for mouse glyceraldehyde-3-phosphate dehydrogenase (GAPDH); forward, 5’-CAT CCT GCC AGC TCC ACC GC-3’ and reverse, 5’-GGG AGG AAG GGC CCT GGT GT-3’ for mouse COX-2; forward, 5’-GTG GTG ACA AGC ACA TTT GG-3’ and reverse, 5’-GGC TGG ACT TTT CAC TCT GC-3’ for mouse inducible nitric oxide synthase (iNOS); forward, 5’-CAG GCT CCG AGA TGA ACA ACA AAA-3’ and reverse, 5’-TGG GGA ACT CTG CAG ACT CAA ACT-3’ for mouse IL-1β; forward, 5’-CCG GAG AGG AGA CTT CAC AG-3’ and reverse, 5’-TGG TCT TGG TCC TTA GCC AC-3’ for mouse IL-6. Perkin-Elmer GeneAmp PCR System 2400 is used and cDNA was prepared using reverse transcriptase originating from Murine-Moloney leukemia virus (Promega, Madison, WI).
Cell-cycle analysis
Cells were treated with gastrin and PBS as a control. After trypsinization, cells were fixed in 70% ethanol for 30 minutes at 4°C. The cells were washed twice with PBS, and then incubated for 30 minutes under dark condition at 37°C in 1 mL of PBS containing 100 µg propidium iodide and 100 µg RNase A. The flow cytometric analysis was done and the proportion of cells was assessed by the histograms generated using the computer program, Mod-Fit.
Small interfering RNA
Small interfering RNA (siRNA) was purchased from Bioneer (Seoul, Korea) and transfected into HCT116 cells using lipofectamine 2000 according to the manufacturer’s procedure (Invitrogen, Grand Island, NY).
Immunohistochemical staining
After deparaffinization and rehydration, antigen retrieval was performed by boiling in citrate buffer (pH 6.0). After cooling, slides were incubated for 60 minutes at room temperature with the following rabbit or mouse monoclonal antibodies: F4/80 (1:500; eBioscience, San Diego, CA) or CD3 (1:300; Dako, Denmark) or Ki67(1:2000; Abcam, Cambridge, UK), respecively. All slides were developed with diaminobenzidine followed by hematoxylin counterstaining. Scoring was estimated as the percentage of positively stained cells of whole tissue and performed by the first author, who was blinded as to the primary antibodies and the treatment groups.
Animals and study protocol
C57BL/6J-APC (Min/+) mice were obtained from Jackson Laboratories (Bar Harbor, ME) in a pure C57BL/6 background. Min mice were determined as previously described by PCR using tail DNA extracted. The sequences of PCR primers used were as follows: 5’-GCC ATC CCT TCA CGT TAG -3’ for mouse wild type; 5’-TTC CAC TTT GGC ATA AGG C -3’ for mouse common type; 5’-TTC TGA GAA AGA CAG AAG TTA -3’ for mouse mutant type. Five-weeks C57BL/6J-APC (Min/+) mice were fed sterilized commercial pellet diets (Biogenomics) and sterile water ad libitum, and housed in an air-conditioned biohazard room at a temperature of 24°C. One group of 8 mice, the first group (group 1), was given intraperitoneum (i.p.) injections of phosphate buffered saline (PBS, iNtRON Biotechnology) 3 times per week for 12 weeks. The second group (group 2) was given i.p. injections of gastrin (10 µg/kg, Sigma-Aldrich) 3 times per week for 12 weeks. Animals were handled in an accredited animal facility in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care International policies. Following euthanasia, small intestine was resected, opened longitudinally, washed with sterile PBS. Polyps on intestinal segments were counted, and their sizes were measured with caliper.
Cytokine array
Cytokine array was performed mouse cytokine antibody array 3 (2 membrane arrays) with accessories, for simultaneous detection of 62 cytokines in 2 samples from RayBiotech (Norcross, Calif, USA). It detects: a novel receptor tyrosine kinase Axl, B lymphocyte chemoattractant (BLC), cluster of differentiation 30 ligand (CD30 L), CD30 T, CD40, cytokine response gene (CRG)-2, cutaneous T cell attracting cytokine (CTACK), chemokine (C-X-C motif) ligand 16 (CXCL16), eotaxin, eotaxin-2, Fas ligand, fractalkine, granulocyte colony-stimulating factor (GCSF), granulocyte-macrophage colony stimulating factor (GM-CSF), interferon-gamma (IFN-γ), insulin like growth factor binding protein (IGFBP)-3, IGFBP-5, IGFBP-6, IL-1, IL-10, IL-12 p40/p70, IL-12 p70, IL-13, IL-17, IL-1, IL-2, IL-3, IL-3 Rb, IL-4, IL-5, IL-6, IL-9, keratinocyte-derived chemokine (KC), leptin, leptin R, lipopolysaccharide-induced CXC chemokine (LIX), L-selectin, lymphotactin, monocyte chemotactic peptide (MCP)-1, MCP-5, macrophage-colony stimulating factor (M-CSF), macrophage inducing gene (MIG), macrophage inflammatory protein (MIP)-1, MIP-1, MIP-2, MIP-3, MIP-3, platelet factor (PF)-4, P-selectin, regulated upon activation, normal T cell expressed, and secreted (RANTES), stem cell factor (SCF), stromal cell-derived factor (SDF)-1, soluble tumor necrosis factor receptor I (sTNF RI), sTNF RII, thymus and activation regulated chemokine (TARC), T-cell activation (TCA)-3 (TCA-3, a chemotactic cytokine), thymus-expressed chemokine (TECK, a CCR9 ligand), tissue inhibitor of metalloproteinases (TIMP)-1, tumor necrosis factor-a (TNF-a), thrombopoietin, VCAM-1, vascular endothelial cell growth factor (VEGF). After blocking the array membranes for 30 minutes, the membranes were incubated with 1 mL of serum at room temperature for 2 hours. After washing with buffer, we added primary biotin-conjugated antibody to each membrane, for incubation at room temperature for 2 hours. After washing with buffer and addition of horseradish peroxidase-conjugated streptavidin to each membrane, we exposed them to detection buffer, using a luminescent image analyzer system (LAS-4000, Fuji Film; Tokyo, Japan). Density was expressed as the percentage of the detected value from the sample versus the background result, using a gelpro32 program (Media Cybernetics, L.P.).
Luciferase reporter activity assay
Cells were seeded at a concentration achieving 50% confluence in 12-well plates for 24 hours prior to transfection. Cells were transiently transfected with 0.2 µg/well of a translucent Tcf/Lef-Luc reporter vector, which was designed to measure the transcriptional activity of Tcf/Lef-responsive genes. After transfection, cells were treated with gastrin in serum-free medium for the indicated time periods, and the cell lysis was carried out with the reporter lysis buffer (Promega). After mixing the cell extract with a luciferase substrate (Promega), the luciferase activity was measured by the luminometer (Perkin Elmer). The β-galactosidase assay was done according to the supplier’s instructions (Promega β-galactosidase enzyme assay system) for normalizing the luciferase activity. All values are expressed as the percentage of activity relative to basal activity.
Statistical analysis
Results are expressed as the mean ±S.D. The data were analyzed by one-way analysis of variance (anova), and the statistical significance between groups was determined by Duncan’s multiple range test. Statistical significance was accepted with a P<0.05.
RESULTS
Gastrin significantly increased intestinal polyposis in APCMin/+ mice
APCMin/+ mice developed multiple intestinal polyps in whole small intestine and colon due to apc mutation accompanied with β-catenin accumulation. As shown in Fig. 1A, control group were fed a normal diet and injected with PBS intraperitoneally 3 times per week for 12 weeks (Group 1), whereas 10 µg/kg of gastrin were injected at the i.p. 3 times per week for 12 weeks as Group 2. Fig. 1B shows the representational gross appearance of small intestine, small sized polyps developed in the whole small intestine with mass-forming type features without any prevalent locations (arrows). However, repeated injections of gastrin (Group 2) led to the significantly increased development of small intestine polyposis (P <0.005, arrows). As shown in 1:1 magnified pathology observation, intestinal polyps were developed more than two times in Group 2 compared to those of Group 1 and the size of each intestinal polyp was apparently increased in Group 2 compared to size of Group 1 polyp, similar to gross photo observation, suggesting that gastrin administration significantly increased the incidence of intestinal polyposis as well as size of each polyp than no-treated group (P <0.005). On pathology, increased polyp size was related to increases in glandular proliferation, concluding that gastrin exerted significant trophic actions in APCMin/+ mice. Accompanied with intestinal crypt proliferation, cellular infiltrations were noted in both group, but more apparently in Group 2, signifying that gastrin might be associated with the change of tumor microenvironment. Therefore, we progressed to measure the changes of immunologic activity according to each group, adopting F4/80 as macrophage marker and CD3 as T-cell marker. As shown in Fig. 1C, the mean expressions of F4/80 were significantly increased in Group 2 compared to Group 1 (P <0.05), but did not differ in CD3, suggesting that both tumor microenvironmental change and trophic action relevant to gastrin administration contributed to increased intestinal polyposis in APCMin/+ mice.
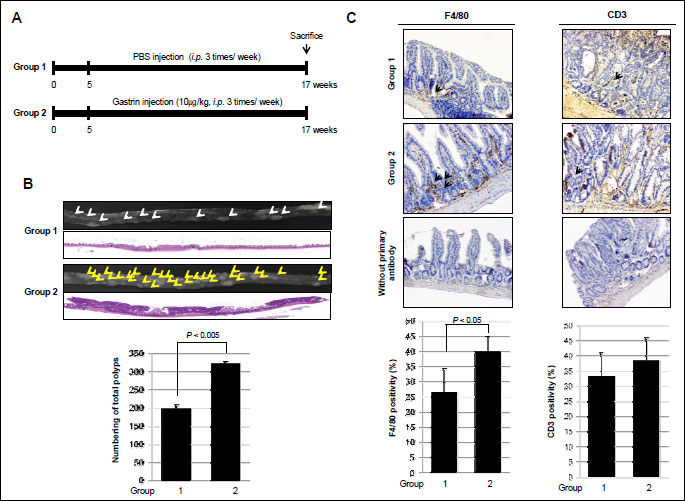
(A) The schematic overview of the experimental protocol of an animal model for small intestine polyposis using APCMin/+ mice. The experimental animal (n=8) were divided into 2 groups: control group and gastrin group were injected with PBS and 10 µg/kg of gastrin, i.p., three times per week for 12 weeks.
(B) Representational gross and microscopic pathologies according to group: Group 1 as control and Group 2 as gastrin administration. Representative pathologic pictures (magnification × 1) (upper panel). Count of polyps in the whole small intestine done according to group (lower panel). Arrows showed intestinal polyp.
(C) Immunohistochemical staining with F4/80 or CD3 antibodies was performed (×100). Results are expressed as means ±S.D. (n=8). Slides also were stained without primary antibody as a negative control (magnification × 100). Arrows showed positive staining cells.
Tumor microenvironmental changes contributed to increased intestinal polyposis after gastrin
To confirm the proliferative effects of gastrin and its responsible genes on fostering tumor microenvironment, Raw 264.7 macrophage cells were treated with gastrin for 12 hours. Gastrin showed proliferative effects on Raw 264.7 cells as seen with significantly increased cellular proliferation following dose-dependent manner (P <0.05, Fig. 2A). In order to assess the involvement of the cell cycle regulatory protein in these macrophage proliferations, we conducted Western blotting using antibodies recognizing cyclin D1 and CDK4, based on our preliminary experiment that gastrin induced S phase increment. Treatment of Raw 264.7 cells with gastrin significantly increased the expression of cyclin D1 and CDK4 compared to non-treated group (Fig. 2B). We repeated immunohistochemistry to compare the proliferative status after gastrin (Fig. 2C), which showed statistically significantly increased Ki-67 expression after gastrin (P <0.005, Fig. 2C). Next, in order to clarify the genes engaged in macrophage activations relevant to gastrin administration, we performed the cytokine array before and after gastrin treatment in macrophage cell lines. Using the murine cytokine antibody array designed for simultaneous detection of 62 cytokines, we repeated measurement thrice to pull out significant spots differed between before and after 100 nM gastrin administration (Fig. 3A), after which IL1β, IL3-Rβ, SDF-1α, TARC and TCA-3 were identified to be significantly increased in the gastrin treated group compared to non-treated group (Fig. 3B). For validating purpose after these cytokine arrays, we checked the expression of IL-6, IL-1β, COX-2, and iNOS expressions, in RT-PCR and qPCR, key cytokines reported to represent inflammatory reactions relevant to gastrointestinal tumorigenesis (12). Gastrin significantly increased the expressions of iNOS, COX-2, IL-6 as well as IL-1β (P <0.05, Fig. 3C).
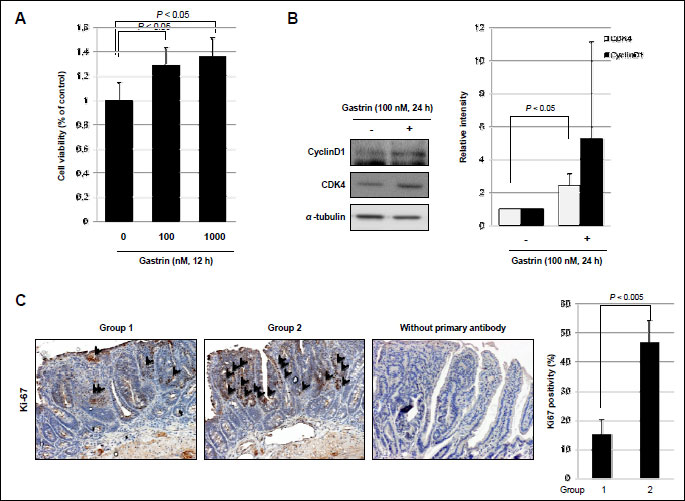
(A) To measure proliferation, Raw 264.7 cells treated with gastrin for 12 h were subjected to MTT assay.
(B) The expression of cyclin D1 and CDK4 was observed in Raw 264.7 cells treated with gastrin (100 nM/ml). All experiments were independently repeated at least 3 times.
(C) Immunohistochemical staining with or without Ki-67 antibody was performed (×100). Results are expressed as means ±S.D. (n=8).
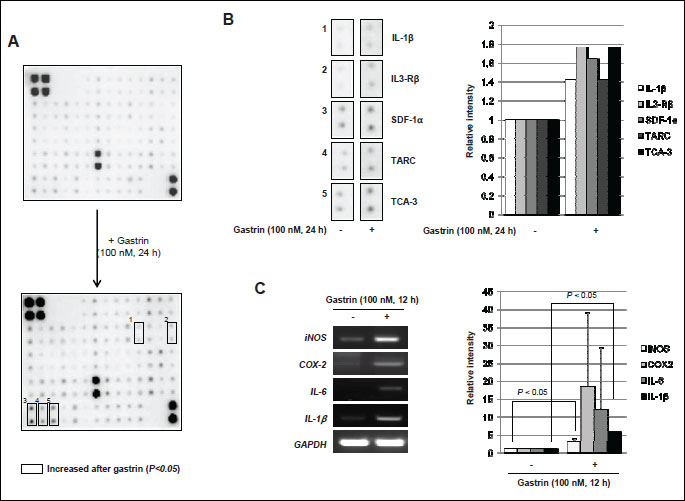
(A) For each of the two groups, upper data is the non-treated group, lower data is the gastrin (100 nM) treated group for 24 h. Square boxes indicated increased cytokines by gastrin.
(B) IL-1β, IL3-Rβ, SDF-1α, TARC and TCA3 were increased in density in the gastrin treated group than non-treated group. Right panel shows the relative intensity of increased each cytokine.
(C) RT-PCR for the proof of inflammatory cytokines with gastrin treatment. Cells were treated with gastsrin (100 nM) for 12 h, left panel shows RT-PCR results for check the expression of iNOS, COX-2, IL-6 and IL-1β. All these data are representative of three independent experiments.
Gastrin increases intestinal epithelial cell proliferation through on its receptor mediation
In order to elucidate the trophic effect of gastrin on epithelial cells, first, MTT assay was done in HCT116 cells. Gastrin significantly evoked proliferating action in a dose dependent manner (Fig. 4A). To determine the proliferative effect of gastrin on cell-cycle progression, serum-starved HCT116 cells were stained with propidium iodide and the DNA content was determined by flow cytometry in the absence and presence of gastrin. In the presence of gastrin, S phase population (24 h 10.63% in gastrin absence vs. 26.32% in gastrin presence) was significantly increased in HCT116 colon cells (Fig. 4B). Cyclin D1 and CDK4 reflecting S phase of cell cycle were checked by Western blot, before and after 10 nM and 100 nM gastrin. As seen in Fig. 4C, the expressions of cyclin D1 and CDK4 were significantly increased after gastrin administration. In addition, another cancer cell lines also were increased cyclin D1 expression after gastrin exposure. Especially human epithelial colorectal adenocarcinoma cells such as CACO-2 certainly showed high levels of cyclin D1 and CDK4 with gastrin and human gastric adenocarcinoma cells such as AGS and human pancreas epithelioid carcinoma cells such as PANC-1 were increased cyclin D1 expression in gastrin treated condition (Fig. 6). To further whether gastrin imposed trophic effects through its receptor, CCK-B, the effects of YM022, CCK-B receptor antagonist, were repeatedly investigated in HCT116 cells. Cell proliferations were significantly increased with gastrin in a dose-dependent manner (P <0.05), but proliferation were inhibited in the presence of YM022 in spite of gastrin (P <0.005, Fig. 5A). To determine the involvement of gastrin receptor in these trophic effects of gastrin, we transfected HCT116 cells with either control (siMock) or small interfering RNA of CCK-B-R (siCCK-B-R). siCCK-B-R transfecting cells lost the proliferating action of gastrin. As shown in Fig. 5B, cells transfected with siMock showed that gastrin significantly increased S phase population (24 hours 15.35% without gastrin vs. 25.79% with gastrin), but cells transfected with siCCK-B-R showed no change in S phase population (16.0% vs. 15.71%) in spite of gastrin administration (Fig. 5B), about which Western blot of cyclin D1 was followed. Increased expression of cyclin D1 was seen after gastrin, but not seen in siCCK-B-R cells (Fig. 5C) in spite of gastrin administration. Since the increased β-catenin nuclear trnaslocation and resulting enhanced Tcf/Lef-DNA binding is one of core driving force for intestinal polyposis in APCMin/+ mice (13), we have measured the expressions of β-catenin between siMock and siCCK-B-R cells and the expression of β-catenin after gastrin administration was decreased in siCCKB-R compared to siMock (Fig. 5D). Finally a Tcf/Lef responsive luciferase reporter assay was done to explore the β-catenin downstream transcription activity. A Tcf/Lef responsive luciferase reporter assay showed that the ablation of gastrin receptor, Tcf/Lef activation was not seen even after gastrin, confirming the proliferating action of gastrin is through gastrin-CCK-B-R-mediated β-catenin activation (Fig. 5E).
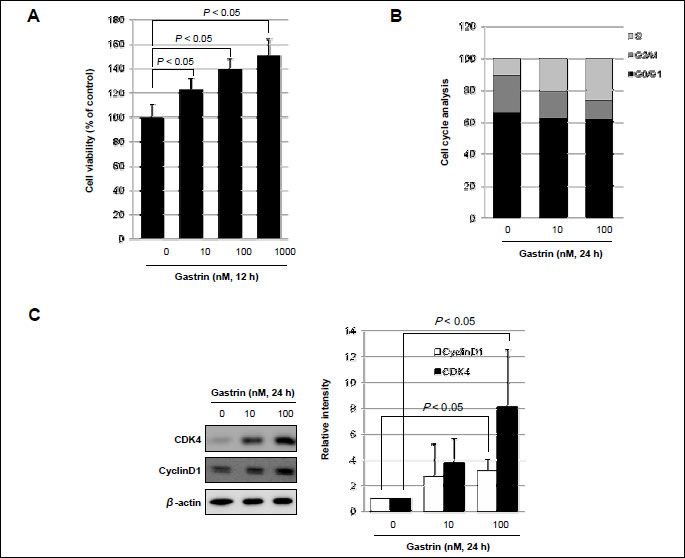
(A) To measure proliferation, HCT116 cells were treated with gastrin for 12 h and then evaluated using a MTT assay.
(B) Flow cytometry analysis of HCT116 cells synchronized at the G1/S transition by serum starvation and released into the cell cycle in the absence or presence of gastrin at different concentrations. The percentage of cells in different phases of the cell cycle was determined by using the Mod-Fit program.
(C) Equal amounts of total protein extracted from HCT116 cells incubated in the absence (-) or presence (+) of gastrin at different concentrations was subjected to Western blot analysis, using the indicated antibodies for cyclinD1 and CDK4 by Western blot.
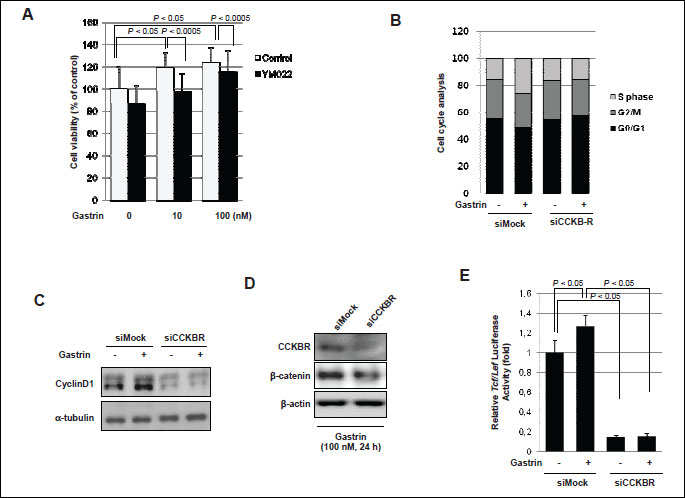
(A) Cells were treated with gastrin or combination of gastrin and YM022 as CCK-B-R-antagonist for 12 h and then measured MTT assay.
(B) Cells were transfected with nonspecific or CCK-B receptor specific siRNA. After serum starvation, flow cytometry analysis of synchronized cells was performed to measure the cell cycle in the absence or presence of gastrin for 24 h. The percentage of cells in different phases of the cell cycle was determined by using the Mod-Fit program.
(C) Cells were exposed with gastrin for 24 h in the absence or presence of CCK-B receptor and soluble protein extracts made were subjected to SDS-PAGE and Western blotting using the indicated antibodies.
(D, E) Cells were transfected with either siMock or siCCK-B-R and Western blot of β-catenin (D) and Tcf/Lef luciferase assay was done (E), respectively. Relative luciferase activity was measured and normalized with-galactosidase activity.
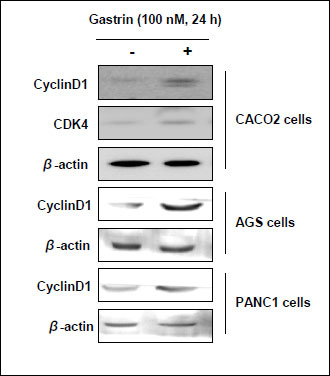 |
Fig. 6. Expressions of cyclinD1 and CDK4 before and after the treatment with 100 nM of gastrin were investigated in various cell types such as CACO-2 colon cells, AGS gastric cells, and PANC-1 pancreatic acinar cells. |
DISCUSSION
Under the hypothesis how gastrin showed trophic action in CRC, current study demonstrated the growth-stimulating actions of gastrin can be ascribed to the binding of gastrin with CCKB receptor, then, led to increased cellular proliferative activity through either induction of canonical β-catenin signaling pathway or downstream inflammatory mediators yielding tumor microenvironment to facilitate intestinal polyposis. Besides of β-catenin-mediated trophic effect of gastrin, gastrin fostered tumor microenvironment to facilitate increased potential of intestinal tumorigenesis and tumor angiogenesis. The polypeptide hormone gastrin secreted from G cells of stomach induces gastric acid secretion, thereby maintaining the appropriate acidic pH for digestion. Since increased gastrin expression is frequently seen with H. pylori infection and longer administration of PPIs, both rendering the risk of gastric tumorigenesis, hypergastrinemia itself has been regarded as a significant risk for GI tumorigenesis including gastric cancer (14). However, though hypergastrinaemia has been shown to promote development of gastric adenocarcinoma in mice, the Zollinger-Ellison syndrome, enterochromaffin-like (ECL) cells hyperplasia, and carcinoid tumors in human, it is still unclear whether gastrin is really a central player or a just secondary phenomenon in GI tumorigenesis or neither.
Gastrin is a diverse transcriptional activator mediating gene expression that is associated with cell division, invasion, angiogenesis as well as anti-apoptotic activity, all of which are also pivotal in the gain of malignant potential. The source of gastrin can be either endocrine through gastric mucosal secretion or of tumor origin or through de novo activation of the gastrin gene following early mutational events, including apc gene mutations. Gastrin has been known to increase proliferation of GI mucosal cells through transcriptional activation of a series of genes (15, 16). In stomach, gene expression of which is induced by gastrin stimulation, is REG1α, which affects the proliferation status of the stem-cell niche and is also overexpressed in gastric cancer and in GI tract, gastrin also promotes proliferation by activating the transcription of cyclin D1 in gastric cancer cells through a mechanism involving both β-catenin and the transcription factor CREB (cAMP response element-binding protein) pathways, which correlates with the finding that gastrin, as well as being a downstream target of β-catenin, stabilizes the expression of β-catenin and activates β-catenin-dependent transcription. Other increasingly expressed genes related to proliferating action of gastrin are EGFR ligands, amphiregulin and EGF, TGF-α, and peroxisome proliferator-activated receptor-γ (PPAR-γ) (17), among which in this our study, we have confirmed the cyclin D1, β-catenin, and its activator, Tcf/Lef as the trophic actions of gastrin in intestinal tumorigenesis.
Remarkable findings drawn in our investigation were that gastrin also significantly contributed to foster tumor microenvironment relevant to macrophage activations and subsequent chemotaxis. Cancer and inflammation are connected by two pathways of the intrinsic pathway and the extrinsic pathway; the intrinsic pathway of carcinogenesis is activated by genetic events, including the activation of various types of oncogene by mutation, chromosomal rearrangement or amplification, and the inactivation of tumor-suppressor genes and the extrinsic pathway included inflammatory or infectious conditions augment the risk of developing cancer at certain anatomical sites. Gastrin exerts inflammatory effects that induce pro-inflammatory cytokines, chemokines, and the adhesion molecules in diverse ways. In detail, role of gastrin in gastric carcinogenesis in H. pylori-infected humans (14) included the following villains participating in tumorigenesis; COX-2 stimulation relevant to inflammation (18, 19), interaction between leukocytes and endothelial cells through P-selectin and VCAM-1 (20), the activation of Rho-GTPase and caspase-3 (21), increased bacterial load and inflammatory activities by H. pylori-hypergastrinemia (22), and TNF-dependent chronic persistent inflammation (23). Since the epithelial cells and surrounding inflammatory mediators form a major part of the tumor microenvironment, inflammatory conditions usually precede development of malignancy and a tumor-promoting inflammatory milieu prevails in tumor tissue (24). The genes including IL-1β, IL-3Rβ, SDF-1α now called CXCL12, TARC (CCL17), and TCA-3 we identified with gastrin challenge were all the genes reported to be implicated in tumor angiogenesis, platelet chemotaxis, allergic reaction, and sepsis derived from macrophage (25-27). IL-6, IL-1β, COX-2, and iNOS, the genes all were increasingly expressed after gastrin, are principally implicated genes in either inflammatory bowel disease as well as colorectal carcinogenesis. However, to prevent growth effects of gastrin, some investigator was suggested using gastrin-vaccination. It might inhibit going gastrointestinal cancer and pancreatic cancer because growth effect of gastrin was limited to the gastrin’s bioactivity against epitope by vaccine-induced antibodies (28, 29).
The starting motive to execute the current study came from our previous investigation (29) that novel application of proton pump inhibitor (PPI) for the prevention of colitis-induced colorectal carcinogenesis in spite of hypergastrinemia relevant to PPI treatment. The question to be raised was the possible proliferative fears of colon cancer risk with hypergastrinemia caused by PPI rather than cancer prevention. However, the use of PPIs in clinical practice does not measurably increase the risk of colorectal cancer (30, 31), rendering hope that the trophic actions of hypergastrinemia also could be blocked with PPI. Results presented in current study reinforce the implication of gastrin in intestinal tumorigenesis as well the intervention of clinical candidates to target this hormone. As evidenced in large scaled cohort study that longstanding use of PPI for the treatment of acid related diseases did not increase the risk of GI tumorigenesis in spite of hypergastrinemia (30, 31) and the remark of Watson SA et al. in “Nature Reviews Cancer” (7) that though gastrin can be active participants or bystander in gastric carcinogenesis, highlighted areas ripe for future investigation will enable investigators to define the optimal combination of clinical candidates to target tropic action of this hormone as well as tumor microenvironment. Since we have already published data that PPI could exert immunomodulation, anti-inflammatory and cytoprotective effects as well as anti-mutagenic activities in spite of hypergastrinemia and additional investigation that both PPI and hypergastrinemia cooperated to augment anti-tumorigenic activities, optimal blocking hypergastrinemia can be the way of either hindering tumor proliferation or correcting tumor microenvironment.
Abbreviations: CCK-B receptor, cholecystokinin-B receptor; COXs, cyclooxygenases; PMSF, phenylmethylsulfonyl fluoride; PBS, phosphate buffered saline; SDK-1α, stromal cell-derived factor-1α.
Acknowledgements: This study was supported by a grant from the Ministry of Education and Science Technology (2010-0002052), Korea.
E.-H. Kim and K.B.Hahm contributed to the study design, data analysis and manuscript preparation. Y.-M. Han, J.-M. Park, and S.-H. Park performed all experiments. All authors have read and approved the final manuscript for publication.
Conflict of interests: None declared.
REFERENCES
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61: 69-90.
- Lofberg R, Brostrom O, Karlen P, Tribukait B, Ost A. Colonoscopic surveillance in long-standing total ulcerative colitis - a 15-year follow-up study. Gastroenterology 1990; 99: 1021-1031.
- Muto T, Bussey HJ, Morson BC. The evolution of cancer of the colon and rectum. Cancer 1975; 36: 2251-2270.
- Smith AM, Watson SA. Gastrin and gastrin receptor activation: an early event in the adenoma-carcinoma sequence. Gut 2000; 47: 820-824.
- Rozengurt E, Walsh JH. Gastrin, CCK, signaling, and cancer. Annu Rev Physiol 2001; 63: 49-76.
- Kovac S, Xiao L, Shulkes A, Patel O, Baldwin GS. Gastrin increases its own synthesis in gastrointestinal cancer cells via the CCK2 receptor. FEBS Lett 2010; 584: 4413-4418.
- Watson SA, Grabowska AM, El-Zaatari M, Takhar A. Gastrin - active participant or bystander in gastric carcinogenesis? Nat Rev Cancer 2006; 6: 936-946.
- Hartwich A, Konturek SJ, Pierzchalski P, et al. Helicobacter pylori infection, gastrin, cyclooxygenase-2, and apoptosis in colorectal cancer. Int J Colorectal Dis 2001; 16: 202-210.
- Smith AM, Watson SA. Review article: gastrin and colorectal cancer. Aliment Pharmacol Ther 2000; 14: 1231-1247.
- Watson SA, Smith AM. Hypergastrinemia promotes adenoma progression in the APC(Min-/+) mouse model of familial adenomatous polyposis. Cancer Res 2001; 61: 625-631.
- Schmitz F, Otte JM, Stechele HU, et al. CCK-B/gastrin receptors in human colorectal cancer. Eur J Clin Invest 2001; 31: 812-820.
- Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140: 883-899.
- Hu R, Khor TO, Shen G, et al. Cancer chemoprevention of intestinal polyposis in APCMin/+ mice by sulforaphane, a natural product derived from cruciferous vegetable. Carcinogenesis 2006; 27: 2038-2046.
- Konturek PC, Konturek SJ, Bielanski W, et al. Role of gastrin in gastric cancerogenesis in Helicobacter pylori infected humans. J Physiol Pharmacol 1999; 50: 857-873.
- Vigen RA, Chen D, Syversen U, Stunes K, Hakanson R, Zhao CM. Serum gastrin and gastric enterochromaffin-like cells during estrous cycle, pregnancy and lactation, and in response to estrogen-like agents in rats. J Physiol Pharmacol 2011; 62: 335-340.
- Takaishi S, Tu S, Dubeykovskaya ZA, et al. Gastrin is an essential cofactor for helicobacter-associated gastric corpus carcinogenesis in C57BL/6 mice. Am J Pathol 2009; 175: 365-375.
- Ptak-Belowska A, Pawlik MW, Krzysiek-Maczka G, Brzozowski T, Pawlik WW. Transcriptional upregulation of gastrin in response to peroxisome proliferator-activated receptor gamma agonist triggers cell survival pathways. J Physiol Pharmacol 2007; 58: 793-801.
- Konturek PC, Kania J, Kukharsky V, Ocker S, Hahn EG, Konturek SJ. Influence of gastrin on the expression of cyclooxygenase-2, hepatocyte growth factor and apoptosis-related proteins in gastric epithelial cells. J Physiol Pharmacol 2003; 54: 17-32.
- Guo YS, Cheng JZ, Jin GF, Gutkind JS, Hellmich MR, Townsend CM. Gastrin stimulates cyclooxygenase-2 expression in intestinal epithelial cells through multiple signaling pathways. Evidence for involvement of ERK5 kinase and transactivation of the epidermal growth factor receptor. J Biol Chem 2002; 277: 48755-48763.
- Ibiza S, Alvarez A, Romero W, Barrachina MD, Esplugues JV, Calatayud S. Gastrin induces the interaction between human mononuclear leukocytes and endothelial cells through the endothelial expression of P-selectin and VCAM-1. Am J Physiol Cell Physiol 2009; 297: C1588-C1595.
- He H, Yim M, Liu KH, Cody SC, Shulkes A, Baldwin GS. Involvement of G proteins of the Rho family in the regulation of Bcl-2-like protein expression and caspase 3 activation by gastrins. Cell Signal 2008; 20: 83-93.
- Chuang CH, Sheu BS, Yang HB, Kao AW, Cheng HC, Yao WJ. Hypergastrinemia after Helicobacter pylori infection is associated with bacterial load and related inflammation of the oxyntic corpus mucosa. J Gastroenterol Hepatol 2004; 19: 988-993.
- Damin DC, Santos FS, Heck R, et al. Effects of the gastrin-releasing peptide antagonist RC-3095 in a rat model of ulcerative colitis. Dig Dis Sci 2010; 55: 2203-2210.
- Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol 2013; 33(Suppl. 1): S79-S84.
- Uberti B, Dentelli P, Rosso A, Defilippi P, Brizzi MF. Inhibition of beta1 integrin and IL-3Rβeta common subunit interaction hinders tumour angiogenesis. Oncogene 2010; 29: 6581-6590.
- Werner L, Guzner-Gur H, Dotan I. Involvement of CXCR4/CXCR7/CXCL12 Interactions in inflammatory bowel disease. Theranostics 2013; 3: 40-46.
- Yamashita U, Kuroda E. Regulation of macrophage-derived chemokine (MDC, CCL22) production. Crit Rev Immunol 2002; 22: 105-114.
- Rehfeld JF, Goetze JP. Gastrin vaccination against gastrointestinal and pancreatic cancer. Scand J Gastroenterol 2006; 41: 122-123.
- Rahma OE, Khleif SN. Therapeutic vaccines for gastrointestinal cancers. Gastroenterol Hepatol (NY) 2011; 7: 517-564.
- Yang YX, Hennessy S, Propert K, Hwang WT, Sedarat A, Lewis JD. Chronic proton pump inhibitor therapy and the risk of colorectal cancer. Gastroenterology 2007; 133: 748-754.
- Robertson DJ, Larsson H, Friis S, Pedersen L, Baron JA, Sřrensen HT. Proton pump inhibitor use and risk of colorectal cancer: a population-based, case-control study. Gastroenterology 2007; 133: 755-760.
A c c e p t e d : August 12, 2013