THE HUMAN ETHER-A’-GO-GO RELATED GENE (hERG) K+ CHANNEL BLOCKADE BY THE INVESTIGATIVE SELECTIVE-SEROTONIN REUPTAKE INHIBITOR
CONA-437: LIMITED DEPENDENCE ON S6 AROMATIC RESIDUES
INTRODUCTION
Drug induced prolongation of the rate-corrected QT interval (QTC-I) on the electrocardiogram (ECG) is associated with an increased risk of the potentially fatal arrhythmia, Torsade de Pointes (TdP); (1-3). As the incidence of TdP for QTC-I prolonging drugs is generally rather low (2), surrogate markers of QTC-I prolongation and TdP are generally used when testing the pro-arrhythmic propensity of novel chemical entities. The predictive accuracy of surrogate markers is a matter of great importance to drug development and safety (3, 4). A core component of pre-clinical cardiac safety testing, consistent with the S7B guidelines released by the International Conference on Harmonisation (ICH) is an in vitro electrophysiological assay to assess inhibitory effects of drugs on recombinant hERG K+ channels (4, 5). hERG (human ether-a-go-go-related gene) is the cloned counterpart of channels responsible for the rapid delayed rectifier K+ channel current ‘IKr’, which plays a key role in ventricular action potential repolarisation (6, 7). It is now widely accepted that the majority of cases of drug-induced QT prolongation are due to blockade of this channel by therapeutically and structurally diverse drugs (3, 4, 8). The high susceptibility of the hERG channel to pharmacological blockade is attributable in part to the channel’s large inner cavity and in part to the presence of distinct aromatic amino-acid residues (Y652 and F656) in the S6 helices that facilitate drug binding (4, 8, 9).
Amongst the non-cardiac drugs now known to be associated with adverse cardiac events (10, 11) through modifying cardiac electrophysiology (12) and linked to acute pharmacological inhibition of hERG channels are antidepressants including the serotonin-selective reuptake inhibitors (SSRIs) fluoxetine (13-15), citalopram (14) and fluvoxamine (16). Citalopram and fluvoxamine have been shown to produce acute hERG channel current (IhERG) inhibition that develops much more rapidly on membrane potential depolarisation than that produced by archetypal high affinity methanesulphonanilide hERG inhibitors such as dofetilide and E-4031 (14, 17, 18)). Fluvoxamine also appears to be particularly notable and unusual amongst identified hERG inhibitors (16) in that mutation of S6 aromatic residues only partially attenuates the potency of its very rapidly developing hERG channel blockade (16). The extent to which fluvoxamine is representative of other SSRIs in this regard is unclear, however, as fluoxetine inhibition of IhERG has been shown to be markedly reduced by mutation of F656 (15). CONA-437 [4-(3-methoxy-4-methylsulfanyl-phenoxy)-pyridin-3-ylmethyl]-dimethyl-amine is an investigative SSRI that preliminary tests have indicated exerts rapidly developing IhERG inhibition (19). The present study was conducted in order to characterise in detail the time-, voltage- and protocol-dependence of acute IhERG inhibition by CONA-437 and to establish the sensitivity of the drug’s hERG channel inhibition to mutation of the Y652 and F656 S6 aromatic residues. The results of the study establish CONA-437 to be a gating-dependent inhibitor of hERG channels that shows a comparatively limited dependence of inhibition on interactions with S6 aromatic amino-acid residues.
MATERIALS AND METHODS
Cell culture
Human embryonic kidney 293 cells stably expressing hERG
Human embryonic kidney (HEK) 293 cells stably expressing hERG1a (Kv11.1a), were obtained from Dr. Craig. T. January (University of Wisconsin) (20). The creation of HEK 293 cell lines stably expressing the S6 mutations F656A and Y652A to hERG1a has been described previously (16). Cells were maintained using minimum essential medium with Earle’s salts (MEM) supplemented with 10% foetal calf serum, 2 mM L-glutamine, 1 mM sodium pyruvate, 1× non-essential amino acids and 0.4 mg.ml–1 Geneticin (G-418) and kept at 37°C in a humidified atmosphere of 5% CO2. For patch clamp experiments, cells were plated onto glass coverslips placed in 30 mm Petri dishes and used within 72 hours of plating.
Chinese hamster ovary cells stably expressing Nav1.5
Chinese hamster ovary (CHO) cells stably transfected with the human SCN5A gene, which encodes Nav1.5 channels, were obtained from ChanTest Corporation. The cell line was grown in Ham’s F-12 with L-glutamine with 10% foetal bovine serum, 2% Penicillin-Streptomycin, and 0.5% Geneticin, and was maintained at ~37°C in a humidified atmosphere containing 5% CO2. The cells were passaged every 3–5 days based on confluence. On the day of the experiment, 80–100% confluent cells were harvested from a 175 cm2 culture flask using Detachin™. After 10 minutes of exposure to Detachin™ at 37°C, the cells were centrifuged for 2 minutes at 1000 RPM. The supernatant was removed and the cell pellet was reconstituted in 5–8 ml of serum free media with 2.5% of 1 M HEPES and placed on the Qstirrer™ and allowed to recover. After a ~30 minute recovery period, experiments were initiated.
H9C2 cells
The rat cardiac H9C2 cell line was utilized in a L-type calcium channel assay (ATCC; Cat # CRL-1446). H9C2 cells were grown in DMEM containing 10% (v/v) heat-inactivated foetal bovine serum (FBS). Cells were expanded, harvested using TrypLE, resuspended in the same media containing 10% (v/v) DMSO and were frozen at a concentration of 5 million per ml at a rate of –1°C/minute (until –80°C).
hERG electrophysiology
Cells were continuously superfused with extracellular solution (ECS) of the following composition (mM): NaCl 130; KCl 4; CaCl2 2; MgCl2 1; glucose 10; HEPES 5; (titrated to pH 7.4 with NaOH). In ECS containing elevated potassium ([K+]o), elevated K+ was compensated for by a corresponding reduction in external Na+, such that the concentration of KCl was 94 mM and NaCl was 50 mM (21, 22). The patch pipette (intracellular) solution (ICS) contained (in mM): KCl 130; MgATP 5; MgCl2 1; HEPES 10; EGTA 5; (titrated to pH 7.2 with KOH). Liquid junction potential values were calculated of +4.8 mV and –1.8 mV in standard ECS/ICS and high K+ ECS/ICS (JPCalc); (23). As these values were small, no corrections were performed. When filled with ICS, the patch pipettes had a tip resistance of 1.5–4 MΩ. Whole-cell patch clamp recordings of membrane currents were made using either a Multiclamp 700A or an Axopatch 200B amplifier (Axon Instruments Molecular Devices Corporation, Foster City, CA). Currents were filtered at 1 kHz, digitised at 5 kHz (unless otherwise stated) and data acquired using either a Digidata 1322A (used with a Multiclamp 700A amplifier) or a Digidata 1200 (used with an Axopatch 200B amplifier). The pClamp suite of software (versions 8 and 9.2; Axon Instruments Molecular Devices Corporation) was used to generate voltage clamp protocols, acquire data and analyse current traces. Series resistance compensation was routinely applied to at least 75% and tips of pipettes were coated in a parafilm and mineral oil emulsion to reduce stray capacitance. On achieving the whole-cell configuration, pulses were repeatedly applied until stable evoked current responses were obtained; experiments then commenced. A control period was recorded in ECS and then CONA-437 was applied until steady state effects were achieved. All experiments were carried out at 35 ±1°C, unless otherwise stated.
Nav1.5 electrophysiology
Nav1.5 current was elicited and recorded using the automated QPatch HT™ system. The suspended cells in the Qstirrer™ were transferred to 48 individual recording chambers on a QPlate 48™ containing extracellular recording saline composed of (in mM): NaCl 138, KCl 5.3, CaCl2 1.3, MgCl2 0.5, glucose 5.6, HEPES 5, MgSO4 0.4, KH2PO4 0.44, NaHCO3 4.2, Na2HPO4 0.34 adjusted to pH 7.4 ±0.1 with NaOH. Membrane currents were measured at room temperature (~22°C) using the QPlate 48™ electrode array, filled with intracellular recording saline composed of (in mM): 130 KCl, 1 MgCl2, 10 HEPES, 5 Mg-ATP, and 5 EGTA; adjusted to pH 7.2 ±0.1 with KOH.
Nav1.5 current (INa) was elicited with a voltage step to –20 mV for 30 ms from a holding potential of –80 mV. Test pulses were delivered at a frequency of 1 Hz. At least 5 minutes of control recordings were performed prior to the first exposure of the cell to test compound. Four drug concentrations were tested on each cell, each exposure lasting 5 minutes or until steady-state effects were observed. The Class Ic antiarrhythmic drug propafenone was employed as a positive control.
Cell based functional L-type calcium channel assay
Calcium channel activity was measured in the cardiac H9C2 cell line which was maintained in Dulbecco’s Modified Eagle Medium containing 10% (v/v) heat-inactivated foetal bovine serum and glutamine (2 mM). Cells were plated into 384-well black, clear-bottom tissue culture treated plates (Greiner, Germany) at a density of 5000 cells per well (100 µl) and were left to grow for 72 hours prior to the start of the experiment.
Media was removed from the plates and 20 µl of Hank’s balanced salt solution (Invitrogen, USA) containing 20 mM HEPES and Calcium-5 dye (Molecular Devices, USA) was added to each well. The plates were returned to the incubator for 1 hour to allow dye loading. Subsequently, 5 µl of compound or vehicle was added to each well. Following a further 15 min incubation at room temperature, the plate was transferred to a FLIPR® Tetra fluorescent plate reader. 25 µl of a KCl buffer (KCl, 140 mM; MgCl2, 1 mM; HEPES, 20 mM; glucose, 10 mM, CaCl2, 10 mM) was added to each well using the FLIPR to depolarize the cells. The real-time change in fluorescence was measured every second for a total of 120 s. The dihydropyridine L-type Ca2+ channel blocker nifedipine was employed as a positive control.
Data analysis
Data were analysed using Clampfit v9.2 (Axon Instruments Molecular Devices Corporation), QPatch assay software v5.0, Excel 2000 (Microsoft) and Origin v.6.0 (Microcal) software. Data are presented as mean ± S.E.M. with the exception of IC50 and nH, which are expressed as mean and 95% confidence intervals (CI). n denotes the number of cells recorded from or wells (plate-based Ca2+ assay). Statistical comparisons were made using a two-tailed Student’s t-test or one-way analysis of variance (ANOVA) with a Bonferroni post test (* indicates P≤0.05, ** indicates P≤0.01 and *** indicates P≤0.001).
Drugs and reagents
CONA-437 [4-(3-methoxy-4-methylsulfanyl-phenoxy)-pyridin-3-ylmethyl]-dimethyl-amine was dissolved in DMSO to prepare a stock solution of 10 mM. Further dilutions were then made in DMSO to give the desired test concentrations with a final DMSO concentration of 0.1%. Dofetilide was prepared as a 10 mM stock solution in DMSO and diluted down to a final concentration of 10 µM. Solutions were either made up fresh each experimental day or small aliquots were stored at –80°C and defrosted only once. CONA-437 and dofetilide were both synthesised in house at Pfizer Inc. Propafenone and nifedipine were purchased from Sigma-Aldrich. Tissue culture reagents were obtained from Invitrogen or Sigma-Aldrich.
RESULTS
Dose-dependence of CONA-437 inhibition of IhERG is temperature-independent
A ‘step-ramp’ protocol identical to that used in prior IhERG pharmacology studies from our laboratories (24, 25) was used to evoke IhERG in control conditions and in the presence of CONA-437, as illustrated in Fig. 1a. Cells were voltage-clamped at –80 mV and depolarised to +20 mV for 1 s followed by a repolarising ramp back to –80 mV at a rate of 0.5 mV/ms. The voltage command was applied every 4 s. IhERG was measured both at the end of the depolarising step and as peak current during the repolarising ramp phase of the voltage protocol. The effects of CONA-437 on IhERG when recording at 35 ±1°C were compared to those on currents recorded at room temperature (~22°C). To establish a concentration-response relationship, increasing concentrations of CONA-437 were cumulatively applied to the bath until steady-state effect was achieved (24). A supra-maximal concentration of dofetilide (10 µM) was also applied at the end of each experiment as a positive control (Fig. 1b) (24). Dofetilide is known to become trapped in IKr/hERG channels with poor recovery of block at negative voltages (26). CONA-437 washout was therefore not investigated in this study. For each concentration of CONA-437, steady-state percentage inhibition was evaluated when 3 consecutive sweeps were superimposed on one another (24). Mean percentage block values were calculated for both depolarising step and peak IhERG at each concentration. There was no significant difference between mean % block of depolarising step and peak IhERG currents at each concentration of CONA-437 (data not shown). Concentration-response curves were constructed for peak IhERG, and a Hill equation fitted to the data, yielding an IC50 of 1.34 µM (95% CI: 0.98–1.83 µM) and a Hill coefficient (nH) of 0.9 at 35 ±1°C and an IC50 of 0.99 µM (95% CI: 0.78–1.24 µM) and nH of 0.8 at room temperature (Fig. 1c). The derived IC50 values at the two temperatures were not statistically significantly different from one another (P=0.84).
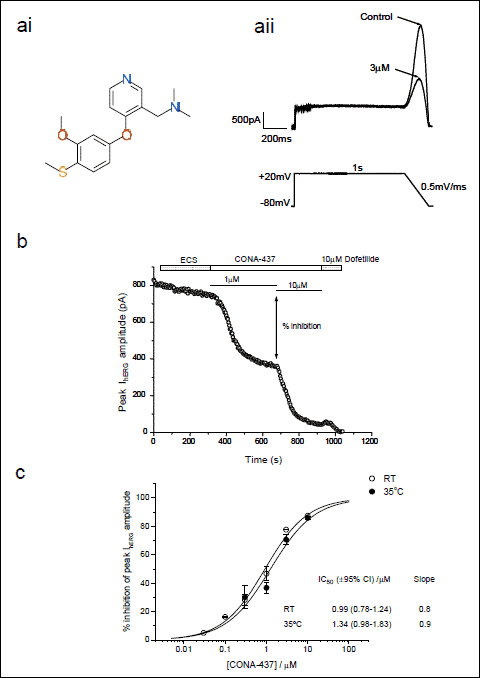 |
Fig. 1. Dose-dependence of CONA-437 IhERG inhibition is temperature-independent. (a) (ai) shows chemical structure of CONA-437. (aii) ‘Step-ramp’ voltage command used to evoke hERG currents. Peak hERG current amplitude in control conditions and in the presence of 3 µM CONA-437 was measured during the repolarising phase as indicated in the example traces shown. (b) This graph illustrates the effects of cumulative concentrations of CONA-437 (1 and 10 µM) on peak hERG amplitude with time. Dofetilide (10 µM) was added at the end of the experiment as a positive control. (c) Cumulative concentration–response curves to CONA-437 at RT and at 35 ±1°C. Data are shown fitted to a Hill equation of the form y=[(A1–A2)/(1+(x/C)nH)]+A2 where A1 and A2 are 0 and 100% inhibition, respectively, C is the IC50 concentration and nH is the Hill coefficient (n=2–7 cells per concentration). |
Comparison of potency of IhERG inhibition by CONA-437 between different voltage waveforms
There is evidence that the observed potency of some hERG inhibiting drugs in electrophysiological experiments is influenced by the voltage protocol used (25, 27-29). In order to determine whether or not this is the case for CONA-437, we conducted experiments using different voltage commands to stimulate hERG channels. Thus, in addition to the step-ramp protocol employed in Fig. 1, we used a ‘step-step’ protocol where cells were depolarised from a holding potential of –80 mV to +20 mV for 2 s followed by a 4 s step to –40 mV to elicit outward IhERG tails: this was applied every 12 s (Fig. 2a; left-hand panel). We also applied a physiological ventricular action potential (VAP) waveform recorded from guinea pig papillary muscle to elicit hERG currents (Fig. 2a; right-hand panel). The effects of CONA-437 on the peak IhERG tails were measured and concentration-response relationships generated, as illustrated in Fig. 2b. We observed small, although not statistically significant, differences in potency using differing voltage protocols (VAP, IC50: 0.72 µM, (95% CI: 0.61–0.84), nH 0.81; “step-step” protocol, IC50: 2.42 µM, (95% CI: 1.97–2.97), nH 0.79), compared to that generated using the “step-ramp” protocol. IhERG tails elicited by the step-step protocol in Fig. 2a were fitted with a bi-exponential function to obtain fast and slow time-constants of deactivation (τf and τs, respectively) in the absence and presence of 3 µM CONA-437. τf was 161.4±32.9 ms in control and 226.8 ±34.3 ms in CONA-437 (p<0.05), whilst τs was similar in control and CONA-437 (1222 ±80 ms and 1138 ±82 ms respectively; no significant difference). Thus, the fast component of deactivation became slower in the presence of CONA-437, without any change to the slower component.
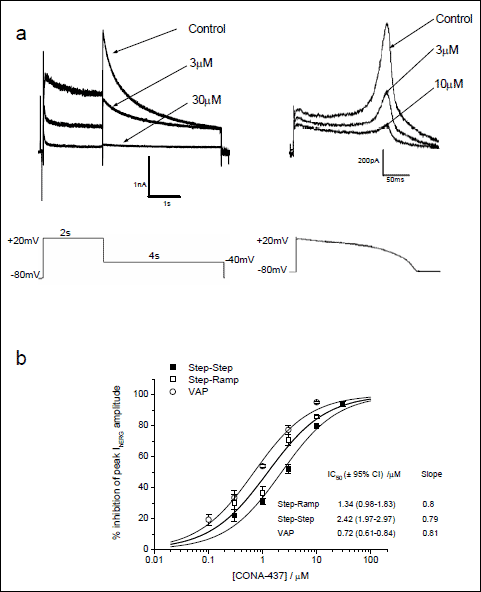 |
Fig. 2. Potency of CONA-437 is influenced by different voltage waveforms. (a) This panel illustrates an example of currents evoked using either the “step-step” voltage protocol or a ventricular action potential (VAP) waveform. Voltage commands were applied every 12 s and multiple concentrations of CONA-437 were cumulatively bath-applied to each cell as in Fig. 1b. Example current traces elicited using these waveforms are shown, and the effect of CONA-437 on peak hERG tail currents was measured. (b) Concentration-response curves using the ‘step-step’ and VAP protocols are shown with the ‘step-ramp’ dose-response (at 35±1°C) curve for comparison. As in Fig. 1c, data were fitted to a Hill equation where % inhibition was constrained between 0 and 100%. Values for IC50 and nH are indicated (n=2–13 cells per concentration). |
CONA-437 inhibition of hERG current is weakly voltage-dependent
The effect of CONA-437 on voltage-dependence of IhERG activation was investigated by applying depolarising voltage steps of 2 s duration from a holding potential of –80 mV to a range of test potentials starting at –60 mV and increased in 10 mV increments (Fig. 3a). Some IhERG blocking compounds exhibit a clear voltage-dependence illustrated by a shift in channel activation and V1/2(act) values. It has been reported in some studies that a time-dependent shift in channel activation may occur in the absence of drug (16, 30). Consequently, sufficient time (at least 6 minutes) was allowed to elapse to ensure stable current-voltage relationships to be recorded, thus allowing the study of possible effects of CONA-437 on voltage-dependent activation, independent of time-dependent changes.
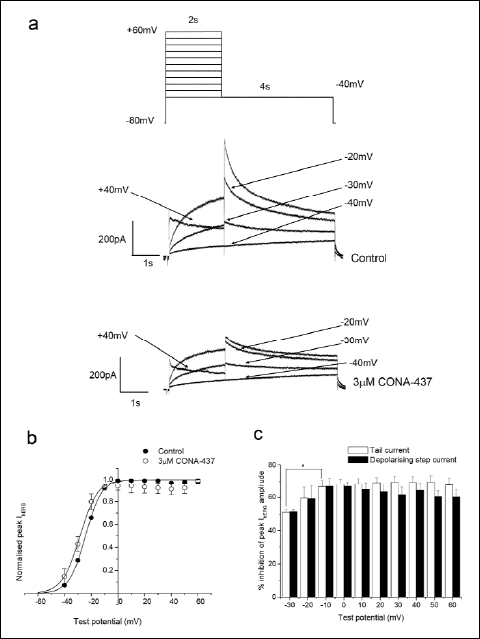 |
Fig. 3. CONA-437 inhibition of hERG currents is weakly voltage dependent. (a) Upper panel shows the voltage command used. The lower panels show representative examples of current traces recorded in the absence and presence of 3 µM CONA-437. For clarity not all current traces are shown. (b) Activation curves for tail currents in control and in the presence of 3 µM CONA-437. Peak tail currents for each individual cell were normalised to the maximum obtained in the absence of drug, and fitted with a Boltzmann function of the form y=[(A1–A2)/(1+e(x–V1/2)/k]+A2 where A1 and A2 are 0 and 1 respectively, V1/2 is the half-activation voltage and k is the slope factor. The V1/2 and k values indicated are the mean from 4 cells. (c) Magnitude of IhERG inhibition by 3 µM CONA-437 at various test potentials. Inhibition of depolarising step currents is indicated by the solid bars and tail currents by the open bars. |
We measured depolarising step IhERG amplitudes and peak IhERG tails upon repolarisation to –40 mV in the absence and presence of 3 µM CONA-437. Representative examples of current traces are shown in Fig. 3a. Current-voltage (I-V) relationships for both depolarising step and peak tail currents were plotted; Fig. 3b shows, the I-V relationship for peak IhERG tails. The peak tail current data obtained from individual cells were fitted to a Boltzmann function and V1/2(act) and slope factors were derived. The mean V1/2(act) was –24.3 ±0.3 mV under control conditions and in the presence of CONA-437 was significantly shifted to –29.3 ±1.5 mV (n=4, P=0.04) whilst the slope factors in control and drug were not significantly different: 6.1±0.1 mV and 5.8±0.4 mV, respectively (P=0.59). Fig. 3c shows the magnitude of inhibition of peak tail and depolarising step currents by 3 µM CONA-437 at different test potentials. At –30 mV, significant block of tail currents was observed (40.4 ±10.6%; n=4). The extent of block was significantly enhanced as the membrane became more depolarised and reached a plateau level at –10 mV (66.6±7.9%; P<0.05) suggesting that block is weakly voltage-dependent. Furthermore, the extent of inhibition of depolarising step currents also exhibited weak voltage-dependence.
Time-dependence of hERG inhibition by CONA-437
In order to investigate the time-dependence of CONA-437 block of IhERG, we used a long duration voltage-command to ascertain whether inhibition was maintained during sustained depolarisation. Cells were stepped to 0 mV for 10 s from a holding potential of –80 mV and a control current was recorded in the absence of CONA-437. Membrane potential was then held at –80mV whilst CONA-437 was applied, and after 5 minutes in CONA-437 in the absence of pulsing the protocol was then repeated (Fig. 4a, upper panel). Block of IhERG was observed during the step to 0 mV (Fig. 4a; lower panel) and the extent of block did not change with application of subsequent long depolarisations (data not shown). IhERG block developed rapidly within the first 1 second of the pulse (Fig. 4b) and could be fit to a single exponential function. A single exponential fit to the development of block in each of the 4 experiments yielded a mean τ value of 54.7±9.5 ms (n=4). We used an ANOVA to examine whether the development of block was time-dependent and found that at later time points (>1s) the degree of block was significantly greater suggesting that block was time-dependent. Analysis of the data demonstrated that block exhibited significant time-dependence (P=0.001). The extent of block at various time points during the entire 10 seconds depolarising step was calculated for 3 µM CONA-437 and was found to remain constant between 1 and 10 s (Fig. 4a, lower panel). Time-dependent inhibition of IhERG during this protocol was accompanied by acceleration of development of the current on depolarisation: in five further experiments the time-to-peak of IhERG following depolarisation to 0 mV was 479.2 ±69.8ms in control and 201.9±3.0 ms in CONA-437. This may reflect acceleration of IhERG activation in the presence of the drug and/or the consequences of time-dependent inhibition.
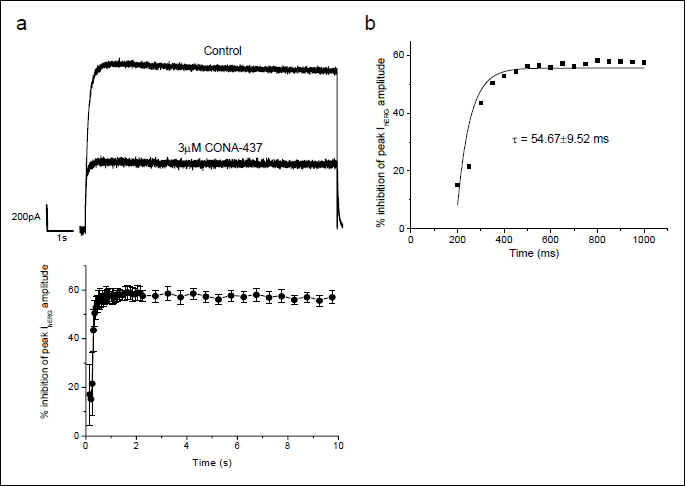
(a) Cells were depolarised from the holding potential of –80 mV to 0 mV for 10 s. This voltage command was repeated until a steady-state response was observed. 3 µM CONA-437 was then bath-applied for 5 minutes whilst the channels were voltage clamped at
–80 mV. The voltage command was then again repeated in the presence of CONA-437. The representative traces in the upper panel indicate the last pulse prior to, and the first pulse during application of 3 µM CONA-437. The lower panel shows mean development of block over time by 3 µM CONA-437 from 4 experiments. Full block was observed within less than 1 s and was maintained for the 10 s duration.
(b) Development of block during the first second of the pulse from a representative cell. Each individual experiment was fit by a single exponential yielding an overall mean tau of 54.7±9.5 ms.
To investigate further the time-dependence of block by CONA-437 at the shorter pulse durations, cells were depolarised to +40 mV for either 30 or 200 ms from a holding potential of –80 mV and IhERG peak tail currents were observed on repolarisation to –40 mV. This protocol was applied under control conditions and after a 5 minute application of 3 µM CONA-437 whilst cells were voltage-clamped at –80 mV. Fig. 5a shows the currents obtained and the corresponding voltage commands. The amplitude of tail currents evoked at –40 mV was measured in the absence and presence of 3 µM CONA-437 and the effects of drug expressed as percentage inhibition of peak tail currents (Fig. 5b). We observed that 3 µM CONA-437 produced a significant attenuation of IhERG even after only a 30 ms depolarising step (35.8±5.1%, n=5, P=0.001) and that inhibition was significantly increased as pulse duration was increased (P<0.02).
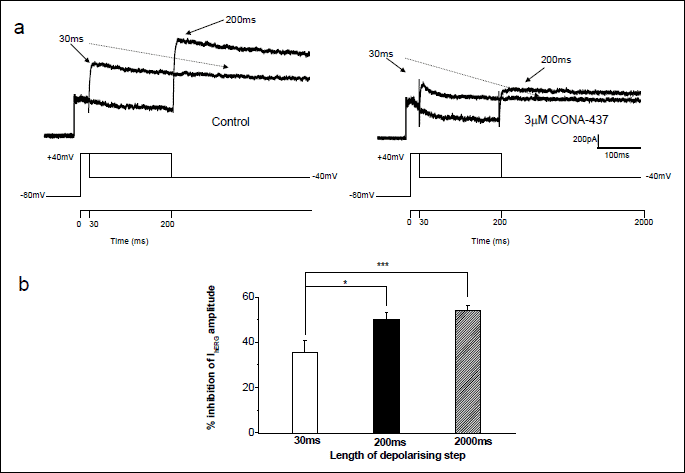
(a) Cells were briefly depolarised from a holding potential of –80 mV to +40 mV for either 30 or 200 ms before repolarising to –40 mV to elicit tail currents. Peak tail currents were measured in control conditions and after equilibration of cells in 3 µM CONA-437, whilst cells were voltage-clamped at –80 mV. The representative traces show the increase in peak tail current with duration of the depolarising step in the absence (left hand traces) and presence (right hand traces) of CONA-437.
(b) Summary of mean data from these experiments (n=4–12 cells) where mean % inhibition of peak tail currents is plotted as a function of pulse duration. With a 30 ms step, 35.8±5.1% inhibition was achieved. As pulse duration was lengthened, inhibition was significantly increased (200 ms: 50.0±2.39%; P=0.018).
Effects of CONA-437 on IhERG inactivation
Data from the experiments described above provide evidence that CONA-437 blockade of IhERG in a manner consistent with rapid gating-dependent inhibition, with the presence of significant inhibition with brief (30 ms) depolarisation also raising the possibility of a component of closed channel block. To investigate further the state-dependence of CONA-437 block of IhERG we examined the agent’s effect on inactivation kinetics of IhERG using two additional voltage protocols. With the first of these, we examined whether CONA-437 had any effects on the voltage-dependence of hERG current inactivation. Cells were depolarised to +40 mV for 500 ms from a holding potential of –80 mV, 2 ms pulses were then applied to test potentials between –140 mV and +30 mV in 10 mV increments, followed by a second 500 ms step to +40 mV. IhERG elicited during the final depolarising step to +40 mV was measured and normalised to the maximum current observed. The normalised values were then plotted as a function of test potential to generate steady-state inactivation relations for each cell in the absence and presence of CONA-437 (3 µM) and V1/2(inact) values calculated. Fig. 6a upper panel shows representative currents in control and CONA-437, focusing on current elicited during the second step to +40 mV following the ladder of brief repolarising steps (shown as an inset to Fig. 6a). Mean inactivation relations in the absence and presence of CONA-437 (3 µM) are also shown in Fig. 6a: CONA-437 had no statistically significant effect on the mean V1/2(inact) values (–59.3 ±9.2 mV and –59.2 ±4.5 mV, n=4, respectively) or on the slope factors for the relation (18.7 ±1.3 mV and 20.6 ±1.2 mV, respectively). In experiments to examine the effects of CONA-437 on the time course of IhERG inactivation, we used a voltage protocol which allowed us to measure time constants of inactivation at different test potentials. Cells were depolarised to +50 mV for 500 ms from a holding potential of –80 mV and then briefly (2 ms) stepped to –100 mV followed by a subsequent step to test potentials ranging from –50 mV to +50 mV. Representative current traces in the absence and presence of 3 µM CONA-437 are shown in Fig. 6b (upper panel). For each test potential, the time-course of inactivation of IhERG elicited during the second depolarising step was fitted to a single exponential function yielding inactivation time constants at each test potential, in the presence and absence of drug. We found that CONA-437 significantly accelerated the time course of inactivation at potentials more negative than, and including, +10 mV (Fig. 6b, lower panel).
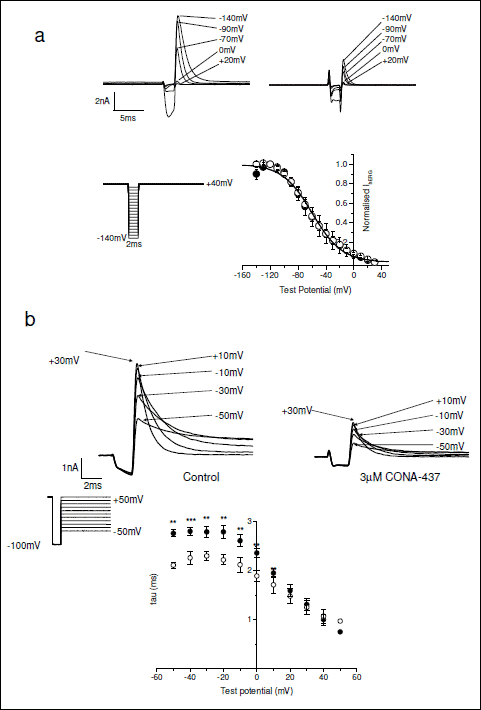 |
Fig. 6. Effects of CONA-437 on hERG current inactivation. (a) Following a 500 ms step to +40 mV, 2 ms pulses were applied to potentials between –140 mV and +30 mV in 10 mV increments, followed by a second 500 ms step to +40 mV (inset). Example of current recordings was displayed at the top (control on the left, and 3 µM CONA-437 on the right). Current amplitudes elicited during the final step to +40 mV were measured, normalised and plotted as a function of the preceding test potential to yield a steady-state inactivation curve. This experiment was conducted under control conditions (in filled circles) and in the presence of 3 µM CONA-437 (in open circles). The mean V1/2(inact) under control conditions was –59.3 ±9.2 mV (slope 18.7 mV) and in the presence of CONA-437, –59.2±4.5 mV (slope 20.6 mV). (b) Example traces in the upper panel illustrate the effects of CONA-437 on inactivation kinetics. Cells were depolarised to +50 mV for 500 ms and then briefly stepped to –100 mV followed by a subsequent step to test potentials ranging from –50 mV to +50 mV (inset). For clarity not all current traces are shown. The outward currents elicited during this final step were then fitted to a single exponential function to yield a time constant at each test potential. Time constants were then plotted versus test potential as shown in the lower part of this panel (n=4 cells). |
Using S6 helix mutants to investigate block of the hERG channel by CONA-437
In order further to understand the mechanism of IhERG block by CONA-437 we examined the effects of mutations of the pore-S6 aromatic amino acid residues, Tyr652 and Phe656 to alanine residues (Y652A and F656A). For these experiments we used HEK 293 cell lines stably expressing either hERG-Y652A or hERG-F656A as previously described (16, 24). The Y652A mutant was tested using the standard ‘step-step’ protocol as described in Fig. 2 and we used a drug concentration which would produce very extensive blockade of wild-type (WT) hERG channels (applying 30 µM CONA-437; >10-fold the IC50 value for WT IhERG in Fig. 2). Representative current traces, as shown in Fig. 7a, indicate the effects of 30 µM CONA-437 on WT-hERG channels (left-hand panel) and on Y652A channels (right-hand panel). For the WT-hERG channels 30 µM CONA-437 produced 94 ±1.4% inhibition of IhERG (n=3) compared to 48±6.1% inhibition in cells transfected with the Y652A channel (n=6), an IC50 value was estimated for the block of Y652A channels of 33.6 µM, an approximate three-fold shift in potency. We also examined the effects of CONA-437 on F656A hERG channels. For these experiments we used 94 mM external K+ in order to maximise current carried by the low expressing F656A mutant, as previously reported (16, 24). Currents were evoked using a modified ‘step-step’ protocol where cells were repolarised from +20 mV to –120 mV for 500 ms prior to returning membrane potential to –80 mV; inhibition of peak IhERG was measured for inward tail currents at –120 mV. We found that measuring tail currents at –120 mV in high external K+ (94 mM) dramatically attenuated block of WT-hERG channels, 30 µM CONA-437 caused approximately 50% inhibition of IhERG (data not shown). To determine whether the inward tails or high external K+ ([K+]e) is responsible for attenuated block of the WT-hERG channels, we carried out similar experiments in ECS (4 mM K+). 30 µM CONA-437 caused 73.7 ±3.8% inhibition of IhERG (data not shown). This attenuation of block in high K+ is similar to that observed previously for other drugs including fluvoxamine, E-4031, amiodarone and dronedarone (31, 32) and is consistent with drug displacement with increased inward flux of K+ ions.
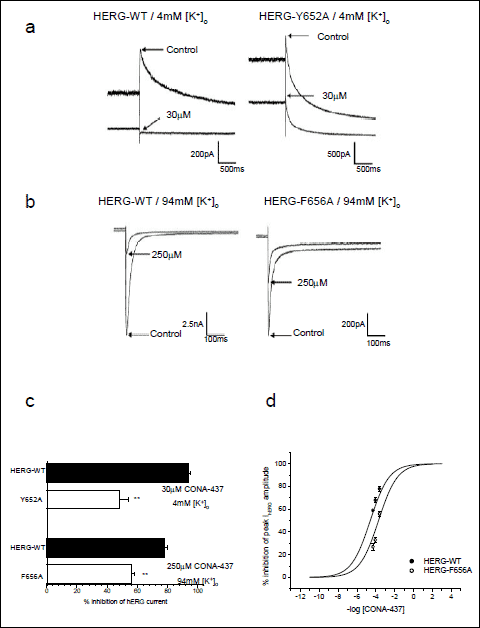 |
Fig. 7. Partial attenuation of CONA-437 block by F656A and Y652A. (a) Representative current traces showing the effects of a maximal concentration of CONA-437 (30 µM) on WT channels (left) and on Y652A channels (right). Experiments were performed in the presence of 4 mM external K+ and the voltage protocol is shown as an inset. (b) Currents were evoked using a modified ‘step-step’ protocol (see inset). Representative current traces shown indicate the effects of a maximal concentration of CONA-437 (250 µM) on WT channels (left) and on F656A channels (right). Experiments were performed in the presence of 94 mM external K+ to maximise the low expression of the F656A mutant (hence, inward tail currents). (c) Magnitude of IhERG inhibition by CONA-437 for the mutants tested, compared to the effects on the WT channel. (d) Concentration - response curve to CONA-437 for hERG-WT and hERG- F656A channels in 94 mM external K+. Data are shown fitted to a Hill equation (as described in the legend to Fig. 1). |
Due to the effect of raised [K+]e on CONA-437 blockade of inward IhERG, for subsequent experiments we used a higher CONA-437 concentration of 250 µM. The representative current traces in Fig. 7b illustrate the effects of 250 µM CONA-437 on WT channels (left-hand panel) and on F656A channels (right-hand panel). In the WT channels 250 µM CONA-437 produced 77 ±2.1% inhibition of IhERG (n=6) compared to 56±2.1% inhibition in cells transfected with the F656A channel (n=8). The data for experiments using Y652A and F656A cell lines are summarised in the bar-chart in Fig. 7c. We generated a concentration-response curve to CONA-437 in 94mM external K+ for both WT-hERG and F656A channels. We observed a significant decrease in potency of CONA-437, as shown in Fig. 7d (right-hand panel), with IC50 values of 41.4 µM (95% CI: 29.1–58.9 µM) and 176 µM (95% CI: 143–217 µM) on hERG-WT and F656A mutant channels, respectively (an approximate 4-fold change in potency).
Effects on Nav1.5 and L-type Ca2+ channels
Some hERG blocking compounds exert mixed ion channel blocking effects (MICE) that, if on inward cation currents (L-type Ca2+ current, INa), could feasibly offset hERG-mediated drug actions on cardiac repolarisation; indeed, a recent study has provided evidence that comparison of concentration-response data on hERG with data on Cav1.2 and Nav1.5 improves predictivity of hERG data in respect of arrhythmia risk (33). Consequently, we performed additional experiments to test effects of CONA-437 on these two channels. CONA-437 at up to 30 µM did not produce any statistically significant effect on INa (11.9 µ3.8% change at 30 µM, n=4, Fig. 8a; P>0.05 against no change), or L-type Ca2+ channel activity (at 30 µM 103.5 ± 1.8% of control activity, n=4, Fig. 8b; P>0.05 against no change to control activity). By contrast, the positive control propafenone produced a concentration-dependent inhibition of Nav1.5 current (IC50 of 1.3 µM and nH of 0.9; Fig. 8a), and nifedipine demonstrated an inhibition of L-type Ca2+ channel activity in a concentration-dependent manner (with an IC50 of 8.7 nM and nH of 1.1; Fig. 8b).
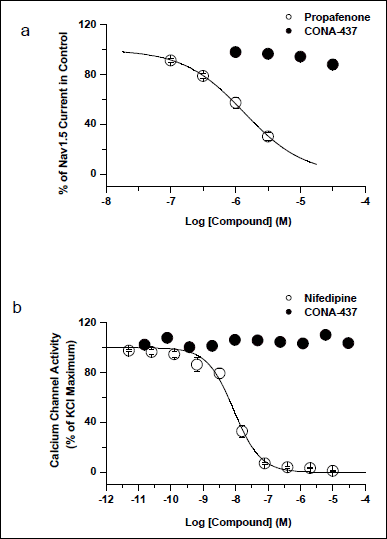 |
Fig. 8. Effects of CONA-437 on INa and L-type Ca2+ channel activity. (a) Effects of CONA-437 and the positive control propafenone on Nav1.5 currents were plotted against test concentrations. CONA-437 did not have any significant effect on Nav1.5 current at all concentrations tested (n=4), whereas the positive control propafenone produced a concentration-dependent inhibition of Nav1.5 with an IC50 of 1.30 µM. (b) Summary data of L-type Ca2+ channel activity by CONA-437 and the positive control of nifedipine were plotted. While the positive control showed a concentration-dependent inhibition of L-type Ca2+ channel activity with an IC50 of 8.7 nM, CONA-437 displayed no effect at up to 30 µM. |
DISCUSSION
Serotonin can produce marked modulation of neuronal activity and neuronal targets for serotonin continue to be identified (34). In this study we have focused not on serotonin itself, but rather on a novel investigative SSRI, CONA-437, demonstrating and characterising the mechanism of acute block of hERG channels by this compound. We found that, under similar conditions, CONA-437 exhibits a similar potency against hERG to a previously reported SSRI, fluvoxamine (2.42 µM and 3.8 µM, respectively) (16), a potency which is broadly comparable to those seen for other SSRIs - citalopram (14) and fluoxetine (15, 35), also studied in mammalian expression systems. Fluoxetine has also been reported to inhibit hERG channel trafficking (15, 35). Data for fluvoxamine on hERG trafficking are not available and the focus of the present study (on acute pharmacological actions on IhERG) precluded the study of CONA-437 in this regard. It has been suggested that both experimental temperature and voltage-clamp waveform profile can influence the observed potency of IhERG blockade (25, 27, 28). However, recording temperature did not significantly alter potency of CONA-437 (22°C: 0.99 µM; 35°C: 1.34 µM). Moreover, the use of different voltage protocols to elicit hERG currents did not have a statistically significant effect on potency (VAP 0.72 µM versus step-ramp 1.34 µM; VAP 0.72 µM versus step-step 2.42 µM): the observed shifts (~3-fold) were small in comparison to some agents previously studied (25, 27, 28). With tritiated tryptamine as serotonin transporter substrate, CONA-437 inhibits the human serotonin reuptake inhibitor expressed in HEK 293 cells with an IC50 of ~7 nM (Pfizer Inc, unpublished), which is markedly (~100-fold) less than the hERG IC50 with any protocol studied here; however, the compound’s effects on other monoamine transporters occur at concentrations at which substantial hERG blockade would be anticipated (IC50 for human norepinephrine transporter of 6.5 µM and for human dopamine transporter of >40 µM; Pfizer Inc, unpublished). Structurally diverse compounds can have off-target effects that involve unexpected actions on Na+ or Ca2+ channels (36, 37). For drugs that produce hERG/IKr K+ channel inhibition, accompanying pharmacological effects on Na+ or Ca2+ channels can, in principle, mitigate hERG/IKr blockade (33). However, our experiments showed that CONA-437 at up to 30 µM had no significant effect on Nav1.5 current or L-type Ca2+ channel activity and thus the drug’s effect on IKr is not likely to be balanced by concomitant actions on opposing inward currents (33).
Strong protocol-dependence of IhERG blockade has been reported for drugs that exhibit marked use-dependent blocking kinetics (25, 29). For example, under similar conditions to the present study cisapride was found to exhibit IC50 values that varied ~10-fold between ventricular AP and step protocols (25). CONA-437 differs from cisapride in exhibiting much more rapidly developing blockade on membrane depolarisation and this characteristic of the drug’s action is likely to minimise the influence on inhibitory potency of different voltage stimulus waveforms. The observation that CONA-437 produces significant IhERG inhibition with very brief (30 ms) membrane depolarisation is comparable to prior data for fluvoxamine (16) and indicates considerably faster onset of action than reported for either citalopram or fluoxetine (13, 14). Collectively, our data provide evidence that, similar to fluvoxamine (16), CONA-437 is likely to exhibit a mixed state-dependence of channel block. IhERG inhibition by CONA-437 was weakly voltage-dependent: a ~5 mV leftward shift in the activation curve was observed and there was a significant accentuation of the magnitude of inhibition at more depolarised potentials. These findings would suggest that channel opening influences CONA-437 binding. Although the lack of a significant shift in voltage-dependence of inactivation suggests that CONA-437 did not act to stabilise hERG channel inactivation, the onset of inactivation appeared to be significantly accelerated by CONA-437 (Fig. 6). This suggests either that the transition rate between the open and inactive states is accelerated upon binding of CONA-437 to the channel, or that the apparent acceleration of inactivation reflects rapid development of open channel block during depolarisation. Thus, our data on WT IhERG recorded in standard ECS suggest that CONA-437 exhibits rapid open hERG channel block, although a component of closed channel block cannot be ruled out, since very rapidly developing open channel block and closed channel block cannot readily be distinguished from one another (16, 38). The attenuation of CONA-437 blocking potency of IhERG high [K+]e may, however, suggest a predominantly open channel blocking mechanism: it is both suggestive of drug binding in/close to the ion conduction pathway and is consistent with reduced ability of the drug to bind to open channels through electrostatic repulsion or ‘knock off’ of CONA-437 from its binding site, when the direction of K+ flux is inward and enhanced through raised [K+]e.
A striking aspect of the data in the present study is that IhERG blockade by CONA-437 was only modestly affected by mutation of the S6 residues Y652A and F656A. These residues in the S6 region of the channel constitute key components of the canonical drug binding site on the hERG channel as, for almost all hERG-blocking drugs tested to date, mutation of one or both of these residues has caused either a complete or a dramatic attenuation of blockade (8, 9). For example, the IhERG blocking potency of the high affinity blocker MK-499 was decreased by 94- and 650- fold by Y652A and F656A respectively, with cisapride and terfenadine also reported to be highly sensitive (39). The potency of chloroquine was reduced approximately 500-fold by each mutation (40) and a number other drugs including (amongst others) fluoxetine (15), lidoflazine (21), clemastine (22), E-4031 and dofetilide (41), ibutilide and clofilium (42), are highly sensitive to mutation of one or both of these residues.
The SSRI fluvoxamine has been considered to be an exception amongst IhERG blockers in that Y652A and F656A mutations only partially attenuate IhERG carried by these mutant channels at drug concentrations that produce profound inhibition of WT IhERG (16). In this study, we estimated the IC50 of CONA-437 for the block of Y652A and F656A channels to be raised ~3–4 fold compared to WT-hERG. This reduction in potency is rather modest compared to that occurring for the majority of hERG blockers studied previously, suggesting that - as for the structurally distinct SSRI fluvoxamine - neither Y652 nor F656 is an obligatory component of the drug binding site. Obligatory components of the drug binding site on the hERG channel for CONA-437 therefore remain to be elucidated. Interestingly, both fluvoxamine and a dronedarone (a benzofuran IhERG blocker, the action of which is also relatively insensitive to mutation of Y652 and F656 (32)) share in common with CONA-437 the property of attenuated IhERG block in high [K+]e conditions. This may indicate that binding in/near the ion conduction pathway (most likely in the channel’s inner cavity, rendered accessible on channel gating) can occur without a critical dependence on either Y652 or F656. A detailed understanding of the molecular determinants of IhERG blockade is of prime importance if, in the future, hERG inhibition is to be avoided early during the drug design and development process (8). CONA-437 may therefore provide a useful pharmacological tool for future studies, in order better to understand how some agents can exert rapidly developing IhERG inhibition by interacting with the channel without a critical dependence on the Y652 or F656 residues.
Acknowledgements: The authors would like to thank Lesley Arberry for excellent technical assistance.
Conflict of interests: This work was funded by Pfizer Global Research and Development.
REFERENCES
- Shah RR. Drug-induced QT interval prolongation-regulatory guidance and perspectives on hERG channel studies. Novartis Found Symp 2005; 266: 251-280.
- Yap YG, Camm AJ. Drug induced QT prolongation and torsades de pointes. Heart 2003; 89: 1363-1372.
- Gintant GA. Preclinical Torsades-de-Pointes screens: advantages and limitations of surrogate and direct approaches in evaluating proarrhythmic risk. Pharmacol Ther 2008; 119: 199-209.
- Hancox JC, McPate MJ, El Harchi A, Zhang YH. The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol Ther 2008; 119: 118-132.
- Friedrichs GS, Patmore L, Bass A. Non-clinical evaluation of ventricular repolarization (ICH S7B): results of an interim survey of international pharmaceutical companies. J Pharmacol Toxicol Methods 2005; 52: 6-11.
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell 1995; 81: 299-307
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, an inward rectifier in the voltage-gated potassium channel family. Science 1995; 269: 92-95.
- Sanguinetti MC, Mitcheson JS. Predicting drug-hERG channel interactions that cause acquired long QT syndrome. Trends Pharmacol Sci 2005; 26: 119-124.
- Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature 2006; 440: 463-469.
- Witchel HJ, Hancox JC, Nutt DJ. Psychotropic drugs, cardiac arrhythmia, and sudden death. J Clin Psychopharmacol 2003; 23: 58-77.
- Pacher P, Kecskemeti V. Cardiovascular side effects of new antidepressants and antipsychotics: new drugs, old concerns? Curr Pharm Des 2004; 10: 2463-2475.
- Sala M, Coppa F, Cappucciati C, et al. Antidepressants: their effects on cardiac channels, QT prolongation and Torsade de Pointes. Curr Opin Investig Drugs 2006; 7: 256-263.
- Thomas D, Gut B, Wendt-Nordahl G, Kiehn J. The antidepressant drug fluoxetine is an inhibitor of human ether-a-go-go related gene (HERG) potassium channels. J Pharm Exp Ther 2002; 300: 534-548.
- Witchel HJ, Pabbathi VK, Hofmann G, Paul AA, Hancox JC. Inhibitory actions of the selective serotonin re-uptake inhibitor citalopram on HERG and ventricular L-type calcium currents. FEBS Lett 2002; 512: 59-66.
- Rajamani S, Eckhardt LL, Valdivia CR, et al. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol 2006; 149: 481-489.
- Milnes JT, Crociani O, Arcangeli A, Hancox JC, Witchel HJ. Blockade of HERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br J Pharmacol 2003; 139: 887-898.
- Kiehn J, Lacerda AE, Wible B, Brown AM. Molecular physiology and pharmacology of HERG. Single channel currents and block by dofetilide. Circulation 1996; 94: 2572-2579.
- Snyders DJ, Chaudhary A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Mol Pharmacol 1996; 49: 949-955.
- Alexandrou AJ, Milnes JT, Leaney JL, et al. The impact of voltage-clamp protocol on the apparent potency of a rapidly acting human ether-a-go-go related gene (hERG) potassium channel blocker. J Physiol 2005; 567P: PC25.
- Zhou Z, Gong Q, Ye B, et al. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J 1998; 74: 230-241.
- Ridley JM, Dooley PC, Milnes JT, Witchel HJ, Hancox JC. Lidoflazine is a high affinity blocker of the HERG K+ channel. J Mol Cell Cardiol 2004; 36: 701-705.
- Ridley JM, Witchel HJ, Hancox JC. Witchel HJ. Clemastine, a conventional antihistamine, is a high potency inhibitor of the HERG K+ channel. J Mol Cell Cardiol 2006; 40: 107-118.
- Barry PH. JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods 1994; 51: 107-116.
- Alexandrou AJ, Duncan RS, Sullivan A, et al. Mechanism of hERG K+ channel blockade by the fluoroquinolone antibiotic moxifloxacin. Br J Pharmacol 2006; 147: 905-916.
- Milnes JT, Witchel HJ, Leaney JL, Leishman DJ, Hancox JC. Investigating dynamic protocol-dependence of hERG potassium channel inhibition at 37°C: cisapride versus dofetilide. J Pharmacol Toxicol Methods 2010; 61: 178-191.
- Carmeliet E. Voltage- and time-dependent block of the delayed rectifier K+ current in cardiac myocytes by dofetilide. J Pharmacol Exp Ther 1992; 262: 809-817.
- Kirsch GE, Trepakova ES, Brimecombe JC, et al. Variability in the measurement of hERG potassium channel inhibition: effects of temperature and stimulus pattern. J Pharmacol Toxicol Methods 2004; 50: 93-101.
- Yao JA, Du X, Lu D, Baker RL, Daharsh E, Atterson P. Estimation of potency of HERG channel blockers: impact of voltage protocol and temperature. J Pharmacol Toxicol Methods 2005; 52: 146-153.
- Stork D, Timin EN, Berjukow S, et al. State dependent dissociation of HERG channel inhibitors. Br J Pharmacol 2007; 151: 1368-1376.
- Ferreira S, Crumb WJ, Jr., Carlton CG, Clarkson CW. Effects of cocaine and its major metabolites on the HERG-encoded potassium channel. J Pharmacol Exp Ther 2001; 299: 220-226.
- Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Lett 1997; 417: 43-47.
- Ridley JM, Milnes JT, Witchel HJ, Hancox JC. High affinity HERG K+ channel blockade by the antiarrhythmic agent dronedarone: resistance to mutations of the S6 residues Y652 and F656. Biochem Biophys Res Comm 2004; 325: 883-891.
- Kramer J, Obejero-Paz CA, Myatt G, et al. MICE models: superior to the HERG model in predicting Torsade de Pointes. Sci Rep 2013; 3: 2100. doi: 10.1038/srep02100.
- Palus K, Chrobok L, Lewandowski MH. Depolarization of the intergeniculate neurons by serotonin - in vitro study. J Physiol Pharmacol 2013; 64: 773-778.
- Wible BA, Hawryluk P, Ficker E, Kuryshev YA, Kirsch G, Brown AM. HERG-Lite: a novel comprehensive high-throughput screen for drug-induced hERG risk. J Pharmacol Toxicol Methods 2005; 52: 136-145.
- Baylie RL, Cheng H, Langton PD, James AF. Inhibition of cardiac L-type calcium channel current by the TRPM8 agonist, (-)-menthol. J Physiol Pharmacol 2010; 61: 543-550.
- Szentandrassy N, Papp F, Hegyi B, Bartok A, Krasznai Z, Nanasi PP. Tetrodotoxin blocks native cardiac L-type calcium channels but not Cav1.2 channels expressed in HEK cells. J Physiol Pharmacol 2013; 64: 807-810.
- Mitcheson JS. Drug binding to HERG channels: evidence for a ‘non-aromatic’ binding site for fluvoxamine. Br J Pharmacol 2003; 139: 883-884.
- Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. A structural basis for drug-induced long QT syndrome. Proc Natl Acad Sci USA 2000; 97: 12329-12333.
- Sanchez-Chapula JA, Navarro-Polanco RA, Culberson C, Chen J, Sanguinetti MC. Molecular determinants of voltage-dependent human ether-a-go-go related gene (HERG) K+ channel block. J Biol Chem 2002; 277: 23587-23595.
- Kamiya K, Niwa R, Mitcheson JS, Sanguinetti MC. Molecular determinants of HERG channel block. Mol Pharmacol 2006; 69: 1709-1716.
- Perry MD, deGroot MJ, Helliwell R, et al. Structural determinants of HERG channel block by clofilium and ibutilide. Mol Pharmacol 2004; 66: 240-249.
A c c e p t e d : April 22, 2014
and Prof. Jules Hancox, School of Physiology and Pharmacology, and Cardiovascular Research Laboratories, School of Medical Sciences, University Walk, Bristol, BS8 1TD, United Kingdom; e-mail: jules.hancox@bristol.ac.uk