IN VITRO TESTING THE POTENTIAL OF A NOVEL CHIMERIC IGG VARIANT FOR INHIBITING COLLAGEN FIBRILS FORMATION IN RECURRENT HEREDITARY GINGIVAL FIBROMATOSIS: CHIMERIC ANTIBODY IN A GINGIVAL MODEL
INTRODUCTION
Gingival fibromatosis (GF) comprises a rare clinical condition characterized by abnormally diffuse or localized growth of the maxillary and mandibular gingiva. The condition among others occurs as a side effect of some medications. It may be related to hereditary factors or in some cases the aetiology of the enlargement remains unknown (1-3). Hereditary gingival fibromatosis (HGF) may manifest as an autosomal-dominant or, less commonly, an autosomal-recessive mode of inheritance, either as an isolated disorder or as part of a syndrome (2, 4-6). Autosomal dominant forms of GF, which are usually non-syndromic, have been genetically linked to the chromosomes 2p21-p22 and 5q13-q22. Recently, a mutation in the Son-of-Sevenless (SOS-1) gene has been suggested as a possible cause of isolated (non-syndromic) gingival fibromatosis. SOS-1 is a guanine nucleotide-exchange factor that functions in the transduction of signals that control cell growth and differentiation. A single base insertion mutation (SOS-1 g.126,142-126,143insC) introduces a frameshift, creating a premature stop codon, abolishing 4 functionally important proline-rich SH3 binding domains normally present in the carboxyl-terminal region of the SOS-1 protein (7). The resultant protein chimera has enhanced activity, since it lacks the carboxyl-terminal domain that normally exerts negative allosteric control (8). This mutation causes HGF in humans, and similar SOS-1 deletion constructs are functional in animal models, and a transgenic mouse construct with a comparable SOS-1 chimera produces a skin hypertrophy phenotype (7, 9-11). A mutation in SOS-1 gene has been associated with increased fibroblast proliferation and collagen matrix synthesis in HGF (12).
Although the overgrown tissue does not cause a pain, it may eventually cover most of the teeth. The condition may hinder oral cavity hygiene and cause bone loss, result in speech disturbance, difficulties with mastication, and, in extreme cases, it may distort the jaws (11, 13, 14). A classic feature of GF is that epithelial rete pegs extend deep into the connective tissue compared with normal tissues, and contains fibrotic connective tissue with various levels of inflammation (10, 15-18).
Although the mechanisms that trigger fibrosis in the course of GF are extensively described, this topic is still not fully elucidated (19-22). The most attractive concept is that molecular matrix remodeling programs are initiated in epithelial cells that enable these now motile and transitioning cells to invade through basement membranes and migrate into connective tissue stroma where they contribute to new extracellular matrix (ECM) production (21). Epithelial-to-mesenchymal transition (EMT) is a process in which epithelial cell-cell and cell-extracellular matrix interactions are weakened as epithelial cells trans-differentiate into fibrogenic fibroblast-like cells. Although a normal process in development, EMT underlies pathological processes in mature tissues including progression of tumors of epithelial origin, and heart, lung, liver, and kidney fibrosis (20). Additionally, in this process are involved several pro-fibrogenic cytokines and receptors, such as connective tissue growth factor (CTGF) and transforming growth factor β1 (TGFβ1) which are considered the main inducers of EMT (23, 24). GF is characterized by elevated levels of pro-fibrogenic cytokines, including TGFβ1 and CTGF. Thus, another concept is that growth factors trigger fibrosis via direct regulation of molecules involved in collagen and other ECM components synthesis. At present, appropriate oral hygiene and scaling are sufficient for localized and minimal enlargements, while cases of massive gingival overgrowth require surgical correction (25). On the other hand, the inaccessibility of alternative, effective, and less invasive methods indicates on the importance of discerning studies testing novel tools that might become valuable to GF treatment in the future.
Recently, it has been proposed that the extracellular process of collagen fibril formation may serve as a valid target for reducing collagen deposits in fibrotic tissues. It has been demonstrated that employing a monoclonal antibody that targets the C-terminal telopeptide of the α2 chain of collagen I blocks critical collagen I-collagen I interaction, thereby reducing the amount of collagen deposits in vitro and in models representing growth of fibrotic tissues (26). Furthermore, recently Fertala et al. shown efficient inhibition of collagen fibril formation in vivo and in cultures of various cells associated with the fibrotic process that develops after trauma by using a novel chimeric antibody variant (chIgG) (27).
The objective of this study was testing in in vitro conditions the inhibitory potential of a novel chimeric chIgG in a highly recurrent diffuse hereditary gingival fibromatosis.
MATERIAL AND METHODS
The study was carried out in accordance with the Declaration of Helsinki and the consent of the patient and healthy subjects was obtained prior to the study as approved by the Bioethical Committee of the Jagiellonian University, Medical College in Cracow, Poland.
Gingival biopsies were collected from the anterior region of the mandibular gingiva of the patient diagnosed with non-syndromic, diffuse, hereditary gingival fibromatosis with a high rate of lesion recurrence. The diagnosis was based on an intraoral examination, the patient's medical/family history, histological evaluation of the gingival biopsy and routine blood tests. Twelve months earlier the patient had undergone surgery with complete excision of the lesion. Gingival fibroblast cultures established from three healthy subjects qualified to the orthodontic treatment were used as controls in tests performed with chimeric anti-collagen antibody. The chimeric antibody variant (chIgG) tested in this study was constructed and kindly provided by dr. A. Fertala from Department of Orthopaedic Surgery, Jefferson Medical College, Thomas Jefferson University, Philadelphia, PA, USA.
Histology of gingival biopsies from hereditary gingival fibromatosis
Two gingival biopsies (1 × 2 mm, 2 × 2 mm) obtained from the anterior region of the mandibular gingiva of HGF patient were fixed in 10% formalin in a phosphate buffered saline (PBS) for 24 hours at 4°C. The tissue was dehydrated, paraffin embedded and serially sectioned (thickness 7 µm). A histo-morphological evaluation was conducted in gingival sections stained according to the haematoxylin-eosin method and using a fluorescent microscope (Nikon Eclipse Ti, Japan) connected to NIS-Elements F 3.0 software (Nikon Inc.). Heidenhain's trichrome stain was used to visualize collagen fibers in tissue sections.
Cell culture
Gingival biopsies from patient with hereditary gingival fibromatosis and from controls were used to establish fibroblast cultures. In brief, following the enzymatic separation of the epithelial layer the remaining connective tissue was digested with 0.1% collagenase I (Invitrogen). After digestion, the cell pellet was plated in T25 flask in Dulbecco's Modified Eagle Medium (DMEM, PAA GmbH) supplemented with 10% heat-inactivated foetal bovine serum (FBS, PAA), penicillin/streptomycin (50 u/ml), gentamicin (50 u/ml), and nystatin (10 µg/ml, PAA), at 37°C in a humidified atmosphere containing 5% CO2. The homogeneity of the gingival fibroblast culture was confirmed after passage 1 by immunofluorescent staining (vimentin+, cytokeratin–). Gingival fibroblast cultures at passage 3–5 were used for the experiments. Cell viability was confirmed by Trypan blue staining and amounted to 97–99%.
Inhibition of collagen deposit formation in fibroblast cultures in the presence of the anti-a2Ct chIgG.
Western blot
In brief, for Western blot assays, 50 × 103 cells per well were seeded into a 24-well plate. Cell cultures were overlayed with a fresh medium supplemented with 40 µg/ml of L-ascorbic acid phosphate magnesium salt n-hydrate (Wako Pure Chemical Co.) and a mixture of 70-kDa and 400-kDa Ficoll (Sigma-Aldrich) added at concentrations of 40 mg/ml and 25 mg/ml, respectively. Subsequently, chIgG was added to the medium at a concentration of 70 µg/ml. Cell cultures were maintained for 6 days and the medium was changed every 2 days. The optimal concentration of chIgG for cell culture utility and the cytotoxic assays were standardized previously (27). Cell lysates were collected by employing a lysis buffer consisting of 1% SDS, 1% sodium deoxycholate, 0.1% Triton X-100, 10 mM EDTA, 3% β-mercaptoethanol and a mixture of protease inhibitors (Thermo Scientific). The lysates (60 µg of protein, a Bradford protein assay) were electrophoresed in polyacrylamide gels followed by their transfer onto nitrocellulose membranes. Collagen I was detected using a polyclonal anti-collagen I antibody, as described previously (27). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was detected with an anti-GAPDH antibody (Santa Cruz Biotechnology Inc.).
Immunofluorescent staining
For the immunostaining assays, 15 × 103 cells per well were seeded into an 8-chamber slide. Further procedures were performed identical to the Western blot assays. The medium was supplemented with chIgG (70 µg/ml). Cell cultures were maintained for 6 days and the medium was changed every 2 days. The cells were fixed with ice-cold methanol, rinsed with PBS and then collagen fibrils were immunolocalized with the primary polyclonal rabbit anti-collagen I antibodies, followed by the secondary anti-rabbit IgG antibodies conjugated to Alexa Fluor 594 (Invitrogen). The nuclei of the examined cells were stained with 4',6-diamidino-2-phenylindole (DAPI). The immunofluorescent reaction was analyzed with a fluorescent microscope (Eclipse E600, Nikon Inc.). Microphotographs were obtained with a digital camera (DS-Qi1Mc, Nikon Inc.) controlled by the NIS-Elements software (Nikon Inc.).
RESULTS
Histological evaluation of gingival biopsies from patient with hereditary gingival fibromatosis
A microscopic evaluation revealed epithelial acanthosis with singular elongated or enlarged rete pegs extending into underlying connective tissue stroma (Fig. 1A-1C). Locally abundant, irregular collagen bundles were visualized within the connective stroma using Heidenhain's trichrome stain (Fig. 1D-1F). Inflammatory cells were distributed throughout almost the entire connective tissue (Fig. 1A-1C). Inflammatory infiltrates were characterized by the prevalence of oval-shaped cells with a round, eccentrically located nucleus and an abundant basophilic cytoplasm. The nuclear chromatin of those cells was coarse and distributed in a pattern sometimes resembling the spokes of a wheel (cartwheel nucleus). The other predominant cell population found was represented by large mononuclear cells with a vesicular, elongated or kidney-shaped nucleus with a clearly visible nuclear membrane as well as an abundant, and eosinophilic cytoplasm. Two predominant populations of cells that infiltrated the overgrown gingival tissue were identified as plasmocytes and histiocytes, respectively. The third and less numerous population was represented by lymphocytes.
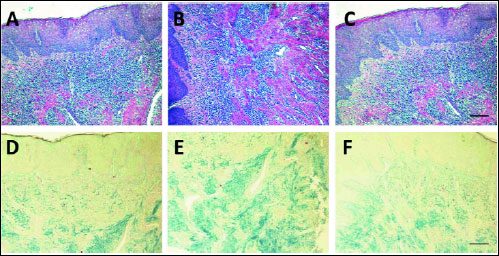 |
Fig. 1. Histological presence of hereditary gingival fibromatosis: epithelium and lamina propria visible in sections stained with haematoxylin and eosin, original magnification 100 × (A–C); collagen fibrils stained with Heidenhain's trichrome, magnification 100 × (D–F). Bars = 100 µm. |
The inhibitory effect of chimeric anti-α2Ct chIgG on collagen fibril formation
Collagen fibril inhibition was measured using Western blot and immunostaining assays. In the case of Western blot we used cell lysates from fibroblast cultures maintained for 6 days in the presence of therapeutic chIgG at a concentration of 70 µg/ml. As shown in Fig. 2, chIgG almost completely blocked collagen fibril formation, while the same effect was not observed after cells were treated with a non-inhibitory hIgG or in the absence of IgG. Microscopic analysis revealed relatively abundant collagen I fibers in the presence of control hIgG (Fig. 3A). After treatment with therapeutic chIgG fibril deposits had been reduced to almost zero (Fig. 3B). Gingival fibroblast cultures established from healthy subjects were used as controls (Fig. 3C-3D).
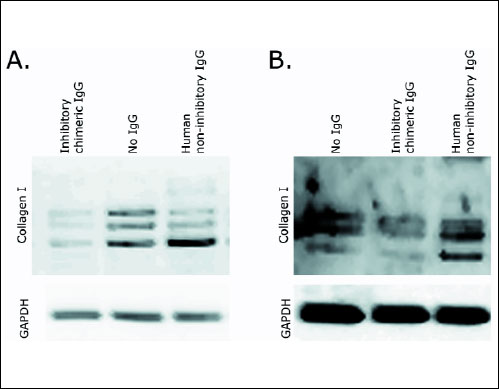 |
Fig. 2. Western blot assays of collagen I extracted from primary gingival fibroblast cultures derived from a patient with recurrent, diffuse, hereditary gingival fibromatosis, A); and from controls, B). Cells were cultured with the inhibitory chIgG variant of the anti-α2Ct antibody (A - left; B - middle); cells cultured with hIgG (A - right; B - right) or in the absence of IgG (A - middle; B - left). Experiments were repeated three times and representative images of Western blots are presented. Symbols: inhibitory chimeric IgG, inhibitory chIgG variant; human non-inhibitory IgG, hIgG; No IgG, sample not treated with any IgG variant; Collagen I, partially processed pro-α1 chains and cross-linked forms of α1 chains; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; indicates uniform gel loading and provides a reference for the α1 collagen chain. |
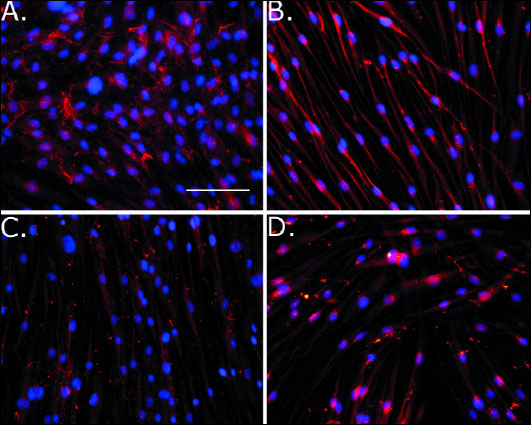 |
Fig. 3. Microscopic assays of collagen fibrils deposited in fibroblast cultures derived from a patient diagnosed with recurrent, diffuse, hereditary gingival fibromatosis (A-B), or in fibroblast cultures from controls (C-D). Cells were cultured in the presence of non-inhibitory hIgG, (A, C) or with the inhibitory chIgG variant of the anti-α2Ct antibody (B, D). Experiments were performed three times, and representative images are presented. Magnification 200 ×. Bars = 100 µm. |
DISCUSSION
GF is a condition characterized by progressive enlargement of the gingiva. A common feature of the condition is a problem with maintaining oral hygiene, speech disturbances, and difficulties with mastication (13, 14, 28). If left untreated, the enlarged tissue may eventually cover most of the teeth and because of the difficulties in maintaining oral hygiene, cause bone loss. The condition may also induce caries development and other inflammatory periodontal disorders (11, 13, 23). Additionally, oral infections stemming from gingival overgrowth in patients with unbalanced normal microbiota of oral cavity may lead to activation of Th17 cells and macrophages which upon induction produce excessive levels of proinflammatory cytokines. This process, in turn, can potentially impair systemic health of the patients (29, 30). Epithelial acanthosis with its potential cosmetic implications and tendency to provide a niche for further growth of microorganism, the build-up of plaque, and gingival inflammation, is still challenge for periodontists to diagnose and manage effectively.
Hereditary gingival fibromatosis may be transmitted as a Mendelian trait, and both autosomal dominant and autosomal recessive transmission have been reported (31). Linkage studies have localized loci for isolated nonsyndromic autosomal dominant forms of gingival fibromatosis to chromosome 2p21-p22 (32) and to chromosome 5q13-q22 (33). In addition to nonsyndromic forms, GF has been associated with a variety of syndromic manifestations. Hart et al. performed sequence analysis of 6 known candidate genes, as well as 10 additional, previously uncharacterized genes within the refined interval, and identified a mutation in the SOS-1 gene, that segregated with the HGF phenotype in a large group of members of a Brazilian family (7). It has been suggested that the g.126,142-126,143insC mutation produces the truncated protein chimera p.K1084fsX1105 that causes HGF through a gain-of-function mechanism. However, why the HGF phenotype appears localized to the gingiva is unknown. The keratinized masticatory mucosa is recognized to be developmentally unique, and different tissue-specific signaling pathways in this unique tissue may be responsible for the limited tissue distribution of the GF phenotype. Although the sites-specific localization of this phenotype is remarkable, the gingiva is also the only site affected in drug-induced and most syndromic forms of GF. The SOS-1 mutation in individuals with HGF described by Hart et al. identified the first genetic mutation for isolated HGF (7). Identification of the specific genetic basis in HGF should help to elucidate the pathogenic mechanism by which gingival overgrowth occurs (34). Otherwise, hereditary gingival fibromatosis displays genetic heterogeneity, and mutations in other genes are likely involved (35, 36). In turn, Lee et al. (12) analyzed histological, morphological, and proliferation characteristics in monolayer and three-dimensional cultures of fibroblasts with the SOS-1 g.126,142-126,143insC mutation. Histological assessment of HGF gingiva indicated increased numbers of fibroblasts (30%) and increased collagen (10%). Cell proliferation studies demonstrated increased growth rates and 5-bromo-2-deoxyuridine incorporation for HGF fibroblasts. Flow cytometry showed greater proportions of HGF fibroblasts in the G2/M phase. Subsequently, HGF derived fibroblasts in three-dimensional culture showed greater cell proliferation, higher cell density, and alteration of surrounding collagen matrix. The observations from this study clearly shown that increased fibroblast numbers and collagen matrix changes are associated with mutation of the SOS-1 gene in vitro and in vivo (12). Increased fibroblasts proliferation and extracellular matrix deposition in HGF was also reported by others (16, 17, 37-40).
Recently, it has been proposed that the extracellular process of collagen fibril formation may serve as a valid target for reducing the mass of collagen-rich fibrotic tissues. It has been postulated that targeting the specific process of fibril formation may offer not only an effective but also a safe way to reduce collagen synthesis in fibrotic tissues. Since fibril formation is driven by telopeptide mediated collagen-collagen interaction, an anti-α2Ct antibody was generated that specifically binds to the telopeptide of the α2 chain of human collagen I, thereby blocking its critical binding interaction (26, 41). Consequently, the anti-α2Ct antibody was tested as the inhibitor of localized fibrosis in a keloid-like model and these initial tests indicated the great potential of the anti-α2Ct antibody to reduce the mass of the formed fibrotic tissue (26). However, the prototypic anti-α2Ct antibody originates from mice and it belongs to the IgA class. As a consequence, its utility as a therapeutic agent in human patients is limited. Subsequently, the same group engineered a chimeric antibody variant (chIgG), consisting of variable domains derived from the prototypic IgA-type anti-α2Ct antibody and the constant domains of the human IgG (hIgG). The novel antibody was tested for its ability to inhibit collagen fibril formation in vivo and in cultures of various cells associated with the fibrotic process that develops in response to trauma (27). According to above evidences we proposed herein to study a novel approach to treating gingival overgrowth lesions using a novel chimeric IgG. To test the inhibitory potential of a novel anti-collagen IgG we used gingival fibroblasts from a patient diagnosed with diffuse, hereditary non-syndromic gingival fibromatosis with the recurrence of about 12 months after complete excision of the lesion. Histological evaluation of gingival sections revealed epithelial acanthosis with singular elongated or enlarged rete pegs extending into the underlying connective tissue stroma. Abundant, irregular collagen bundles were found locally in the stromal part of the gingiva. We observed an approximately 2.7-fold increase in the expression of procollagen I and a roughly 2-fold increase in proliferation at 72 hours in fibroblast cultures derived from this patient as compared to controls (data not shown). The tests conducted in the present study shown almost complete inhibition of collagen fibril formation, and this effect was not detected in cells supplemented with non-inhibitory hIgG or in the absence of IgG (Fig. 2). This observation was confirmed by immunofluorescent microscopic analysis of cell cultures treated with non-inhibitory hIgG (Fig. 3A), or therapeutic chIgG, respectively (Fig. 3B).
As shown by others a mutation in SOS-1 gene has been associated with increased fibroblasts proliferation and collagen matrix synthesis in HGF (12). Increased proliferation was also noted in fibroblasts used in the current study that were isolated from hereditary gingival overgrowth (unpublished data). Therefore another attempt to counteract gingival overgrowth is employment of chemicals that inhibit gingival cells (fibroblasts) proliferation. An attractive example is a chemotherapeutic agent mitoxantrone (MTX) that was tested using dental pulp stem cells (DPSCs) and human dermal fibroblasts (HDFs) (42). MTX is successfully used in clinical medicine, e.g. it limits the frequency of relapse and is effective in slowing the progression of several variants of multiple sclerosis (MS) (43). MTX mechanisms of action involve DNA intercalation through hydrogen bonding as well as inhibition of topoisomerase II which induces double strand breaks (DSBs) of DNA (44). It has been shown that treatment of DPSCs and HDFs with clinical concentrations of MTX resulted in stress-induced premature senescence (SIPS) that is a permanent cell cycle arrest in response to DSBs observed in some cell types. It was also found that lower concentration of MTX induced SIPS, while higher concentration of MTX induced programmed cell death, which was accompanied by increased expression of proapoptotic protein puma and activation of effector caspases 3/7 in both cell types. The results presented by Seifrtova et al. are interesting and such approach would seem promising also with respect to counteract gingival overgrowth. However, first, relevant studies are needed that would clarify the effect of MTX on gingival fibroblasts derived from HGF lesions, cells susceptibility and response to this drug as well as its mechanisms of action.
Another interesting approach concerns employment of bone marrow mesenchymal stem cells (BMSCs) and/or mesenchymal stem cells (MSCs) of oral origin to therapy of a number of periodontal diseases. DPSCs and periodontal ligament stem cells (PDLSCs) are of particular interest. An abundant source of both cell types are teeth (45, 46) and MSCs are normally obtained from teeth removed during routine dental care. It has been shown that DPSCs and PDLSCs induced apoptosis of some cells in in vitro conditions (47, 48). Moreover, transplanted BMSCs in response to appropriate pharmacological induction differentiated into cementoblasts, osteoblasts, osteocytes and fibroblasts of the regenerated tissue. Therefore replacement of abnormal fibroblasts from overgrown gingival tissue by allogeneic and/or autologous drafts of fibroblasts of MSCs origin, by DPSCs or PDLSCs would be of some importance in supporting therapy (regenerative phase) of gingival overgrowth lesions. Although MSCs from dental sources such as the dental pulp, the periodontal ligament and the gingiva represent a high potential for tissue regeneration, its biology and "behaviour" within regenerating tissues is yet not fully explored, indicating the need for more research to speed the path to clinical applications.
Histological evaluation of gingival sections in the current study revealed infiltrates of immune cells throughout almost the entire connective tissue i.e. subepithelial connective tissue, around vessels and capillaries, and within the connective stroma. Epithelial acanthosis that lead to partial or complete coverage of teeth provides a niche for microorganisms growth and the build-up of plaque and gingival inflammation. If left untreated such a condition may induce inflammatory diseases of the periodontium. The risk of gingival inflammation is higher in patients with unbalanced composition of normal oral microbiota, e.g. lower amount of Lactobacilli spp. and decreased production of H2O2 that negatively correlates with activation of Th17 cells, and increased levels of IL-17 as well as TNFα (29). Activation of Th17 cells and macrophages leading to excessive production of proinflammatory cytokines and receptor activator of nuclear factor kappa B ligand (RANKL) may cause activation of osteoclasts resulting in bone loss (30). In turn, the interaction between inflamed gingiva, dental plaque and the host response to it may affect fibrotic process in the gingiva.
In conclusion, at present, an effective management of patients with gingival fibromatosis clearly requires the active involvement of both dental and medical professionals to minimize the possibility of complications. Since the pathogenesis of gingival enlargement is not well-understood treatment is still largely limited to maintaining an improved level of oral hygiene, professional scaling, root planning and the surgical excision of overgrown tissue (24, 49). Non-surgical treatment is a far less invasive technique than surgical approaches and it is considered to be the first treatment option for GF. However, such a mode is insufficient in a relatively prevalent group of patients with diffuse and/or recurrent gingival overgrowth. The long-term benefits of surgery, particularly in the case of these patients, cannot be predicted and they are exposed to the risk of consecutive surgical procedures within relatively short time intervals, which may affect the quality of a patient's life. It has been shown that conventional gingivectomy with laser excision resulted in a better treatment outcome with a reduced rate of recurrence when compared to conventional gingivectomy with flap surgery (50). Although this mode of treatment seems to be more effective than routine surgery, the method still remains highly invasive for the patient. Therefore, since the sole mode of treatment of GF consists of invasive, surgical methods and the long-term benefit of surgery in a number of patients cannot be predicted, discerning studies testing novel tools that might be of value in the future (alternative or supporting) treatment of gingival overgrowth are of great importance. Finally, we report in the present study that the chimeric IgG variant appears to be an effective tool in counteracting collagen fibril accumulation in a fibroblast model derived from diffuse, hereditary gingival fibromatosis. Notably, a novel anti-collagen IgG was effective in culture of cells that derive from gingival lesion with a high propensity to recurrence (high proliferation index). Employing cell cultures from standardized group of patients with recurrent HGF as well as standardising relevant 3D (tissue-like) models will be crucial for further tests of the antibody.
Acknowledgements: We would like to thank Dr. Andrzej Fertala for his contribution to the chimeric antibody-based studies.
This work was supported by a grant from the National Science Centre, Poland (2012/07/B/NZ6/03524) (to K.G.), a Statutory Grant of the Jagiellonian University, Poland (K/ZDS/002418) (to M.C.G.), and a TEAM grant from the Foundation for Polish Science, Poland (DPS/424-329/10) (to J.P.). The Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University is a beneficiary of structural funds from the European Union (POIG.02.01.00-12-064/08).
Conflict of interests: None declared.
REFERENCES
- Seymour RA, Ellis JS, Thomason JM. Risk factors for drug-induced gingival overgrowth. J Clin Periodontol 2000; 27: 217-223.
- Coletta RD, Graner E. Hereditary gingival fibromatosis: a systematic review. J Periodontol 2006; 77: 753-764.
- Tavargeri AK, Kulkarni SS, Sudha P, Basavprabhu SP. Idiopathic gingival fibromatosis - a case report. J Indian Soc Pedod Prev Dent 2004; 22: 180-182.
- Martelli-Junior H, de Oliveira Santos, Bonan PR, et al. Minichromosome maintenance 2 and 5 expressions are increased in the epithelium of hereditary gingival fibromatosis associated with dental abnormalities. Clinics (Sao Paulo) 2011; 66: 753-757.
- Bozzo L, de Almedia OP, Scully C, Aldred MJ. Hereditary gingival fibromatosis. Report of an extensive four-generation pedigree. Oral Surg Oral Med Oral Pathol 1994; 78: 452-454.
- Martelli-Junior H, Lemos DP, Silva CO, Graner E, Coletta RD. Hereditary gingival fibromatosis: report of a five generation family using cellular proliferation analysis. J Periodontol 2005; 76: 2299-2305.
- Hart TC, Zhang Y, Gorry MC, et al. A mutation in the SOS1 gene causes hereditary gingival fibromatosis type 1. Am J Hum Genet 2002; 70: 943-954.
- Corbalan-Garcia S, Margarit SM, Galron D, Yang SS, Bar-Sagi D. Regulation of Sos activity by intramolecular interactions. Mol Cell Biol 1998; 18: 880-886.
- Sibilia M, Fleischmann A, Behrens A, et al. The EGF receptor provides an essential survival signal for SOS-dependent skin tumor development. Cell 2000; 102: 211-220.
- Sharma S, Goyal D, Shah G, Ray A. Familial gingival fibromatosis: a rare case report. Contemp Clin Dent 2012; 3: S63-S66.
- Chaturvedi R. Idiopathic gingival fibromatosis associated with generalized aggressive periodontitis: a case report. J Can Dent Assoc 2009; 75: 291-295.
- Lee EJ, Jang SI, Pallos D, Kather J, Hart TC. Characterization of fibroblasts with Son of Sevenless-1 Mutation. J Dent Res 2006; 85: 1050-1055.
- Casavecchia P, Uzel MI, Kantarci A, et al. Hereditary gingival fibromatosis associated with generalized aggressive periodontitis: a case report. J Periodontol 2004; 75: 770-778.
- Kantarci A, Black SA, Xydas CE, et al. Epithelial and connective tissue cell CTGF/CCN2 expression in gingival fibrosis. J Pathol 2006; 210: 59-66.
- Trackman PC, Kantarci A. Connective tissue metabolism and gingival overgrowth. Crit Rev Oral Biol Med 2004; 15: 165-175.
- Coletta RD, Almeida OP, Graner E, Page RC, Bozzo L. Differential proliferation of fibroblasts cultured from hereditary gingival fibromatosis and normal gingiva. J Periodontal Res 1998; 33: 469-475.
- Saygun I, Ozdemir A, Gunhan O, Aydintug YS, Karslioglu Y. Hereditary gingival fibromatosis and expression of Ki-67 antigen: a case report. J Periodontol 2003; 74: 873-878.
- Gagliano N, Moscheni C, Dellavia C, et al. Morphological and molecular analysis of idiopathic gingival fibromatosis: a case report. J Clin Periodontol 2005; 32: 1116-1121.
- Radisky DC. Epithelial-mesenchymal transition. J Cell Sci 2005; 118: 4325-4326.
- Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol 2006; 172: 973-981.
- Zavadil J, Bottinger EP. TGF- and epithelial-to-mesenchymal transitions. Oncogene 2005; 24: 5764-5774.
- Kantarci A, Nseir Z, Kim Y-S, Sume SS, Trackman PC. Loss of basement membrane integrity in human gingival overgrowth. J Dent Res 2011; 90: 887-893.
- Keremi B, Lohinai Z, Komora P, et al. Antiinflammatory effect of BPC 157 on experimental periodontitis in rats. J Physiol Pharmacol 2009; 60 (Suppl. 7): 115-122.
- Baptista IP. Hereditary gingival fibromatosis: a case report. J Clin Periodontol 2002; 29: 871-874.
- Gontiya G, Bhatnagar S, Mohandas U, Galgali SR. Laser-assisted gingivectomy in pediatric patients: a novel alternative treatment. J Indian Soc Pedod Prev Dent 2011; 29: 264-269.
- Chung HJ, Steplewski A, Chung KY, Uitto J, Fertala A. Collagen fibril formation. A new target to limit fibrosis. J Biol Chem 2008; 293: 25879-25886.
- Fertala J, Steplewski A, Kostas J, et al. Engineering and characterization of the chimeric antibody that targets the C-terminal telopeptide of the 2 chain of human collagen I: a next step in the quest to reduce localized fibrosis. Connect Tissue Res 2013; 54: 187-196.
- Bittencourt LP, Campos V, Moliterno LF, Ribeiro DP, Sampaio RK. Hereditary gingival fibromatosis: review of literature and a case report. Quintessence Int 2000; 31: 415-418.
- Szkaradkiewicz AK, Karpinski TM, Zeidler A, Wyganowska-Swiatkowska M, Szkaradkiewicz A. Protective effect of oral Lactobacilli in pathogenesis of chronic periodontitis. J Physiol Pharmacol 2011; 62: 685-689.
- Racz GZ, Kadar K, Foldes A, et al. Immunomodulatory and potential therapeutic role of mesenchymal stem cells in periodontitis. J Physiol Pharmacol 2014; 65: 327-339.
- Jorgenson RJ, Cocker ME. Variation in the inheritance and expression of gingival fibromatosis. J Periodontol 1974; 45: 472-477.
- Hart TC, Pallos D, Bowden DW, Bolyard J, Pettenati MJ, Cortelli JR. Genetic linkage of hereditary gingival fibromatosis to chromosome 2p21. Am J Hum Genet 1998; 62: 876-883.
- Xiao S, Bu L, Zhu L, et al. A new locus for hereditary gingival fibromatosis (GINGF2) maps to 5q13-q22. Genomics 2001; 74: 180-185.
- Pierre S, Bats AS, Coumoul X. Understanding SOS (Son of Sevenless). Biochem Pharmacol 2011; 82: 1049-1056.
- Hakkinen L, Csiszar A. Hereditary gingival fibromatosis: characteristics and novel putative pathogenic mechanisms. J Dent Res 2007; 86: 25-34.
- Shi J, Lin W, Li X, Zhang F, Hong X. Hereditary gingival fibromatosis: a three-generation case and pathogenic mechanism research on progress of the disease. J Periodontol 2011; 82: 1089-1095.
- Tipton DA, Howell KJ, Dabbous MK. Increased proliferation, collagen, and fibronectin production by hereditary gingival fibromatosis fibroblasts. J Periodontol 1997; 68: 524-530.
- Tipton DA, Woodard ES, Baber MA, Dabbous MK. Role of the c-myc proto-oncogene in the proliferation of hereditary gingival fibromatosis fibroblasts. J Periodontol 2004; 75: 360-369.
- Almeida JP, Coletta RD, Silva SD, et al. Proliferation of fibroblasts cultured from normal gingiva and hereditary gingival fibromatosis is dependent on fatty acid synthase activity. J Periodontol 2005; 76: 272-278.
- Coletta RD, Almeida OP, Ferreira LR, Reynolds MA, Sauk JJ. Increase in expression of Hsp47 and collagen in hereditary gingival fibromatosis is modulated by stress and terminal procollagen N-propeptides. Connect Tissue Res 1999; 40: 237-249.
- Steplewski A, Fertala A. Inhibition of collagen fibril formation. Fibrogenesis Tissue Repair 2012; 5 (Suppl. 1): S29.
- Seifrtova M, Havelek R, Soukup T, Filipova A, Mokry J, Rezacova M. Mitoxantrone ability to induce premature senescence in human dental pulp stem cells and human dermal fibroblasts. J Physiol Pharmacol 2013; 64: 255-265.
- Vacchelli E, Galluzzi L, Fridman WH, et al. Trial watch: chemotherapy with immunogenic cell death inducers. Oncoimmunology 2012; 1: 179-188.
- Zhao H, Traganos F, Darzynkiewicz Z. Phosphorylation of p53 on Ser15 during cell cycle caused by Topo I and Topo II inhibitors in relation to ATM and Chk2 activation. Cell Cycle 2008; 7: 3048-3055.
- Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA 2000; 97: 13625-13630.
- Seo BM, Miura M, Gronthos S, et al. Investigation of multipotent postnatal stem cells from human periodontal ligament. Lancet 2004, 364: 149-155.
- Zhao Y, Wang L, Jin Y, Shi S. Fas ligand regulates the immunomodulatory properties of dental pulp stem cells. J Dent Res 2012; 91: 948-954.
- Wada N, Menicanin D, Shi S, Bartold PM, Gronthos S. Immunomodulatory properties of human periodontal ligament stem cells. J Cell Physiol 2009; 219: 667-676.
- Kelekis-Cholakis A, Wiltshire WA, Birek C. Treatment and long-term follow-up of a patient with hereditary gingival fibromatosis: a case report. J Can Dent Assoc 2002; 68: 290-294.
- Mavrogiannis M, Ellis JS, Seymour RA, Thomason JM. The efficacy of three different surgical techniques in the management of drug-induced gingival overgrowth. J Clin Periodontol 2006; 33: 677-682.
A c c e p t e d : July 1, 2014