DUAL EFFECT OF ETHANOL ON INWARD RECTIFIER POTASSIUM
CURRENT
IK1 IN RAT VENTRICULAR MYOCYTES
INTRODUCTION
Alcohol intoxication may induce arrhythmias (1-4), even the sudden cardiac death (5, 6). Changes of electrocardiographic parameters in human (namely a prolongation of the P wave, QRS complex, PR, QT and QTc intervals) were also reported, under moderate (7, 8) and even low (9) doses of alcohol.
Ethanol is known to alter the action potential (AP) configuration. Namely, AP shortening, deceleration of the upstroke velocity, and/or decrease of AP amplitude were observed (10-14). Ethanol was documented to inhibit some cardiac voltage-gated ionic currents: the sodium current (INa) (13, 15), the L-type calcium current (ICa) (13), and structural correlate of a-subunit of the fast delayed rectifier potassium current IKr (hERG, human ether-a-go-go-related gene channel) (16, 17). Laszlo et al. (18) revealed that a 120-hour intravenous ethanol infusion in rabbits reduced INa and ICa in subsequently isolated atrial cells but did not significantly alter the examined potassium currents, namely the transient outward current Ito, the sustained outward current Isus, and the inward rectifier current IK1. In our recent study (19), ethanol inhibited INa, ICa and Ito in rat ventricular myocytes; ICa was the most sensitive current to ethanol.
The impact of IK1 on regulation of the resting membrane potential, AP configuration and excitability in cardiomyocytes is well known (20). Modifications of IK1 (including those genetically-based) and IK1 heterogeneity are known to play an important role in the pathogenesis of arrhythmias (21-26). The role of selective IK1 inhibitors in the treatment of atrial fibrillation, the most common arrhythmia related to the alcohol consumption, has been analysed and discussed (27). Unfortunately, the current knowledge about changes of the cardiac IK1 under ethanol is very limited. To our knowledge, only the above mentioned study by Laszlo et al. (18) includes measurements of the ethanol effect on IK1 in cardiomyocytes. The authors concluded that the ethanol infusion (the blood alcohol levels 34-93 mM) did not significantly alter IK1 current density in rabbit atrial myocytes. Other sporadic data related to IK1 were obtained in particular Kir2.x subunits, namely in the IRK1 (Kir2.1) expressed in Xenopus oocytes or human embryonic kidney (HEK) 293T cells, showing a resistance (28) or a decrease of the current (29). These studies were, however, primarily focused on the G-proteins-coupled inwardly rectifying (GIRK) channels and, thus, data related to the effect of ethanol on Kir2.1 are extremely marginal.
Considering the role of IK1 in the cardiac electrophysiology and the limited knowledge of ethanol-induced changes of this current, we decided to examine the effect of ethanol on IK1 in rat ventricular myocytes, at concentrations relevant to current alcohol consumption in humans.
MATERIALS AND METHODS
The experiments were carried out with respect to recommendations of the European Community Guide for the Care and Use of Laboratory Animals; the experimental protocol (No. 4-11-06-2012) was approved by the Local Committee for Animal Treatment at Masaryk University, Faculty of Medicine (permission No. 12175/2001-1020(A)).
Cell isolation
Myocytes were isolated from right ventricles of adult male Wistar rats (250±50 g) anaesthetised by intramuscular administration of a mixture of tiletamin and zolazepam (65 mg kg-1; Zoletil® 100 inj., Virbac, France), and xylazine (20 mg kg-1; Rometar® inj., Spofa, Czech Republic).
The dissociation procedure was previously described in detail (30). In brief, the heart was retrogradely perfused via aorta with 0.9 mM CaCl2 Tyrode solution and then with nominally Ca-free Tyrode solution. During the first digestion step, the perfusion continued with nominally Ca-free Tyrode solution containing collagenase (type S, Yakult Pharmaceuticals, 0.2 mg mL-1), protease (type XIV, Sigma-Aldrich, 0.053 mg mL-1), and EGTA (Sigma-Aldrich; 34 µM). In the second digestion step, protease was omitted. The enzyme solution was then washed out in two steps by a perfusion with the low calcium Tyrode solutions (0.09 and 0.18 mM CaCl2). All solutions were oxygenated with 100% O2 at 37°C.
Solutions and chemicals
Tyrode solution with the following composition was used both during the dissociation procedure and to perfuse myocytes during the measurements (in mM): NaCl 135, KCl 5.4, MgCl2 0.9, HEPES 10, NaH2PO4 0.33, CaCl2 0.9, glucose 10 (pH was adjusted to 7.4 with NaOH). To inhibit ICa and the delayed rectifier potassium current IK in the course of IK1 measurements, CoCl2 (2 mM) and tetraethylammonium chloride (TEA, 50 mM), respectively, were applied. Additionally, 1 µM atropine and 10 µM glybenclamide were administered to avoid a contribution of the acetylcholine-activated potassium current IK(Ach) and the ATP-sensitive potassium current IK(ATP) to the observed IK1 changes despite it is unlikely under our experimental conditions (5 mM ATP in the pipette solution, isolated ventricular cells). In the case of measurements of ICa changes under 8 mM ethanol (Fig. 7), TEA (50 mM) and 4-aminopyridine (4-AP, 3 mM) were applied to inhibit IK and Ito, respectively.
The patch electrode filling solution contained (in mM): L-aspartic acid 130, KCl 25, MgCl2 1, K2ATP 5, EGTA 1, HEPES 5, GTP 0.1, Na2-phosphocreatine 3 (pH 7.25 adjusted with KOH).
CoCl2 (Sigma-Aldrich), atropine (Sigma-Aldrich) and 4-AP (Sigma-Aldrich) were prepared as 1 M, 1 mM and 100 mM stock solutions, respectively, in the deionized water (in the case of 4-AP, pH adjusted to 7.4 with HCl). Glybenclamide (Sigma-Aldrich) was prepared as 100 mM stock solution in dimethyl sulfoxide (DMSO; AppliChem GmbH, Germany). The concentration of DMSO in the final control solution and test solutions was identical (0.01%), moreover, it is unlikely that this concentration of DMSO has any effects on the cardiac IK1 (31, 32). To prepare the TEA-containing stock solution, NaCl in the used Tyrode solution (described above) was replaced equimolarly by TEACl (Sigma-Aldrich). Ethanol (99.5%, TAMDA, Czech Republic) was added to the Tyrode solution to obtain the final concentrations between 0.2 and 200 mM (corresponding to 0.0009 and 0.9%, respectively).
The solutions were applied in a close vicinity of the measured cell via a perfusion system; the time to change the solution was approximately 2 s.
Electrophysiological measurements and evaluation
Single rod-shaped cells with well visible striations were used for the current and AP recordings applying the whole cell patch-clamp technique in the voltage clamp and current clamp mode, respectively. The patch pipettes were pulled from borosilicate glass capillary tubes and heat polished on a programmable horizontal puller (Zeitz-Instrumente, Germany). The resistance of the filled glass electrodes was below 1.5 MΩ to keep the access resistance as low as possible. For generation of experimental protocols and data acquisition, the Axopatch 200B equipment and pCLAMP 9.2 software (Molecular Devices) were used. The series resistance was compensated up to 75%. The measured ionic currents were digitally sampled at 10 kHz and stored on the hard disc. Experiments were performed at room temperature (23±1°C). Experimental protocols are described in Results. The holding potential was -75 mV and the stimulation frequency was 0.2 Hz in all experiments except for data included in Fig. 6 and in Table 2 where the stimulation frequency was 1 Hz.
IK1 was evaluated as the Ba2+-sensitive current at the end of 500-ms pulse, either to -100 mV or to varying voltages between -120 and +10 mV. The evaluated current is expressed as the current density (in pA/pF) all over the manuscript to reduce differences among cells caused by their varying size.
The time course of the effect of 8 and 80 mM ethanol on IK1 was fitted with a double-exponential function y=A1*exp(-(t - t0)/ τ1) + A2*exp(-(t - t0)/τ2) + Aoo. Time course of the wash-out from the effect of 8 mM ethanol was evaluated as the time to 95% recovery of the current to its control level.
Mathematical simulations
To simulate the impact of experimentally assessed changes of the main cardiac ionic currents including IK1 under 0.8, 8 and 50 mM ethanol on AP configuration, a previously published mathematical model of the rat ventricular myocyte was used (33). The numerical solution was performed using the computational software MATLAB v. 7.2 (MathWorks, Inc.). The code of the model is available at: http://www.it.cas.cz/en/cl3/1033/biophysics-cardiac-cells
Statistical analysis
The results are presented as means ± S.E.M. from n cells (Origin, version 8.5.1, OriginLab Corporation). The curve fitting, paired and unpaired t-test, and one-way and repeated measures ANOVA with the Bonferroni post-test were performed using the GraphPad Prism, version 4.0 (GraphPad Software, Inc.); P<0.05 was considered statistically significant.
RESULTS
Effect of ethanol on the cardiac IK1 at a high concentration (80 mM)
Effect of 80 mM (~0.37%) ethanol is shown in Fig. 1. Superimposed representative steady-state recordings of the membrane current (Fig. 1A) elicited by a 500-ms hyperpolarizing test pulse from the holding potential of -75 mV to -100 mV were recorded in control conditions and under the effect of ethanol (15-ms prepulse to -40 mV was applied to inactivate INa). The recordings were performed both in the presence and absence of 100 µM Ba2+ to distinguish IK1 from contaminating currents that, anyway, appeared to be insensitive to ethanol. It follows that ethanol-induced changes of the measured current are exclusively caused by changes of IK1.
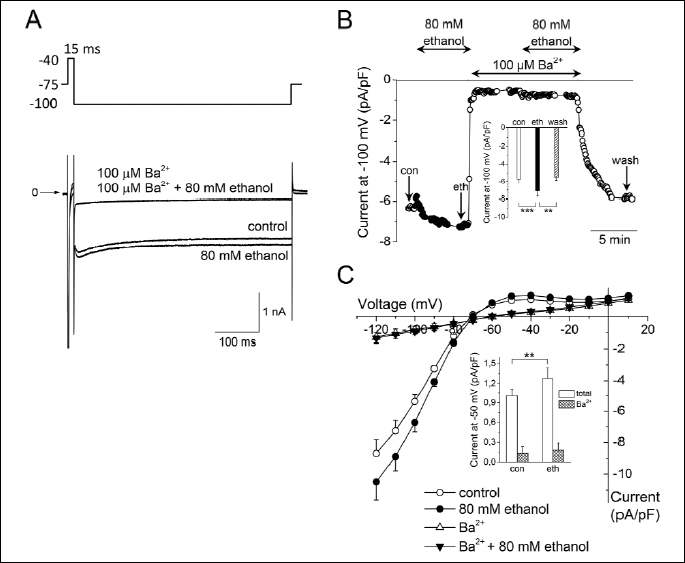
A: Superimposed current traces in control conditions and under the effect of 80 mM ethanol, both in absence and presence of 100 µM Ba2+ to distinguish IK1 from contaminating currents; the experimental protocol is shown above.
B: Changes of the current (expressed as the current density in pA/pF) measured at the end of 500-ms pulse to -100 mV during an experiment; arrows indicate the approximate time of analysis of the steady-state current as presented in the inset (an average from 5 consecutive traces preceding the change of a solution was evaluated). Inset: Averaged steady-state current at the end of 500-ms pulse to -100 mV in control conditions (con), under 80 mM ethanol (eth) and during the wash-out period (wash; n=8); ** and *** - statistical significance at P<0.01 and P<0.001, respectively; the current densities in control and wash-out were comparable (P>0.05).
C: Current-voltage relationship of the current measured at the end of 500-ms pulses in control and under 80 mM ethanol, both in the absence and presence of 100 µM Ba2+ (n=8). The residual current in the presence of 100 µM Ba2+ was not affected by ethanol. Experimental protocol: 500-ms pulses between -120 and +10 mV in 10-mV steps preceded by 15-ms prepulse to inactivate INa, the holding potential of -75 mV. Inset: Averaged steady-state current at the end of 500-ms pulse to -50 mV in control conditions (con), and under 80 mM ethanol (eth), both in the absence of Ba2+ ("total" denotes the total current), and in its presence in the concentration of 100 µM ("Ba2+" denotes the Ba2+-insensitive current); **–statistical significance at P<0.01 (n=8).
Fig. 1B illustrates running changes of the ionic current evaluated at the end of 500-ms hyperpolarizing pulse to -100 mV in the course of the above described experiment. The cell was successively exposed to 80 mM ethanol, 100 µM Ba2+, and a combination of ethanol and Ba2+. The current at -100 mV was significantly increased by 24.5±2.4% at the steady-state application of ethanol (eth) compared to control (con; n=8, P<0.001) and the effect of ethanol was fully reversible during the wash-out period (wash; inset in Fig. 1B).
The average current-voltage relationship in control and under 80 mM ethanol, both in the absence and presence of 100 µM Ba2+ (n=8) is shown in Fig. 1C (for the experimental protocol see legend). Currents were measured at the end of 500-ms pulses. Compared to the above described steady-state changes of the current at -100 mV under ethanol (inset in Fig. 1B), comparable significant changes were observed at physiologically relevant voltages, e.g. the current at -50 mV was significantly increased by 25.9±5.6% (n=8, P<0.01; inset in Fig. 1C). The voltage dependence of the residual current in the presence of Ba2+ was nearly linear and unaffected by ethanol (see also inset in Fig. 1C).
Fig. 2A shows representative traces of the current-voltage relationship of IK1 evaluated as the Ba2+-sensitive current in control conditions and under 80 mM ethanol. Both the inward and outward components of IK1 were significantly increased in the presence of 80 mM ethanol (Fig. 2B); the increase was voltage-independent (Fig. 2C).
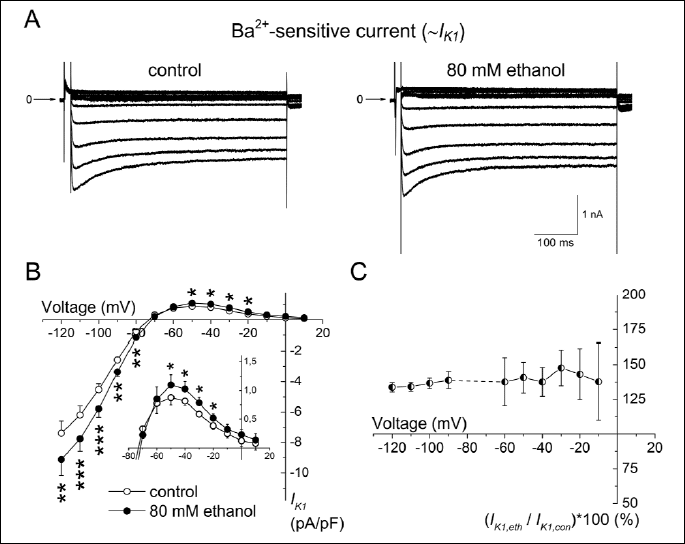
A: Representative records of IK1 evaluated as the Ba2+-sensitive current in the control conditions and in the presence of 80 mM ethanol (the experimental protocol described in legend to Fig. 1C).
B: Current-voltage relationship of IK1 in control and under 80 mM ethanol; inset: detail of changes of the outward component of IK1; *, ** and *** - statistical significance of the difference between IK1 under ethanol vs. in control at P<0.05, P<0.01 and P<0.001, respectively.
C: The relative increase of IK1 under 80 mM ethanol did not show statistically significant voltage dependence.
Dual effect of ethanol on IK1 manifested in the time course of its action
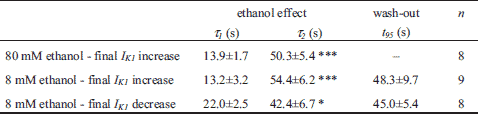
Fig. 3A illustrates in detail the time course of effect of 80 mM ethanol on IK1 evaluated at the end of 500-ms hyperpolarizing pulse to -100 mV in a representative cell (for the experimental protocol see Fig. 1A). Interestingly, the application of 80 mM ethanol regularly induced a distinct transient decrease of IK1, by 9.4±2.1% on average, followed by its increase by 24.5±2.4% (n=8; P<0.05 and 0.001, respectively; visible also in Fig. 1B). This time course of IK1 response to ethanol was approximated by a double-exponential function (for a representative fit see Fig. 3A) with the resulting average time constants τ1 = 13.9±1.7 s (related to the initial decrease) and τ2 = 50.3±5.4 s (related to the following increase; Table 1). The unusual mode of ethanol effect on IK1 (the dual effect) points to two underlying mechanisms (for details see Discussion).
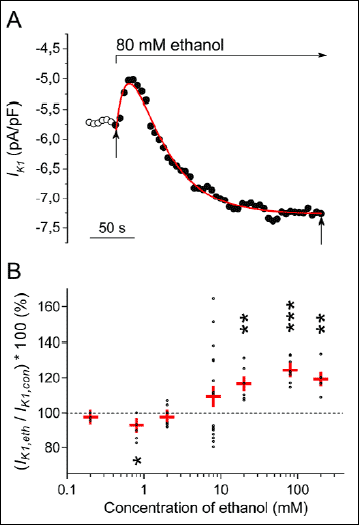 |
Fig. 3. Time- and concentration-dependence of ethanol effect on IK1. A: Transient changes of IK1 during application of 80 mM ethanol in a representative cell. Red line represents a double-exponential approximation of the time course of IK1 changes, vertical arrows indicate the start and the end of the fit. Note similar transient changes under ethanol in Fig. 1B. B: Pooled data of the concentration dependence of ethanol effect on IK1. Within the investigated concentration range (0.2 and 200 mM), a dual effect of ethanol was observed, proceeding from IK1 decrease (significant at a low concentration of 0.8 mM) to IK1 increase (at concentrations ≥20 mM); n=3-18 in particular concentrations. Red crosses represent the mean data ± S.E.M. in individual concentrations; *, ** and *** - statistical significance of the ethanol effect vs. the respective control at P<0.05, P<0.01 and P<0.001, respectively. |
Dual effect of ethanol on IK1 manifested in the concentration-dependence
The concentration-dependence of the effect of ethanol on IK1 was studied in the concentration range between 0.2 and 200 mM (Fig. 3B; the experimental protocol - inset in Fig. 1A). Changes of IK1 were evaluated after reaching the steady state. Surprisingly, the dual effect of ethanol on IK1 appeared also in the concentration-dependence. At very low concentrations ≤0.8 mM (~0.004%), ethanol decreased IK1 (the decrease was statisticaicant at 0.8 mM ethanol; n=5; P<0.05). However, in the ethanol concentrations ≥20 mM (~0.09%), IK1 was conversely significantly increased. Within the concentration range of a transition between the decrease and increase of IK1, i.e. in response to the application of 2 mM (~0.009%) and 8 mM (~0.04%) ethanol, a decrease of IK1 appeared in some cells but an increase in others (at 8 mM: IK1 decrease by 12.1±1.4% in 9 cells, but IK1 increase by 31.5±6.2% in other 9 cells). Both effects of ethanol were fully recovered during the subsequent wash-out (Figs. 4A and 4B).
Effect of ethanol on the cardiac IK1 at a low concentration (8 mM)
Effect of 8 mM (~0.04%) ethanol is shown in Fig. 4. The time courses of IK1 changes in two representative cells responding respectively by the steady-state increase or decrease of the current are illustrated in Fig. 4A (the experimental protocol - inset in Fig. 1A). In accordance with the previous results obtained at 80 mM ethanol (Figs. 1B and 3A), the steady-state increase of IK1 under 8 mM ethanol by 30.2±6.9% was preceded by a transient IK1 decrease by 12.8±2.0% at the beginning of the application (n=9; Fig. 4A, upper panel). Parameters describing the time course of these biphasic changes in the presence of 8 mM ethanol were not significantly different from those obtained under 80 mM ethanol (Table 1).
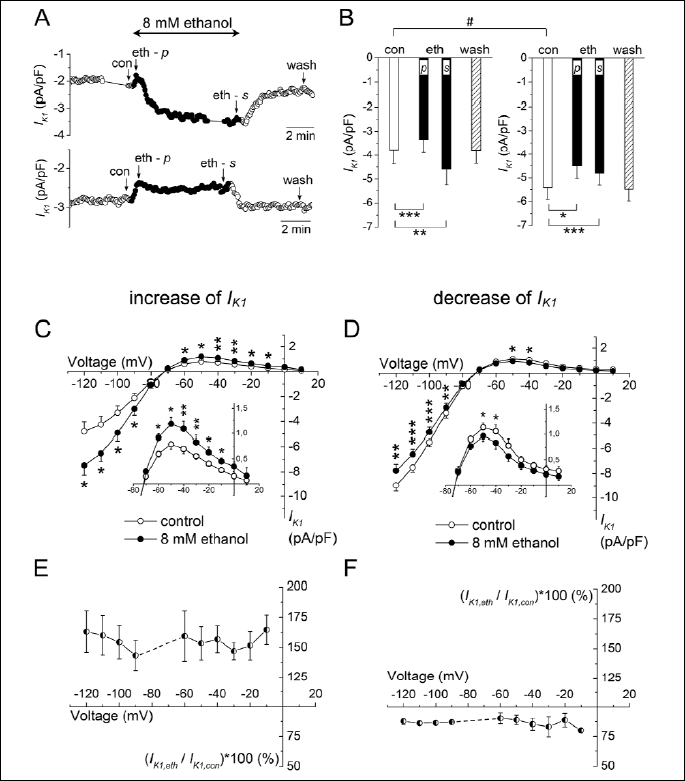
A: Time course of changes of IK1 evaluated at the end of 500-ms pulse to -100 mV (the experimental protocol - inset in Fig. 1A) under the effect of ethanol (full circles) and the subsequent wash-out in two representative cells showing either the final increase of IK1 (upper panel) or the final decrease of IK1 (lower panel); con, the control conditions; eth - p, the peak effect of ethanol at the beginning of its application; eth - s, the steady-state effect of ethanol at the end of its application; wash, the wash-out.
B: Averaged steady-state IK1 at the end of 500-ms pulse to -100 mV (for abbreviations see A) in cells with the final IK1 increase (left panel; n=9) or the final IK1 decrease (right panel; n=9); *, ** and *** - statistical significance at P<0.05, P<0.01 and P<0.001, respectively; IK1 densities in control and wash-out were comparable within the respective groups (P>0.05) but significantly different between the two groups of cells (# - statistical significance at P<0.05).
C, D: Current-voltage relationships of IK1 in control conditions (open circles) and under the effect of 8 mM ethanol (full circles) in the case of final IK1 increase (C; n=5) and final IK1 decrease (D; n=5); the experimental protocol described in legend to Fig. 1C; insets: details of changes of the outward component of IK1; *, ** and *** - statistical significance of the difference between IK1 under ethanol vs. in control at P<0.05, P<0.01 and P<0.001, respectively.
E, F: The relative changes of IK1 under 8 mM ethanol did not show any statistically significant voltage dependence.
In the cells showing a persistent decrease of IK1 in the presence of 8 mM ethanol (after ~400 s by 11.8±2.1%; n=9), the course of IK1 changes was also biphasic (Fig. 4A, lower panel; Table 1). The inhibition transiently deepened by 17.2±5.1% (n=9) at the beginning of ethanol application.
Both effects induced by 8 mM ethanol were reversible during the subsequent wash-out (Figs. 4A and 4B) within a comparable time (Table 1, P>0.05).
Intriguingly, the character of ethanol effect seemed to be dependent on the density of IK1 in control conditions. Of all the cells measured under 8 mM ethanol, the final increase of IK1 was present rather in the cells showing a smaller control IK1 density (-3.77±0.60 pA/pF, n=9; Fig. 4B, left panel) whereas the final decrease of IK1 was observed in cells with the higher control IK1 density (-5.42±0.49 pA/pF, n=9; Fig. 4B, right panel). The difference of the control current density in cells showing the opposite final changes of IK1 was statistically significant (P<0.05).
Figs. 4C and 4D show the current-voltage relationship of IK1 in control conditions and under 8 mM ethanol in cells with the final IK1 increase (C) or IK1 decrease (D; the experimental protocol - legend to Fig. 1C). Both the inward and outward components of IK1 were significantly changed. The dual effect of 8 mM ethanol on IK1 was virtually voltage-independent in both directions of the changed current (Figs. 4E and 4F). For instance, the inward current at -100 and the outward current at -50 mV were either comparably increased in a part of cells (by 43.4±11.2% and 42.3±10.3%, respectively; n=5, P>0.05), or decreased in others (by 10.2±2.5% and 15.4±5.2%, respectively; n=5, P>0.05).
Effect of ethanol in clinically relevant concentrations on AP configuration
The recorded alterations of AP configuration induced by ethanol in a moderate concentration of 50 mM (~0.24%) were supplemented by mathematical simulations. APs were elicited by 0.5-ms suprathreshold current pulses of 4-10 nA at the stimulation frequency of 0.2 Hz. Fig. 5A illustrates superimposed representative APs in control conditions and under 50 mM ethanol recorded in a rat right ventricular myocyte. In the presence of ethanol, a shortening of AP duration (APD) was apparent, at 90 % repolarization (APD90) by 7.2±2.2% (n=5). Fig. 5B shows the simulated APs (upper panel) and corresponding IK1 traces (lower panel) generated by a previously published model of the rat ventricular myocyte (33). Parameters of the stimulations were set in agreement with experiments. AP under 50 mM ethanol was simulated after implementing experimentally assessed inhibitions of INa, ICa and Ito by 5.6%, 8.35% and 2%, respectively (medians from data by Bebarova et al. (19)), and the activation of IK1 by 20% (predicted by the fitted curve in Fig. 7). The changes of simulated AP after application of 50 mM ethanol were consistent with those observed in experiments. Interestingly, the shortening of APD90 remained nearly unaltered when the impact of IK1 change alone was simulated (APD90 shortened by 8.5% vs. by 9.7% when changes of all the relevant currents were included as listed above; see the blue dashed AP waveform in Fig. 5B, upper panel). It suggests that the observed shortening of AP duration in the final repolarization under the effect of 50 mM ethanol is predominantly caused by the ethanol-induced changes of IK1.
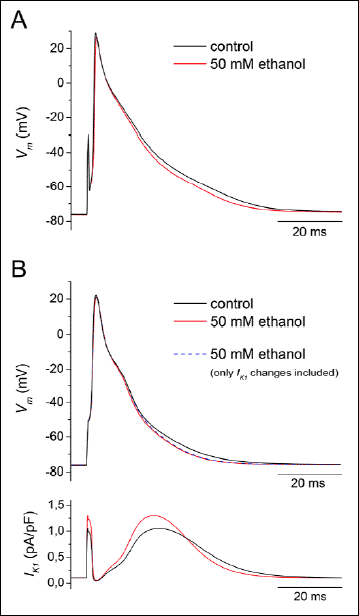 |
Fig. 5. Impact of IK1 changes on the action potential (AP) configuration under 50 mM (~0.24%) ethanol. A: Representative original APs in control conditions and under 50 mM ethanol recorded in a rat right ventricular myocyte (APs were elicited by 0.5-ms suprathreshold current pulses of 4–10 nA at the stimulation frequency of 0.2 Hz); Vm - the membrane voltage. B: Simulated AP waveforms (upper panel) were generated by a previously published model of the rat ventricular myocyte (33; parameters of the stimulation were set in agreement with experiments). Ethanol effect at 50 mM was simulated by implementing experimentally assessed inhibitions of INa by 5.6%, ICa by 8.35% and Ito by 2% (19) along with the activation of IK1 by 20% (Fig. 7). Note that AP shortening in the final repolarization remained nearly unaltered when the impact of IK1 change alone was simulated (see the blue dashed AP waveform). The lower panel illustrates simulated traces of IK1 in control and under 50 mM ethanol. For details see the related text. |
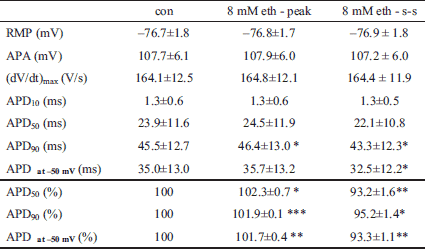
Fig. 6 shows the effect of ethanol on AP configuration at low concentrations (the stimulation frequency was 1 Hz). During application of ethanol at 8 mM (~0.04%), APD90, APD50 (APD at 50% repolarization) and APD at -50 mV (APD at -50 mV) were first prolonged and, subsequently, gradually shortened reaching a steady-state (Fig. 6A, left panel; see also Table 2) in agreement with the previously described effects of 8 mM ethanol on IK1. The modelled APs showed similar changes (Fig. 6A, right panel). Other AP characteristics remained unaltered (Table 2) which confirms the unique effects of 8 mM ethanol on IK1. Under the effect of 0.8 mM (~0.004%) ethanol, we observed only a prolongation of APs both in experiments and in the model (Fig. 6B). Fig. 6C demonstrates the course of APD90 changes during application of 0.8 and 8 mM till reaching the steady-state as simulated in the model.
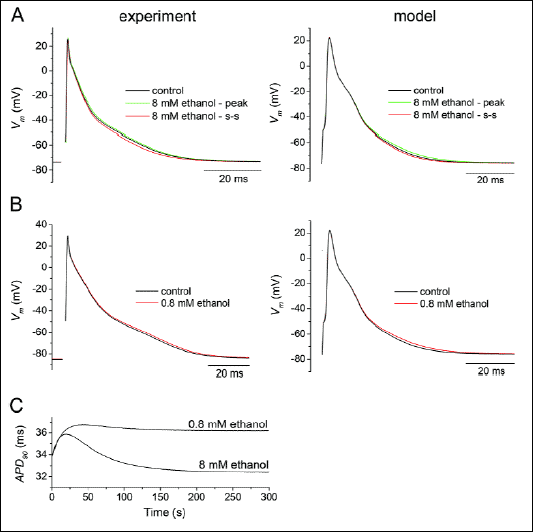 |
Fig. 6. Impact of IK1 changes on the action potential (AP) configuration under 8 and 0.8 mM ethanol at the stimulation frequency of 1 Hz. A: Application of 8 mM (~0.04%) ethanol resulted in a transient AP prolongation (the green line) followed by its shortening reaching a steady-state (the red line), both in experiments and in the model of rat ventricular myocyte (33). Other AP characteristics remained unaltered. Vm - the membrane voltage. B: Under the effect of 0.8 mM (~0.004%) ethanol, APs were prolonged both in experiments and in the model. C: The simulated course of changes of AP duration at 90% repolarization (APD90) during the first 300 s of application of 0.8 and 8 mM ethanol. |
DISCUSSION
In an effort to reveal mechanisms underlying effects of the alcohol consumption on the cardiac electrophysiology, a series of detailed studies has appeared, focused on the effect of ethanol on individual current components in mammalian cardiac cells (13, 15-19). However, a systematic study aimed at the ethanol effect on IK1, one of the principle potassium currents in cardiomyocytes, has been missing so far. The present study performed on rat ventricular cells showed for the first time that: 1) the sensitivity of IK1 to ethanol appears to exceed that of the majority of other currents explored so far (Fig. 7); 2) ethanol affects the cardiac IK1 in a dual way, manifested both in the time course and concentration dependence of its action in voltage clamp experiments. These effects became apparent also in corresponding changes of AP configuration as recorded in experiments and simulated on a computer model (Figs. 5 and 6).
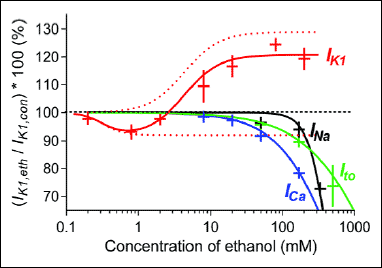 |
Fig. 7. Concentration dependence of the ethanol effect on IK1 in comparison with that on several other cardiac ionic currents. The mean ± S.E.M. values of relative changes of IK1 (red crosses; presented also in Fig. 3B) were fitted by a biphasic sigmoidal curve (the red line); for details see the related text; red dotted lines - the two components of the biphasic fit according to eq. (1). Effects of ethanol on sodium current INa, L-type calcium current ICa, and transient outward potassium current Ito as obtained in our previous study (19) are plotted for comparison. |
Fig. 7 shows the concentration dependence of ethanol effect on IK1 in comparison with that on INa, ICa and Ito investigated under the same experimental conditions previously (19). According to the concentration causing 10%-inhibition (75, 170, and 220 mM in ICa, Ito and INa, respectively), ICa was the most sensitive current to the ethanol effect. The lowest concentration of ethanol measured in that study was 20 mM. Therefore, in the present study, we supplemented the data by a measurement of the ethanol effect on ICa at 8 mM, i.e. at the concentration which considerably affects IK1. However, no significant change of ICa was observed. Thus, considering the above mentioned data on principal cardiac ionic currents in rat ventricular myocytes, IK1 seems to be the one most sensitive to the ethanol effect.
In spite of the fact that 50 mM ethanol affects also other ionic currents in rat ventricular myocytes, the respective shortening of APD in the final repolarization seems to result predominantly from the experimentally assessed ethanol-induced changes of IK1 as suggested by our mathematical simulations (Fig. 5B, upper panel). Under 0.8 and 8 mM ethanol, the changes of AP configuration appear to be completely related to the effects of ethanol on IK1 (Fig. 6).
In contrast to the ethanol-induced inhibition of the three cardiac ionic currents formerly investigated by our group (19), the shape of the ethanol concentration-response curve for IK1 is complex (Fig. 7). It resembles the biphasic (hormetic) dose-response which has been described in numerous toxicological and pharmacological studies over the past two decades (34). In our case, the dose-response is, however, inverted (IK1 is depressed at low concentrations and enhanced at higher concentrations). This unusual dual effect of ethanol on IK1 points to at least two underlying mechanisms.
In an attempt to formulate this idea in mathematical terms we tentatively fitted the ethanol–induced concentration dependent changes of IK1 observed in the current study to a biphasic sigmoid curve of the form (expressed in the logarithmic scale).
IC50 = 0.263 mM and EC50 = 4.365 mM are values of partial inhibition component and activation component, respectively; c denotes concentration of ethanol in mM, Ai (=8%) and Aa (=29%) are the maximal values of inhibition and activation components, hi (=3) and ha (=1.8) are the Hill coefficients. In Fig. 7, the concentration dependence of inhibition and activation terms appearing in eq. (1) are plotted as red dotted lines. It follows that the reducing and enhancing effects of ethanol on the rat ventricular IK1 appears to be saturated at concentrations above 1 and 20 mM, respectively.
Under 8 mM ethanol, two populations of the used ventricular myocytes were distinguishable according to their response to the ethanol effect, one with the resulting activation of IK1 and the other with its inhibition. The final response seems to be dependent on the current density in the control conditions; the cells with significantly lower control IK1 density showed the final activation and vice versa (Fig. 4B-4D). This two populations did not differ in the morphology and in the membrane capacitance (160.6±5.6 pF and 153.6±10.0 pF, respectively; n=9, P>0.05), thus, a contamination by other cell types can be excluded.
The dual effect of ethanol on IK1 was also manifested in the development of IK1 changes during the ethanol application. At low concentrations of ethanol <2 mM, IK1 regularly decreased monotonously (not shown). Surprisingly, 8 mM ethanol induced an initial decrease of IK1 followed by its final steady-state increase or decrease (Fig. 4). At concentrations ≥20 mM, the initial IK1 decrease was always followed by its gradual increase leading to the steady-state increase (Fig. 3A). Fig. 3A also shows that the time course of running IK1 changes under ethanol could be approximated by a double-exponential function with significantly different time constants of particular phases (Table 1). The faster rate of the inhibition compared to the activation further supports the idea of two underlying mechanisms. The faster initial inhibition may be overcompensated (Fig. 4A, upper panel) or partially compensated (Fig. 4A, lower panel) by a slower developed activation resulting in the final steady-state increase or decrease of IK1.
Mechanisms underlying the effect of alcohols on inward rectifier K+ (Kir) channels have been mostly studied in the expressed G-proteins-coupled Kir (GIRK) channels, playing an important role in the central nervous system, less frequently in the G-proteins-independent Kir2.1 (IRK1) channels (29, 35-38). However, distinct similarities between GIRK and IRK channels in the structure of intracellular domain has been described including the possible binding site for alcohols (29, 36). On the other hand, the response of co-expressed subunits cannot be regarded as a simple sum of the individual subunit responses (39). Effects of a substance on an ionic current in cardiac cells may differ from its effects on subunits forming the respective ionic channel heterogeneously expressed in a cell line as have been recently documented by Szentandrassy et al. (40). Hence, analysis of ethanol effects in cardiomyocytes is crucial for the proper understanding of its action.
We faced with the question whether all aspects of the complex manifestation of ethanol effect on IK1 observed in this study could be explicated by an acceptable biophysically based model. Such a model must be compatible with actual findings obtained in expressed channel studies: (i) IK1 channels in cardiac cells are constituted as heterogenic heterotetramers of different pore-forming α-subunits, in the case of ventricles mainly of Kir2.1, less of Kir2.2 and Kir2.3 (41-44) with different sensitivities to blockers or activators (39, 45). (ii) The common important regulator of the channel activity is a membrane anchored anionic phospholipid, phosphatidylinositol-4,5-bisphosphate (PIP2) that affects open channel probability (46-48). (iii) Two types of Kir channels have been reported with different response to ethanol. Beside other regulatory pathways which may affect their activity (49), GIRK channels appeared to be directly activated (in G-proteins-independent way) by primary alcohols up to the size of butanol in a PIP2-dependent manner (29, 38). A hydrophobic alcohol-bound "pocket" in the cytoplasmic domain of GIRK channels has been regarded a putative site for the activation by alcohols stabilizing the open conformation of these channels (29, 36, 38, 50). In contrast to GIRK channels, IRK1 (Kir2.1) channels were shown to be inhibited by short-chain alcohols including ethanol (29). Aryal et al. (29) also showed that mutations in the alcohol-binding pocket of IRK1 channels had no effect on the alcohol-dependent inhibition, suggesting an alternate site involved in the inhibition.
A relatively simple model that is able to simulate all the described unusual features of the effect of ethanol on IK1 (Fig. 8) is based on the following assumptions:
1. The dual effect of ethanol manifested in steady-state concentration responses (Fig. 3B) results from the presence of at least two populations of channels underlying ventricular IK1, one responding to ethanol by the activation, another one by the inhibition. IC50 for the inhibition is less than EC50 for the activation, thus, only inhibition was observed at low ethanol concentrations in all cells (0.8 mM in Fig. 3B).
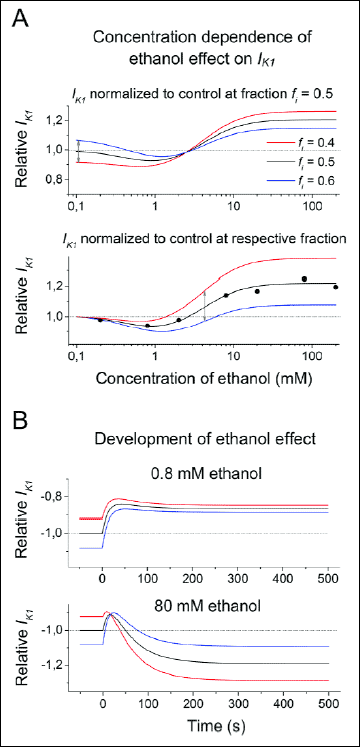 |
Fig. 8. Simulated features of the effect of ethanol on IK1. A: Concentration dependence of the ethanol effect. Upper panel: differences between cells varying in fractions of channels responding to ethanol by inhibition (fi). The curves intersect and, at concentrations bellow 2 mM, the cells with prevailing ethanol-induced activation (fi = 0.4) exhibit lower currents (red line) in comparison with the cells with prevailing ethanol-induced inhibition (fi = 0.6; blue line). Lower panel: differences between cells varying in fi after normalization to the control currents (~in the absence of ethanol) in agreement with experimental results (circles). The difference between concentration-dependent current curves from the cells with different fi is shifted toward higher concentrations (double arrow) revealing a range of concentrations where variations in fi may induce inhibition or activation in different cells. B: Transient changes of IK1 following application of ethanol at concentrations 0.8 mM (upper panel) and 80 mM (lower panel). All values are normalized to the current in the absence of ethanol and equal abundance ratio of channels responding to ethanol by inhibition and by activation (fi = 0.5). The normalized values are plotted with negative sign for better comparison with experimental results. |
2. The both putative populations of channels are assumed to contribute differently to the total conductance of IK1. Since the control conductance of channels responding to ethanol by the inhibition is higher, their contribution to IK1 prevails. However, the ratio of amounts of the channels of both types may differ in different cells, which is described in the model by variations of their fractions (fi stands for the fraction of channels responding to ethanol by the inhibition). This concept can explain why the cells that show the decrease at moderate concentration exhibit greater current under control conditions (~ in the absence of ethanol). Fig. 8A (upper panel) shows simulated concentration responses in cells with different fi. It is apparent that the curves intersect and cells showing the higher ethanol-induced activation exhibit the low current in control and vice versa. The concentration dependent effect illustrated in Fig. 3B summarizes experimental results obtained at individual concentrations from different cells so that all values are normalized to currents measured at the given cells in their control. Applying this normalization in the model (Fig. 8A, lower panel), the difference between currents from the cells with different fraction fi is shifted toward higher concentrations. This might explain our observations that at a certain range of ethanol concentrations some cells show increase and others decrease of the current at the steady state as indicated by the double arrow.
3. Kinetics of the transient changes following ethanol application is assumed to be governed by an interaction of the channel with PIP2. The response to ethanol bond is regarded several times faster in the channels that are inhibited than in channels that are activated in agreement with the time constants presented in Table 1. Interestingly, An et al. (51) described that changes of PIP2 concentrations evoked changes of the current with time constants around 16 and 55 s in Kir2.1 and Kir2.2 channels, respectively, which resembles the time constants of initial decrease and following increase of IK1 after the ethanol application observed in this study. Results from the model (Fig. 8B) show that the cells respond to ethanol by a simple inhibition at very low concentration (0.8 mM). At 80 mM, the transient inhibition is followed by the gradual activation. Again, the cells showing the greatest ethanol-induced steady-state current IK1 are those with the smallest current in control.
The observed changes of the cardiac IK1 under ethanol within concentrations relevant to the current alcohol consumption may be significant considering the important role of IK1 in control of the cardiac excitability and arrhythmogenesis (20, 22, 24). There are several clinical studies describing significant ECG changes in healthy volunteers at various ethanol plasma concentrations, Rossinen et al. (7) at moderate ethanol concentration of 26.1 mM (~0.12%), Cameli et al. (9) even at low ethanol concentration of 0.05%, Lorsheyd et al. (52) at high and very high ethanol concentrations (0.4% and 0.8%, respectively). Surprisingly, more than 20% of voluntaries in the latter study did not show presumptive ECG changes even at the very high ethanol level of 0.8%. These facts imply that the occurence of ECG changes induced by ethanol in healthy persons has to be regarded as highly individual. The risk of ethanol-induced ECG changes and arrhythmias related to IK1 changes may be increased under conditions affecting the cardiac IK1, particularly under an increased sympathetic tone, coexisting heart diseases or usage of drugs (21, 53-58), or in the presence of clinically latent inherited syndromes with gain-/loss-of-function mutations in the gene encoding Kir2.1 (23, 25, 48, 59) or other potassium channels (60).
To summarize, this is the first study of the effect of ethanol on IK1 in ventricular myocytes. Ethanol affected IK1 within concentrations considerably lower than in the case of the majority of other cardiac ionic currents studied so far. The IK1 was increased or decreased depending on the applied ethanol concentration. Both effects were voltage-independent and exhibited different kinetics, the inhibition proceeded faster than the activation. Changes of AP configuration studied under clinically relevant concentrations of ethanol agreed well with the data acquired in the voltage clamp experiments and with the simulated AP waveforms. The results suggest at least two different underlying mechanisms that remain to be clarified.
Acknowledgements: We thank to Prof. P. Braveny for reading the manuscript and valuable comments, and to Mrs. B. Vyoralova for the technical assistance. This work was supported by the grant project NT14301-3/2013 from the Departmental Program for Research and Development of the Ministry of Health of the Czech Republic, and partly by the institutional support RVO: 61388998.
Conflict of interests: None declared.
REFERENCES
- Haissaguerre M, Fleury B, Le Metayer P, et al. Arrhythmia in relation to acute alcoholic intoxication in the absence of obvious cardiopathy. Presse Med 1984; 13: 1723-1726.
- Fuenmayor AJ, Fuenmayor AM. Cardiac arrest following holiday heart syndrome. Int J Cardiol 1997; 59: 101-103.
- Kodama S, Saito K, Tanaka S, et al. Alcohol consumption and risk of atrial fibrillation: a meta-analysis. J Am Coll Cardiol 2011; 57: 427-436.
- Mandyam MC, Vedantham V, Scheinman MM, et al. Alcohol and vagal tone as triggers for paroxysmal atrial fibrillation. Am J Cardiol 2012; 110: 364-368.
- Templeton AH, Carter KL, Sheron N, Gallagher PJ, Verrill C. Sudden unexpected death in alcohol misuse-an unrecognized public health issue? Int J Environ Res Public Health 2009; 6: 3070-3081.
- Chiuve SE, Rimm EB, Mukamal KJ, et al. Light-to-moderate alcohol consumption and risk of sudden cardiac death in women. Heart Rhythm 2010; 7: 1374-1380.
- Rossinen J, Sinisalo J, Partanen J, Nieminen MS, Viitasalo M. Effects of acute alcohol infusion on duration and dispersion of QT interval in male patients with coronary artery disease and in healthy controls. Clin Cardiol 1999; 22: 591-594.
- Aasebo W, Erikssen J, Jonsbu J, Stavem K. ECG changes in patients with acute ethanol intoxication. Scand Cardiovasc J 2007; 41: 79-84.
- Cameli M, Ballo P, Garzia A, et al. Acute effects of low doses of red wine on cardiac conduction and repolarization in young healthy subjects. Alcohol Clin Exp Res 2009; 33: 2141-2146.
- Gimeno AL, Gimeno MF, Webb JL. Effects of ethanol on cellular membrane potentials and contractility of isolated rat atrium. Am J Physiol 1962; 203: 194-196.
- Williams ES, Mirro MJ, Bailey JC. Electrophysiological effects of ethanol, acetaldehyde, and acetate on cardiac tissues from dog and guinea pig. Circ Res 1980; 47: 473-478.
- Carryl OR, Gallardo-Carpentier A, Carpentier RG. Effects of nicotine and ethanol on rat atrial membrane potentials. Alcohol 1992; 9: 87-92.
- Habuchi Y, Furukawa T, Tanaka H, Lu LL, Morikawa J, Yoshimura M. Ethanol inhibition of Ca2+ and Na+ currents in the guinea-pig heart. Eur J Pharmacol 1995; 292: 143-149.
- Tsai CS, Loh SH, Jin JS, et al. Effects of alcohol on intracellular pH regulators and electromechanical parameters in human myocardium. Alcohol Clin Exp Res 2005; 29: 1787-1795.
- Klein G, Gardiwal A, Schaefer A, Panning B, Breitmeier D. Effect of ethanol on cardiac single sodium channel gating. Forensic Sci Int 2007; 171: 131-135.
- O'Leary ME. Inhibition of HERG potassium channels by cocaethylene: a metabolite of cocaine and ethanol. Cardiovasc Res 2002; 53: 59-67.
- Himmel HM. Suitability of commonly used excipients for electrophysiological in-vitro safety pharmacology assessment of effects on hERG potassium current and on rabbit Purkinje fiber action potential. J Pharmacol Toxicol Methods 2007; 56: 145-158.
- Laszlo R, Eick C, Schwiebert M, et al. Alcohol-induced electrical remodeling: effects of sustained short-term ethanol infusion on ion currents in rabbit atrium. Alcohol Clin Exp Res 2009; 33: 1697-1703.
- Bebarova M, Matejovic P, Pasek M, et al. Effect of ethanol on action potential and ionic membrane currents in rat ventricular myocytes. Acta Physiol (Oxf) 2010; 200: 301-314.
- Dhamoon AS, Jalife J. The inward rectifier current (IK1) controls cardiac excitability and is involved in arrhythmogenesis. Heart Rhythm 2005; 2: 316-324.
- Fauconnier J, Lacampagne A, Rauzier JM, Vassort G, Richard S. Ca2+-dependent reduction of IK1 in rat ventricular cells: a novel paradigm for arrhythmia in heart failure? Cardiovasc Res 2005; 68: 204-212.
- Piao L, Li J, McLerie M, Lopatin AN. Transgenic upregulation of IK1 in the mouse heart is proarrhythmic. Basic Res Cardiol 2007; 102: 416-428.
- Schimpf R, Borggrefe M, Wolpert C. Clinical and molecular genetics of the short QT syndrome. Curr Opin Cardiol 2008; 23: 192-198.
- Sekar RB, Kizana E, Cho HC, et al. IK1 heterogeneity affects genesis and stability of spiral waves in cardiac myocyte monolayers. Circ Res 2009; 104: 355-364.
- Tristani-Firouzi M, Etheridge SP. Kir2.1 channelopathies: the Andersen-Tawil syndrome. Pflugers Arch 2010; 460: 289-294.
- Wakili R, Voigt N, Kääb S, Dobrev D, Nattel S. Recent advances in the molecular pathophysiology of atrial fibrillation. J Clin Invest 2011; 121: 2955-2968.
- Ehrlich JR. Inward rectifier potassium currents as a target for atrial fibrillation therapy. J Cardiovasc Pharmacol 2008; 52: 129-135.
- Yamakura T, Lewohl JM, Harris RA. Differential effects of general anesthetics on G protein-coupled inwardly rectifying and other potassium channels. Anesthesiology 2001; 95: 144-153.
- Aryal P, Dvir H, Choe S, Slesinger PA. A discrete alcohol pocket involved in GIRK channel activation. Nat Neurosci 2009; 12: 988-995.
- Bebarova M, Matejovic P, Pasek M, Simurdova M, Simurda J. Effect of ajmaline on transient outward current in rat ventricular myocytes. Gen Physiol Biophys 2005; 24: 27-45.
- Ogura T, Shuba LM, McDonald TF. Action potentials, ionic currents and cell water in guinea pig ventricular preparations exposed to dimethyl sulfoxide. J Pharmacol Exp Ther 1995; 273: 1273-1286.
- Bosch RF, Li GR, Gaspo R, Nattel S. Electrophysiologic effects of chronic amiodarone therapy and hypothyroidism, alone and in combination, on guinea pig ventricular myocytes. J Pharmacol Exp Ther 1999; 289: 156-165.
- Pasek M, Simurda J, Christe G. The functional role of cardiac T-tubules explored in a model of rat ventricular myocytes. Philos Trans A Math Phys Eng Sci 2006; 364: 1187-1206.
- Calabrese EJ. Hormesis and medicine. Brit J Clin Pharmacol 2008; 66: 594-617.
- Kobayashi T, Ikeda K, Kojima H, et al. Ethanol opens G-protein-activated inwardly rectifying K+ channels. Nat Neurosci 1999; 2: 1091-1097.
- Pegan S, Arrabit C, Slesinger PA, Choe S. Andersen's syndrome mutation effects on the structure and assembly of the cytoplasmic domains of Kir2.1. Biochemistry 2006; 45: 8599-8606.
- Howard RJ, Slesinger PA, Davies DL, Das J, Trudell JR, Hartus RA. Alcohol-binding sites in distinct brain proteins: the quest for atomic level resolution. Alcohol Clin Exp Res 2011; 35: 1561-1573.
- Bodhinathan K, Slesinger PA. Molecular mechanism underlying ethanol activation of G-protein-gated inwardly rectifying potassium channels. Proc Natl Acad Sci USA 2013; 110: 18309-18314.
- Schram G, Pourrier M, Wang Z, White M, Nattel S. Barium block of Kir2 and human cardiac inward rectifier currents: evidence for subunitheteromeric contribution to native currents. Cardiovasc Res 2003; 59: 328-338.
- Szentandrassy N, Papp F, Hegyi B, Bartok A, Krasznai Z, Nanasi PP. Tetrodotoxin blocks native cardiac L-type calcium channels but not CaV1.2 channels expressed in HEK cells. J Physiol Pharmacol 2013; 64: 807-810.
- Wang Z, Yue L, White M, Pelletier G, Nattel S. Differential distribution of inward rectifier potassium channel transcripts in human atrium versus ventricle. Circulation 1998; 98: 2422-2428.
- Zobel C, Cho HC, Nguyen TT, et al. Molecular dissection of the inward rectifier potassium current (IK1) in rabbit cardiomyocytes: evidence for heteromeric coassembly of Kir2.1 and Kir2.2. J Physiol 2003; 550: 365-372.
- Dhamoon AS, Pandit SV, Sarmast F, et al. Unique Kir2.x properties determine regional and species differences in the cardiac inward rectifier K+ current. Circ Res 2004; 94: 1332-1339.
- Gaborit N, Le Bouter S, Szuts V, et al. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J Physiol 2007; 582: 675-693.
- Zhang L, Liu Q, Liu C, et al. Zacopride selectively activates the Kir2.1 channel via a PKA signaling pathway in rat cardiomyocytes. Sci China Life Sci 2013; 56: 788-796.
- Takano M, Kuratomi S. Regulation of cardiac inwardly rectifying potassium channels by membrane lipid metabolism. Progr Biophys Mol Biol 2003; 81: 67-79.
- Xie LH, John SA, Ribalet B, Weiss JN. Activation of inwardly rectifying potassium (Kir) channels by phosphatidylinosital-4,5-bisphosphate (PIP2): interaction with other regulatory ligands. Prog Biophys Mol Biol 2007; 94: 320-335.
- Hibino H, Inanobe A, Furutani K, Murakami S, Findlay I, Kurachi Y. Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev 2010; 90: 291-366.
- Witkowski G, Rola R, Szulczyk P. Effect of cyclic adenosine monophosphate on the G protein-dependent inward rectifier K(+)-like channel current in medial prefrontal cortex pyramidal neurons. J Physiol Pharmacol 2012; 63: 457-462.
- Lewohl JM, Wilson WR, Mayfield RD, Brozowski SJ, Morrisett RA, Harris RA. G-protein-coupled inwardly rectifying potassium channels are targets of alcohol action. Nat Neurosci 1999; 2: 1084-1090.
- An HL, Lu SQ, Li JW, et al. The cytosolic GH loop regulates the phosphatidylinositol 4,5-bisphosphate-induced gating kinetics of Kir2 channels. J Biol Chem 2012; 287: 42278-42287.
- Lorsheyd A, de Lange DW, Hijmering ML, Cramer MJ, van de Wiel A. PR and OTc interval prolongation on the electrocardiogram after binge drinking in healthy individuals. Neth J Med 2005; 63: 59-63.
- Karle CA, Zitron E, Zhang W, et al. Human cardiac inwardly-rectifying K+ channel Kir(2.1b) is inhibited by direct protein kinase C-dependent regulation in human isolated cardiomyocytes and in an expression system. Circulation 2002; 106: 1493-1499.
- Zitron E, Kiesecker C, Luck S, et al. Human cardiac inwardly rectifying current IKir2.2 is upregulated by activation of protein kinase A. Cardiovasc Res 2004; 63: 520-527.
- Zitron E, Gunth M, Scherer D, et al. Kir2.x inward rectifier potassium channels are differentially regulated by adrenergic alpha1A receptors. J Mol Cell Cardiol 2008; 44: 84-94.
- Koumi S, Backer CL, Arentzen CE. Characterization of inwardly rectifying K+ channel in human cardiac myocytes. Alterations in channel behavior in myocytes isolated from patiens with idiopathic dilated cardiomyopathy. Circulation 1995; 92: 164-174.
- Sanchez-Chapula JA, Salinas-Stefanon E, Torres-Jacome J, Benavides-Haro DE, Navarro-Polanco RA. Blockade of currents by the antimalarial drug chloroquine in feline ventricular myocytes. J Pharmacol Exp Ther 2001; 297: 437-445.
- Caballero R, Dolz-Gaiton P, Gomez R, et al. Flecainide increases Kir2.1 currents by interacting with cysteine 311, decreasing the polyamine-induced rectification. Proc Natl Acad Sci USA 2010; 107: 15631-15636.
- Mahida S, Hogarth AJ, Cowan C, Tayebjee MH, Graham LN, Pepper CB. Genetics of congenital and drug-induced long QT syndromes: current evidence and future research perspectives. J Interv Card Electrophysiol 2013; 37: 9-19.
- El Harchi A, Zhang H, Hancox JC. The S140G KCNQ1 atrial fibrillation mutation affects 'I(KS)' profile during both atrial and ventricular action potentials. J Physiol Pharmacol 2010; 61: 759-764.
A c c e p t e d : July 1, 2014