EFFECT OF THIOL ANTIOXIDANTS ON LIPOPOLYSACCHARIDE-INDUCED CYCLOOXYGENASE-2 EXPRESSION IN PULMONARY EPITHELIAL CELLS
INTRODUCTION
Lung inflammation due to environmental pollutants, including endotoxins and lipopolysaccharide (LPS), plays an important role in the development and progression of chronic respiratory diseases including asthma (1). Acute bacterial pneumonia is the most common cause of clinical acute lung injury, which is a leading cause of death due to infectious diseases (2, 3). LPS is a major component of the outer membrane of all gram-negative bacteria and the predominant inducer of inflammatory responses to these common respiratory pathogens (4, 5). The epithelium lining of the airways is the first tissue to encounter pathogens and their products, resulting in a local inflammatory response. It is, therefore, a critical component of the innate immune system as the front line of the host defense against invading microorganisms (6). A number of studies recently revealed that the lung epithelium is an important sentinel and effector of innate immunity (7).
Cyclooxygenase (COX)-2 is an essential enzyme involved in inflammation processes and the pathogenesis of other conditions (8). Two isoforms of COX, encoded by distinct genes, have been identified. COX-1 is constitutively expressed in most tissues and considered to be the housekeeping isoform that produces prostaglandins required for the maintenance of normal cell and organ functions. In contrast, COX-2 is primarily an inducible isoform whose expression can be upregulated in many cell types by cytokines and endotoxins. It is highly expressed and believed to produce prostaglandins involved in inflammatory processes (9, 10). The involvement of COX-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases, such as allergic airway inflammation, asthma, and acute lung injury, has been reported (11). COX-2 gene expression is chiefly regulated at the level of transcription and mediated by multiple transcription factors, including nuclear factor-κB (NF-κB), activator protein-1 (AP-1), and signal transducer and activator of transcription-3 (STAT-3) (11, 12).
Reactive oxygen species (ROS), such as superoxide anions, hydroxyl radicals, and hydrogen peroxides, are highly reactive and modify intracellular molecules. Accordingly, ROS are considered as mediators of cellular events or as secondary messengers (13). Increased levels of ROS have been implicated in initiating inflammatory responses through the activation of transcription factors such as NF-κB, AP-1, and STAT-3 (12, 14-16).
Glutathione (GSH) is a ubiquitous, essential tripeptide containing a sulfhydryl group that protects cells against oxidants. GSH, which accounts for 90% of intracellular non-protein thiols, is a key intracellular reducing agent and is implicated in immune modulation and inflammatory conditions (17). N-acetyl-l-cysteine (NAC), a well-known thiol-containing antioxidant, is capable of facilitating intracellular GSH biosynthesis and has the potential to interact directly with oxidants (18-21).
A variety of toxic agents, including asbestos, cigarette smoke, airborne particulate matter, diesel exhaust, and ozone, produce ROS as cell-signaling molecules and stimulate inflammatory responses in the airway and pulmonary epithelial cells (22, 23). However, the involvement of ROS in bacterial LPS-induced pro-inflammatory gene expression has remained elusive. The purpose of the present study was to investigate the role of ROS in LPS-stimulated COX-2 induction and the inhibitory effects of thiol antioxidants on LPS-induced COX-2 expression in human lung epithelial A549 cells.
MATERIALS AND METHODS
Cell culture
A549 cells, human alveolar type II epithelial cells, were obtained from ATCC (CCL-185; Rockville, MD, USA). Cells were grown in 5% CO2 at 37°C in high-glucose Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Cambrex, Walkersville, MD, USA) and an antibiotic-antimycotic mix (Gibco, Grand Island, NY, USA). LPS from Escherichia coli serotype O26:B6, NAC, and GSH (Sigma-Aldrich, St. Louis, MO, USA) were all dissolved in phosphate buffered saline (PBS). The cells were treated with 10 µg/ml of LPS for various time periods.
Experimental protocol
For time-course experiments, the cells (1 × 105 cells/ml) were treated with LPS for 30, 60, 120, and 180 min for determination of intracellular ROS levels. For determination of DNA binding activities of NF-κB, AP-1 and STAT-3, the cells were treated with LPS for15, 30, and 60 min, respectively. The cells were treated with LPS for 1, 2, 3, and 4 hours for measuring mRNA expression of COX-2. The LPS-treated cells were cultured for 2, 4, 6, and 8 h for determination of COX-2 protein levels. To investigate the effect of thiol antioxidants, the cells were pretreated with GSH and NAC (at final concentrations of 2, 5, and 10 mM) for 1 hour prior to LPS stimulation. DNA-binding activities of NF-κB, AP-1, and STAT-3 were determined at 1 h. ROS levels were assessed at 2 h. mRNA and protein levels of COX-2 were determined at 4 h and 8 h, respectively.
Measurement of reactive oxygen species levels
Cells were incubated with 10 µM 5’,6’-carboxy-2’,7’-dichlorodihydrofluorescein diacetate (DCF-DA; Molecular Probes, Eugene, OR, CA, USA) for 30 min. Next, the cells were washed twice and harvested with 1 ml of PBS and seeded in 96-well black plates. The change in dichlorofluorescein (DCF) fluorescence was measured (at excitation and emission wavelengths of 485 and 535 nm, respectively) with a fluorometer (VICTOR 5 multi label counter, Perkin Elmer, Boston, MA, USA) or a laser scanning confocal microscope (Leica TCS-NT, Heidelberg, Germany).
Preparation of whole-cell and nuclear extracts
Whole-cell and nuclear extracts were prepared for western blot analysis and electrophoretic mobility shift assay (EMSA), respectively (24). Briefly, the harvested cells were extracted with a lysis buffer (10 mM Tris-HCl, pH 7.4, 10% Nonidet P-40, and protease inhibitor cocktail) and centrifuged. The supernatants were used for preparing whole-cell extracts. To prepare nuclear extracts, the cells were rinsed with ice-cold PBS, harvested by scraping into PBS, and pelleted by centrifugation at 1500 g for 5 min. The cells were lysed in a buffer containing 10 mM HEPES, 10 mM KCl, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 1.5 mM MgCl2, 0.2% Nonidet P-40, 1 mM dithiothreitol (DTT), and 0.5 mM phenylmethylsulfonylfluoride (PMSF). The nuclear pellet was resuspended on ice in a nuclear extraction buffer containing 20 mM HEPES, 420 mM NaCl, 0.1 mM EDTA, 1.5 mM MgCl2, 25% glycerol, 1 mM DTT, and 0.5 mM PMSF. Protein concentrations were determined using the Bradford assay (Bio-Rad Laboratories, Hercules, CA, USA).
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts (4 µg of protein) were incubated with 32P-labeled double-stranded NF-κB, AP-1, and STAT-3 oligonucleotide in 20 µl of EMSA buffer (10 mM HEPES, pH 7.9, 1 mM EDTA, 1 mM DTT, 60 mM KCl, 7% glycerol, and 2 µg of poly(dI-dC)). For the preparation of each 32P-labeled double-stranded oligonucleotide, each oligonucleotide was annealed and radiolabeled with [γ-32P] dATP by T4 polynucleotide kinase (Promega, Madison, WI, USA). To determine NF-κB, AP-1, and STAT-3 activity, EMSA was performed using the following commercially available oligonucleotides: NF-κB (Promega), 5’-AGTTGAGGGGACTTTCCCAGGC-3’; AP-1 (Promega, Madison, WI, USA), 5’-CGCTTGATGACTCAGCCGGAA-3’; STAT-3 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), 5’-GATCCTTCTGGGAATTCCTAGATC-3’. After 30 min of incubation, the reaction mixture was loaded onto a standard 6% nondenatured polyacrylamide gel in 0.25 × Tris borate/EDTA buffer. Following electrophoresis, the gel was dried and exposed using intensifying screens to a radiography film at –80°C.
Real-time PCR analysis
COX-2 mRNA expression was assessed using real-time PCR. Total RNA was isolated from cells using the TRI reagent (Molecular Research Center, Inc. Cincinnati, OH, USA). Total RNA was converted into cDNA by reverse transcription using a random hexamer and a viral reverse transcriptase (Promega) under the following conditions: 23°C for 10 min, 37°C for 60 min, and 95°C for 5 min. The cDNA was used for real-time PCR with specific primers for COX-2 and β-actin. The COX-2 primer sequences were 5’-TTCAAATGAGATTGTGGGAAAATTGCT-3’ (forward primer) and 5’-AGATCATCTCTGCCTGAGTATCTT-3’ (reverse primer), yielding a 296-bp PCR product; and the β-actin primer sequences were 5’-ACCAACTGGG ACGACATGGAG-3’ (forward primer) and 5’-GTGAGG ATCTTCATGAGGTAGTC-3’ (reverse primer), yielding a 353-bp PCR product. PCR amplification of the cDNA included 40 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 15 s, and extension at 72°C for 45 s. The β-actin gene was amplified in the same reaction to serve as the reference gene.
Western blot analysis
Western blot analysis was carried out as previously described (25). Whole-cell extract (70 µg of protein/lane) was subjected to 8% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes (Amersham Bioscience, Piscataway, NJ, USA) by electroblotting. The membranes were blocked using 5% nonfat dry milk in TBS-T (Tris-buffered saline and 0.2% Tween 20) for 2 hours at room temperature. The membranes were then incubated with antibodies for COX-2 and actin (all from Santa Cruz Biotechnology; diluted in TBS-T containing 5% dry milk) overnight at 4°C. After washing with TBS-T, primary antibodies were detected using horseradish peroxidase-conjugated secondary antibodies and visualized by the enhanced chemiluminescence detection system (Santa Cruz Biotechnology) according to the manufacturer’s instruction. Actin was used as a loading control. For the ratio of COX-2/actin, the protein bands of COX-2 and actin were scanned using a Bio-Rad scanner (GS-700) driven by a volume analysis software and quantified with Molecular Analysis software (version 4).
Statistical analysis
Statistical differences were determined using one-way ANOVA by Newman-Keul’s test. All values are expressed as a mean ± SE of four different experiments. Four samples/group were used in each experiment. A value of P < 0.05 was considered statistically significant.
RESULTS
Reactive oxygen species levels and activation of nuclear factor-κB, AP-1, and STAT-3 in lipopolysaccharide-stimulated cells
Intracellular ROS levels were determined using a fluorometer and expressed as DCF fluorescence (%) (Fig. 1A). Treatment with 10 µg/ml of LPS significantly increased ROS levels in a time-dependent manner in A549 cells. ROS levels were maximum at 2 h and slightly increased until 3 h (close circle). However, ROS levels did not increase in the cells without LPS stimulation during the culture period (open circle). To investigate whether LPS increased the activation of redox-sensitive transcription factors, DNA-binding activities of NF-κB, AP-1, and STAT-3 in the nuclear protein extracts from the cells stimulated with LPS (10 µg/ml) were determined using EMSA (Fig. 1B). NF-κB, AP-1, and STAT-3 activation started at 15 min, and the maximum activation level was detected at 1 hour. Thus, ROS levels were determined at 2 h while the DNA-binding activities of NF-κB, AP-1, and STAT-3 were assessed at 1 hour in the subsequent experiments to study the effects of thiol antioxidants on ROS levels and the activation of transcription factors.
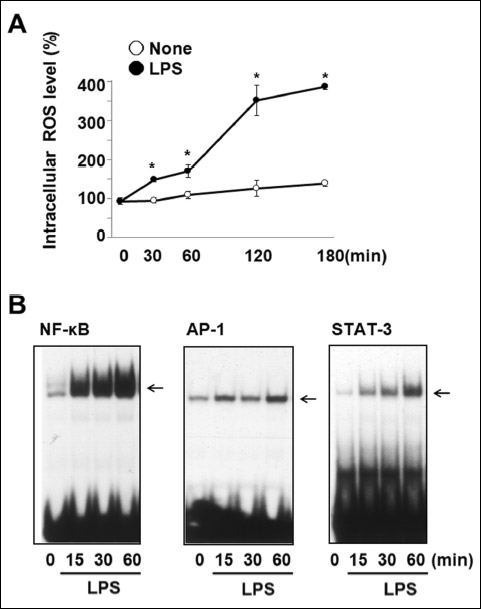 |
Fig. 1. ROS levels and the activation of NF-κB, AP-1, and STAT-3 in LPS-stimulated cells. (A) The cells were stimulated without (open circle) or with (close circle) LPS (10 µg/ml) for 30, 60, 120, and 180 min. Intracellular ROS levels were determined using DCF-DA by fluorometer and expressed as DCF fluorescence (%). Values are means ± SE of four different experiments. *P < 0.05 compared with corresponding none (the cells without LPS stimulation). (B) The cells were stimulated with LPS (10 µg/ml) for 15, 30, and 60 min, and DNA-binding activities of NF-κB, AP-1, and STAT-3 were determined by EMSA. |
Expression of cyclooxygenase-2 in lipopolysaccharide-stimulated cells
Real-time PCR analysis showed that LPS induced the mRNA expression of COX-2 until 4 h in a time-dependent manner (Fig. 2A). COX-2 protein levels in LPS-induced A549 cells were determined by Western blot analysis (Fig. 2B). The highest COX-2 expression level induced by LPS was observed at 8 h while actin level was not affected by LPS. Therefore, the effects of thiol antioxidants on the mRNA and protein levels of COX-2 were studied at 4 h and 8 h.
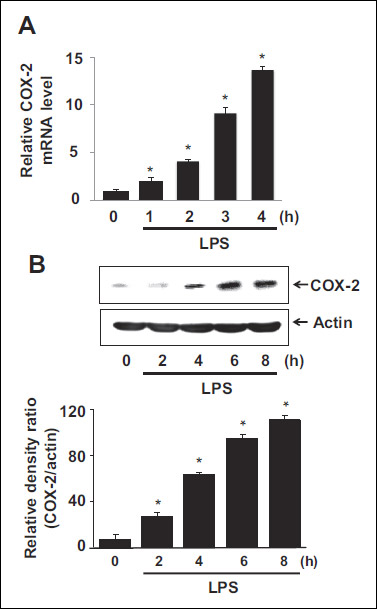 |
Fig. 2. Expression of COX-2 in LPS-stimulated cells. (A) The cells were stimulated with LPS (10 µg/ml) for 1, 2, 3, and 4 h, and mRNA levels of COX-2 were determined using real-time PCR. mRNA levels were normalized to β-actin that served as a loading control. Values are means ± SE of four different experiments. *P < 0.05 compared with 0 h. (B) The cells were stimulated with LPS (10 µg/ml) for 2, 4, 6, and 8 h, and protein levels were determined by western blot analysis, in which actin served a loading control. Following densitometric analysis, the ratio of COX-2/actin was determined. Values are means ± SE of four different experiments. *P < 0.05 compared with 0 h. |
Effects of thiol compounds on intracellular reactive oxygen species levels and the activation of nuclear factor-κB, activator protein-1, and STAT-3 in lipopolysaccharide-stimulated cells
Direct imaging of intracellular ROS levels by confocal laser microscopy showed that LPS stimulation increased ROS levels (Fig. 3A). This increase in ROS levels was attenuated by pretreatment with GSH and NAC. Fluorometer analysis showed similar results as those obtained by confocal laser microscopy imaging. The LPS-induced increase in ROS levels was inhibited by pretreatment with the thiol antioxidants (Fig. 3B).
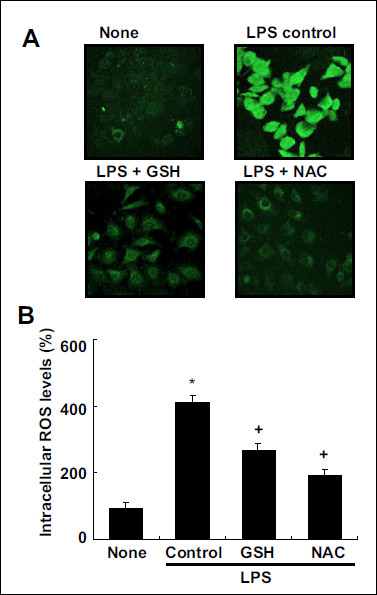 |
Fig. 3. Effects of thiol compounds on intracellular ROS levels in LPS-stimulated cells. The cells were pretreated with 10 mM of GSH and NAC and stimulated with LPS for 2 h. (A) ROS levels were determined using confocal microscopy (A) and fluorometer (B) by measuring the level of the fluorescent DCF at excitation and emission wavelengths of 495 nm and 535 nm, respectively. Values are means ± SE of four different experiments. *P < 0.05 compared with none (the cells without LPS stimulation), +P < 0.05 compared with LPS control (without GSH or NAC pretreatment). |
To determine whether GSH and NAC inhibit the LPS-induced activation of NF-κB, AP-1, and STAT-3, we measured the DNA-binding activities of NF-κB, AP-1, and STAT-3 (Fig. 4). Both GSH and NAC suppressed the LPS-induced activation of NF-κB, AP-1, and STAT-3 in A549 cells in a dose-dependent manner.
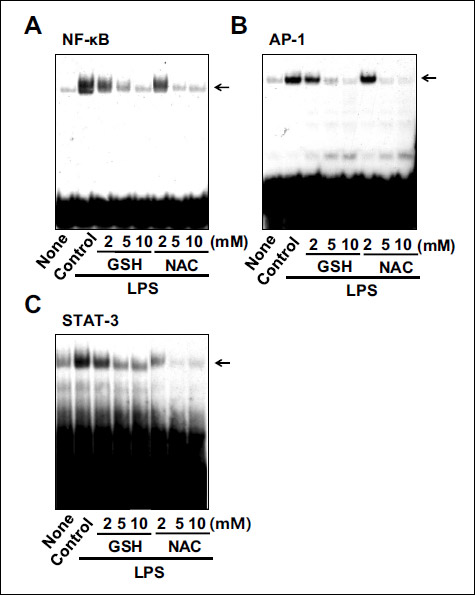 |
Fig. 4. Effects of thiol compounds on the activation of NF-κB, AP-1, and STAT-3 in LPS-stimulated cells. The cells were pretreated with various concentrations of GSH and NAC and stimulated with LPS for 1 hour. The DNA-binding activities of NF-κB, AP-1, and STAT-3 were determined by EMSA. |
Effects of thiol compounds on cyclooxygenase-2 expression in lipopolysaccharide-stimulated cells
The LPS-induced mRNA expression of COX-2 was inhibited by GSH and NAC (Fig. 5A). Pretreatment with 10 mM of GSH and NAC showed the greatest inhibitory effect on COX-2 mRNA expression in LPS-stimulated cells, as determined by real-time PCR. Western blotting revealed that GSH and NAC also attenuated the LPS-induced increase in COX-2 protein levels (Fig. 5B upper panel). Density ratio of COX-2/actin confirmed the suppressive effects of thiol antioxidants on COX-2 protein expression level in LPS-stimulated cells (Fig. 5B lower panel). GSH and NAC inhibited COX-2 expression in a dose-dependent manner at both mRNA and protein levels in LPS-stimulated cells.
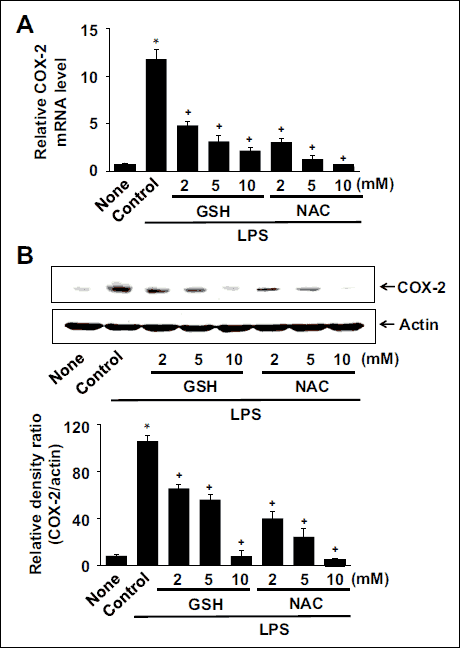 |
Fig. 5. Effects of thiol compounds on the expression of COX-2 in LPS-stimulated cells. (A) The cells were pretreated with various concentrations of GSH and NAC and stimulated with LPS for 4 h. mRNA levels of COX-2 were determined using real-time PCR. The mRNA levels were normalized to β-actin that served as a loading control. Values are means ± SE of four different experiments. *P < 0.05 compared with none (the cells without LPS stimulation), +P < 0.05 compared with LPS control (without GSH or NAC pretreatment). (B) The cells were pretreated with various concentrations of GSH and NAC and stimulated with LPS for 8 h. Protein levels were determined by Western blot analysis, in which actin served a loading control. Following densitometric analysis, the ratio of COX-2/actin was determined. Values are means ± SE of four different experiments. *P < 0.05 compared with none (the cells without LPS stimulation), +P < 0.05 compared with LPS control (without GSH or NAC pretreatment). |
DISCUSSION
The lungs represent one of the largest interfaces between the human body and its environment; thus, it is a major site of interaction with bacteria (26). LPS can enter the lungs during infection by gram-negative bacteria (4, 5, 26). In the present study, we show that bacterial LPS induces COX-2 mRNA and protein expression in human pulmonary epithelial cells. Gloire et al. (16) showed that LPS induces ROS production. Although the source of ROS upon LPS challenge has not been fully understood, Park et al. (27) discovered that in kidney epithelial HEK293T cells, LPS-induced ROS generation and NF-κB activation were mediated by the direct interaction of Toll-like receptor 4 with Nox4, a subunit of NADPH oxidase that transfers electrons from NADPH to O2 to generate O2–·, which is rapidly converted to H2O2 and O2 in cells (27). NADPH oxidase-generated ROS can participate in signal transduction mechanisms linked to immunity and inflammation (28). Since Nox2 and Nox4 of the Nox family are present in human lung epithelial A549 cells (29, 30), LPS-induced ROS production observed in the present study may be related to the activation of NADPH oxidase through LPS-initiated TLR4 signaling in the cells.
Cigarette smoke, airborne particulate matter, diesel exhaust, and ozone induce oxidative stress and activate NF-κB or AP-1 in the airway and pulmonary epithelium (22). In the present study, we found that intracellular ROS production by LPS stimulation also resulted in the activation of NF-κB, AP-1, and STAT-3 in lung epithelial A549 cells. It is well established that NF-κB and AP-1 are redox-sensitive transcription factors that are regulated by oxidative stress. Therefore, the expression of inflammatory mediators can be regulated by the activation of NF-κB and AP-1 in response to ROS (31). NF-κB activation occurs through the interaction with the inhibitory protein IκB, which is rapidly phosphorylated at two conserved serine residues by IκB kinase (IKK), followed by subsequent poly-ubiquitination and degradation through proteasome in response to pro-inflammatory stimuli (32). ROS-mediated activation of IKK leads to the activation of NF-κB (33, 34). IKK activation was involved in the LPS-mediated activation of NF-κB through ROS in mouse macrophages (35-37). AP-1 activity is regulated through the increased synthesis and/or phosphorylation of its components (Jun and Fos proteins); ROS induce the phosphorylation of Jun and Fos to activate AP-1 (33, 34).
While NF-κB and AP-1 are extensively reported to be activated by intracellular ROS, the connection between oxidative stress and STAT-3 activity has not been largely investigated. STAT-3 is a member of a family of transcription factors that are activated upon tyrosine phosphorylation in response to extracellular signals (38). Activated STATs form a dimer through their Src-homology domain II; these are transported into the nucleus, where they bind to cognate DNA sequences and activate gene expression (15). Oxidative stress can trigger STAT-3 tyrosine phosphorylation and nuclear translocation, which correlate with the activation of STAT-3 and its DNA-binding activity (15, 39, 40). The present study showed that LPS-mediated increase in intracellular ROS activated STAT-3 and induced its DNA-binding activity. The mechanism by which LPS-induced ROS production directly activates STAT-3 is not clearly understood. One possible mechanism suggested by Carballo et al. (40) and Waris et al. (15) is that the alteration of redox status could directly change STAT conformation in such a way that its interaction with cytosolic proteins, responsible for nuclear targeting, is increased. Another likely explanation is the ability of oxidants to act as inhibitors of tyrosine phosphatases, thereby inducing STAT-3 nuclear translocation by enhanced tyrosine phosphorylation (15, 40). In addition, the activation of STAT by ROS is likely to be mediated by the activation of multiple kinases such as JAK2 kinase (39).
Since COX-2 gene expression is influenced by its mRNA transcription (11) and the COX-2 promoter contains transcriptional regulatory elements for NF-κB, AP-1, and STAT-3 (41), activation of these transcription factors upregulates the synthesis of COX-2 in A549 cells. Consistently, in our study, when NAC and GSH inactivated NF-κB, AP-1, and STAT-3, LPS-induced COX-2 expression decreased.
Previously, Mitchell et al. (42) reported that COX-1 was expressed constitutively and not affected by LPS treatment while COX-2 was induced by LPS in A549 cells. Several studies showed that LPS induces COX-2 expression, but did not affect COX-1 expression in macrophages and nonciliated bronchiolar secretory cells (43, 44). It is necessary to determine whether COX-1 expression is changed by LPS in the present experimental setting using A549 cells.
COX-2 is known to be degraded by the ubiquitin-proteasome system. Neuss et al. (45) showed that curcumin, an antioxidant agent, stimulated the proteasome-dependent degradation of COX-2 in HeLa cells. However, the effect of other antioxidants on ubiquitin-proteasome system has not been studied. Therefore, it may be helpful to understand the mechanisms how thiol antioxidants reduce COX-2 protein levels by investigating whether thiol antioxidants affect ubiquitin-proteasome system in LPS-treated A549 cells.
Tetsuka et al. (46) reported that antioxidants such as rotenone and pyrrolidine dithiocarbamate inhibited IL-1b-induced COX-2 expression at the post-transcriptional level in rat mesangial cells. They suggested that the change in redox status might regulate eukaryotic initiation factors and/or RNA binding proteins which regulate the translation and/or stability of COX-2 mRNA. In the present study, both GSH and NAC inhibited COX-2 mRNA expression in LPS-stimulated cells. The results show that these antioxidants suppress COX-2 expression at transcriptional level. However, it is essential to determine whether GSH and NAC affect COX-2 expression at post-transcriptional level by determining changes in eukaryotic initiation factors and/or RNA binding proteins.
Previous studies revealed that α-Tocopherol inhibited prostaglandin E2 production and COX-2 activity with no effect on the expression of COX in murine macrophages, Caco2 cells, and brominated diphenyl ether-47-stimulated extravillous trophoblasts (47-49). It is reported that COX activity requires the presence of oxidant hydroperoxides (50). Therefore, it has been proposed that antioxidants may attenuate COX activity by scavenging the oxidant hydroperoxides necessary for COX activation (51). These studies suggest that thiol antioxidants may inhibit LPS-induced COX-2 expression at posttranscriptional level. Further study should be performed to determine whether thiol antioxidants affect COX-2 expression at posttranscriptional and posttranslational levels in LPS-treated A549 cells.
The structure of NAC (Fig. 6) is comprised of sulfhydryl groups that can scavenge free radicals and prevents oxidative damage. In addition, NAC indirectly enhances natural antioxidant defenses by increasing intracellular GSH levels (52). Jafari et al. (53) reported that 1 mM of NAC treatment induces 1.8-fold increase in endogenous GSH biosynthesis in A549 cells at 6 h. In the present study, we pretreated the cells with NAC for 1 h and observed the changes in intracellular ROS levels after LPS stimulation. Therefore, the inhibitory effect of NAC on LPS-induced increase in ROS may be due to the ROS-scavenging effect of NAC, and not due to its effect on GSH biosynthesis.
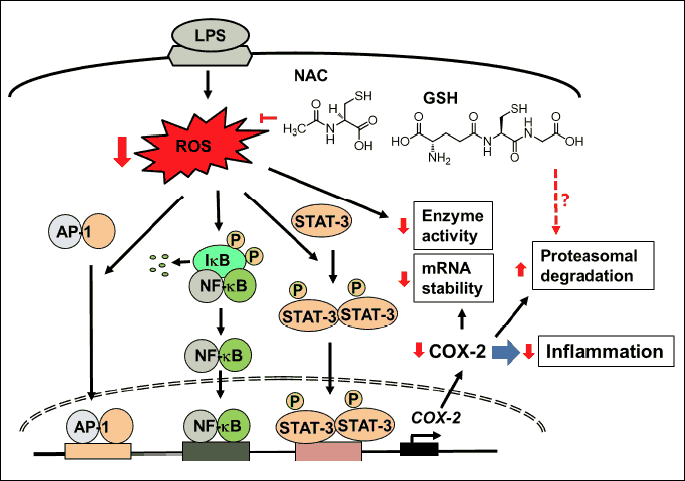
GSH dynamically regulates cellular protein functions by forming reversible disulfide bonds with the proteins apart from creating a reducing intracellular environment and maintaining the proteins in a reduced state (54, 55). Transcription factors such as NF-κB and AP-1 require an active cysteine-SH for their binding to DNA. S-glutathiolation (posttranslational binding of GSH to cellular proteins) of these cysteine-SH residues prevents these transcription factors from binding to DNA, thereby obliterating inflammatory responses due to oxidative stress (56, 57). Since GSH significantly reduced ROS levels in the present study, GSH-induced scavenging ROS might contribute to inhibitory effect of GSH on activation of transcription factors in LPS-stimulated cells. Recently, Mokry et al. (58) showed that treatment with selective phosphodiesterase 4 inhibitor and phosphodiesterase 5 inhibitor suppressed NF-κB activity, lipid peroxidation, and tumor necrosis factor-α level in lung and decreased numbers of circulating leukocytes and eosinophils in experimentally-induced allergic airway inflammation in guinea pigs. Mikolka et al. (59) investigated whether addition of anti-inflammatory agent into the surfactant alleviates inflammation and enhances efficiency of the therapy. They used meconium-induced lung inflammation model using meconium suspension (25 mg/ml, 4 ml/kg body weight) which was instilled into the trachea of young rabbits. They found that surfactant therapy, but particularly combined surfactant with budesonide therapy, reduced markers of oxidative stress and lung edema. In addition, intravenous NAC (10 mg/kg body weight) diminished meconium-induced oxidative stress in adult rabbits (60). These series of studies support that antioxidant therapy may be beneficial for treating pulmonary inflammation by suppressing redox-sensitive transcription factors and inflammatory cytokine expression and by reducing numbers of circulating leukocytes and lung edema.
Previous studies reported that antioxidant such as GSH and NAC inhibited LPS-induced lung inflammation in rats (61, 62). Sun et al. (61) found that intraperitoneal injection of GSH (40 mg/kg body weight) 30 min prior to LPS injection (20 mg/kg) significantly decreased LPS-induced mortality and eliminated lung histological injury in rats. Furthermore, GSH inhibited the expression of COX-2 proteins in LPS-stimulated rat peritoneal macrophages. Mitsopoulos et al. (62) demonstrated that pretreatment of NAC (25 mg/kg body weight, intravenous therapy) 4 h prior to LPS challenge reduced LPS-induced lung injury by reducing lung edema, lipid peroxidation, and pro-inflammatory eicosanoids, thromboxane B2 and leukotriene B4 in lung homogenates. The present findings suggest the inhibitory mechanism of thiol antioxidants on COX-2 expression by directly suppressing transcription factors NF-κB, AP-1, and STAT-3 in pulmonary epithelial A549 cells. Therefore, the present founding may support the inhibitory mechanism of GSH and NAC on pulmonary inflammation shown in in vivo animal models.
The promoter region for COX-2 gene contains binding sites for various transcription factors such as NF-κB, nuclear factor for IL-6 (NF-IL6), nuclear factor of activated T-cells (NFAT), cAMP response element-binding protein (CREB), AP-2, and specificity protein-1 (SP-1) (63-66). Therefore, activation of transcription factors, either alone or in combination, results in increased COX-2 expression. Thus, it is essential to determine whether thiol antioxidants affect transcription factors such as NF-IL6, NFAT, CREB, AP-2, and SP-1 for COX-2 expression in LPS-treated pulmonary epithelial A549 cells for the further study.
The inhibitory effect of thiol antioxidants on LPS-induced COX-2 expression in pulmonary epithelial cells is summarized in Fig. 6. Binding of LPS to the receptor increases intracellular ROS levels which activate transcription factors. ROS induce phosphorylation of inhibitory subunit IkB which is bound to NF-κB. Phosphorylated IkB is degraded and NF-κB is translocated to the nucleus. ROS induce activate AP-1 which is translocated to the nucleus. STAT-3 is phosphorylated by ROS. Activated STAT-3 is translocated to the nucleus. In the nucleus, NF-κB, AP-1, and STAT-3 turn on the expression of COX-2 in pulmonary epithelial cells. Pretreatment of GSH and NAC reduces ROS levels in LPS-stimulated cells. Consequently, they inhibit ROS-mediated activation of transcription factors and COX-2 expression in LPS-stimulated pulmonary epithelial cells. Therefore, pretreatment of GSH and NAC may be beneficial for prevention for LPS-mediated pulmonary inflammation. Since redox status is important for COX-2 mRNA stability and COX-2 enzyme activity, antioxidants treatment may decrease COX-2 activity and COX-2 expression by reducing ROS levels. Another possible mechanism of antioxidants for lowering COX-2 level is that antioxidants may stimulate proteasomal degradation of COX-2 in the stimulated A549 cells. Further study should be performed to determine whether thiol antioxidants affect COX-2 expression at post-transcriptional and post-translational levels in LPS-treated A549 cells.
In conclusion, the present study shows that LPS increases intracellular ROS levels and induces COX-2 expression in human pulmonary epithelial cells. LPS-derived ROS stimulate the activation of transcription factors such as NF-κB, AP-1, and STAT-3. As a result, pretreatment of GSH and NAC, which are classified as sulfur-containing antioxidants, inhibits the activation of transcription factors (NF-κB, AP-1, and STAT-3) related to inflammatory responses and COX-2 overexpression by reducing intracellular ROS levels. This investigation supports that treatment with GSH or NAC may prevent lung inflammatory diseases including pneumonia, asthma, and acute lung injury.
Abbreviations: AP-1, activator protein-1; CREB, cAMP response element-binding protein; COX, cyclooxygenase; DTT, dithiothreitol; EMSA, electrophoretic mobility shift assay; EDTA, ethylenediaminetetraacetic acid; GSH, glutathione; IKK, IkB kinase; LPS, lipopolysaccharide; NAC, N-acetyl-l-cysteine; NF-IL6, nuclear factor for interleukin-6; NF-κB, nuclear factor-κB; NFAT, nuclear factor of activated T-cells; PMSF, phenylmethylsulfonylfluoride; ROS, reactive oxygen species; STAT-3, signal transducer and activator of transcription-3; SP-1, specificity protein-1.
Acknowledgments: This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors. Sangyong Choi (S. Choi) performed the experiments, researched data, and drafted the manuscript. Joo Weon Lim (J.W. Lim) contributed to the analysis. Hyeyoung Kim (H. Kim) conceptualized the experiments and edited the manuscript. All authors read and approved the final version of the manuscript.
Conflict of interests: None declared.
REFERENCES
- Togbe D, Schnyder-Candrian S, Schnyder B, et al. Toll-like receptor and tumour necrosis factor dependent endotoxin-induced acute lung injury. Int J Exp Pathol 2007; 88: 387-391.
- Amin AN, Cerceo EA, Deitelzweig SB, et al. The hospitalist perspective on treatment of community-acquired bacterial pneumonia. Postgrad Med 2014; 126: 18-29.
- Eisner MD, Thompson T, Hudson LD, et al. Efficacy of low tidal volume ventilation in patients with different clinical risk factors for acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 2001; 164: 231-236.
- Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nat Rev Immunol 2003; 3: 169-176.
- Palsson-McDermott EM, O’Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 2004; 113: 153-162.
- Zhang P, Summer WR, Bagby GJ, et al. Innate immunity and pulmonary host defense. Immunol Rev 2000; 173: 39-51.
- N’Guessan PD, Hippenstiel S, Etouem MO, et al. Streptococcus pneumoniae induced p38 MAPK- and NF-k-dependent COX-2 expression in human lung epithelium. Am J Physiol Lung Cell Mol Physiol 2006; 290: L1131-L1138.
- Ahn EK, Yoon HK, Jee BK, et al. COX-2 expression and inflammatory effects by diesel exhaust particles in vitro and in vivo. Toxicol Lett 2008; 176: 178-187.
- Taddei A, Fabbroni V, Pini A, et al. Cyclooxygenase-2 and inflammation mediators have a crucial role in reflux-related esophageal histological changes and Barrett’s esophagus.. Dig Dis Sci 2014; 59: 949-957.
- Vancheri C, Mastruzzo C, Sortino MA, et al. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004; 25: 40-46.
- Park GY, Christman JW. Involvement of cyclooxygenase-2 and prostaglandins in the molecular pathogenesis of inflammatory lung diseases. Am J Physiol Lung Cell Mol Physiol 2006; 290: L797-L805.
- Kim HS, Ye SK, Cho IH, et al. 8-hydroxydeoxyguanosine suppresses NO production and COX-2 activity via Rac1/STATs signaling in LPS-induced brain microglia. Free Radic Biol Med 2006; 41: 1392-1403.
- Koay MA, Christman JW, Segal BH, et al. Impaired pulmonary NF-κB activation in response to lipopolysaccharide in NADPH oxidase-deficient mice. Infect Immun 2001; 69: 5991-5996.
- Rahman I. Oxidative stress, chromatin remodeling and gene transcription in inflammation and chronic lung diseases. J Biochem Mol Biol 2003; 36: 95-109.
- Stokes SE, Winn LM. NF-κB signaling is increased in HD3 cells following exposure to 1,4-benzoquinone: role of reactive oxygen species and p38-MAPK. Toxicol Sci 2014; 137: 303-310.
- Gloire G, Legrand-Poels S, Piette J. NF-κB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 2006; 72: 1493-1505.
- Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacol Ther 1991; 51: 155-194.
- Aruoma OI, Halliwell B, Hoey BM, et al. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med 1989; 6: 593-597.
- Zafarullah M, Li WQ, Sylvester J, et al. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci 2003; 60: 6-20.
- Huang RF, Huang SM, Lin BS, et al. N-acetylcysteine, vitamin C and vitamin E diminish homocysteine thiolactone-induced apoptosis in human promyeloid HL-60 cells. J Nutr 2002; 132: 2151-2156.
- Li J, Quan N, Bray TM. Supplementation of N-acetylcysteine normalizes lipopolysaccharide-induced nuclear factor kB activation and proinflammatory cytokine production during early rehabilitation of protein malnourished mice. J Nutr 2002; 132: 3286-3292.
- Cantin AM, Hartl D, Konstan MW, Chmiel JF. Inflammation in cystic fibrosis lung disease: pathogenesis and therapy. J Cyst Fibros 2015; 14: 419-430.
- Chung SW, Chung HY, Toriba A, et al. An environmental quinoid polycyclic aromatic hydrocarbon, acenaphthenequinone, modulates cyclooxygenase-2 expression through reactive oxygen species generation and nuclear factor kB activation in A549 cells. Toxicol Sci 2007; 95: 348-355.
- Hwang S, Lim JW, Kim H. Inhibitory effect of lycopene on amyloid-beta-induced apoptosis in neuronal cell. Nutrients 2017; 9: E883. doi: 10.3390/nu9080883
- Park, B. Lim JW, Kim H. Lycopene treatment inhibits activation of Jak/Stat3 and Wnt/beta-catenin signaling and attenuates hyper-proliferation in gastric epithelial cells. Nutr Res 2018. http://doi.org/10.1016/j.nutres.2018.07.010
- Chaby R, Garcia-Verdugo I, Espinassous Q, et al. Interactions between LPS and lung surfactant proteins. J Endotoxin Res 2005; 11: 181-185.
- Park HS, Jung HY, Park EY, et al. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-κB. J Immunol 2004; 173: 3589-3593.
- Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 2007; 43: 332-347.
- Amara N, Bachoual R, Desmard M, et al. Diesel exhaust particles induce matrix metalloprotease-1 in human lung epithelial cells via a NADP(H) oxidase/NOX4 redox-dependent mechanism. Am J Physiol Lung Cell Mol Physiol 2007; 293: L170-L181.
- Fink K, Duval A, Martel A, Soucy-Faulkner A, Grandvaux N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-κB in airway epithelial cells. J Immunol 2008; 180: 6911-6922.
- Rahman I, Mulier B, Gilmour PS, et al. Oxidant-mediated lung epithelial cell tolerance: the role of intracellular glutathione and nuclear factor-kB. Biochem Pharmacol 2001; 62: 787-794.
- DiDonato JA, Hayakawa M, Rothwarf DM, Zandi E, Karin M. A cytokine-responsive IkappaB kinase that activates the transcription factor NF-κB. Nature 1997; 388: 548-554.
- Karin M, Takahashi T, Kapahi P, et al. Oxidative stress and gene expression: the AP-1 and NF-κB connections. Biofactors 2001; 15: 87-89.
- Berkelhamer SK, Farrow KN. Developmental regulation of antioxidant enzymes and their impact on neonatal lung disease. Antioxid Redox Signal 2014; 21: 1837-1848.
- Sanlioglu S, Williams CM, Samavati L, et al. Lipopolysaccharide induces Rac1-dependent reactive oxygen species formation and coordinates tumor necrosis factor-alpha secretion through IKK regulation of NF-κB. J Biol Chem 2001; 276: 30188-30198.
- Chen CY, Peng WH, Tsai KD, et al. Luteolin suppresses inflammation-associated gene expression by blocking NF-κB and AP-1 activation pathway in mouse alveolar macrophages. Life Sci 2007; 81: 1602-1614.
- Lin CM, Huang ST, Liang YC, et al. Isovitexin suppresses lipopolysaccharide-mediated inducible nitric oxide synthase through inhibition of NF-kappa B in mouse macrophages. Planta Med 2005; 71: 748-753.
- Darnell JE. STATs and gene regulation. Science 1997; 277: 1630-1635.
- Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol 1998; 275: C1640-C1652.
- Carballo M, Conde M, El Bekay R, et al. Oxidative stress triggers STAT3 tyrosine phosphorylation and nuclear translocation in human lymphocytes. J Biol Chem 1999; 274: 17580-17586.
- Konstantinopoulos PA, Vandoros GP, Karamouzis MV, Gkermpesi M, Sotiropoulou-Bonikou G, Papavassiliou AG. EGF-R is expressed and AP-1 and NF-κB are activated in stromal myofibroblasts surrounding colon adenocarcinomas paralleling expression of COX-2 and VEGF. Cell Oncol 2007; 29: 477-482.
- Mitchell JA, Belvisi MG, Akarasereenont P, et al. Induction of cyclo-oxygenase-2 by cytokines in human pulmonary epithelial cells: regulation by dexamethasone. 1994; 113: 1008-1014.
- Xiao L, Ornatowska M, Zhao G, et al. Lipopolysaccharide-induced expression of microsomal prostaglandin E synthase-1 mediates late-phase PGE2 production in bone marrow derived macrophages. PLoS One 2012; 7: e50244. doi: 10.1371/journal.pone.0050244
- Britt RD, Locy ML, Tipple TE, Nelin LD, Rogers LK. Lipopolysaccharide-induced cyclooxygenase-2 expression in mouse transformed Clara cells. Cell Physiol Biochem 2012; 29: 213-222.
- Neuss H, Huang X, Hetfeld BK, et al. The ubiquitinand proteasome-dependent degradation of COX-2 is regulated by the COP9 signalosome and differentially influenced by coxibs. J Mol Med (Berl) 2007; 85: 961-970.
- Tetsuka T, Baier LD, Morrison AR. Antioxidants inhibit interleukin-1-induced cyclooxygenase and nitric-oxide synthase expression in rat mesangial cells. Evidence for post-transcriptional regulation. J Biol Chem 1996; 271: 11689-11693.
- Park HR, Loch-Caruso R. Protective effect of (±)a-tocopherol on brominated diphenyl ether-47-stimulated prostaglandin pathways in human extravillous trophoblasts in vitro. Toxicol in vitro 2015; 29: 1309-1318.
- Jiang Q, Elson-Schwab I, Courtemanche C, Ames BN. Gamma-tocopherol and its major metabolite, in contrast to alpha-tocopherol, inhibit cyclooxygenase activity in macrophages and epithelial cells. Proc Natl Acad Sci USA 2000; 97: 11494-11499.
- Wu D, Mura C, Beharka AA, et al. Age-associated increase in PGE2 synthesis and COX activity in murine macrophages is reversed by vitamin E. Am J Physiol 1998; 275: C661-C668.
- Kulmacz RJ, Wang LH. Comparison of hydroperoxide initiator requirements for the cyclooxygenase activities of prostaglandin H synthase-1 and -2. J Biol Chem 1995; 270: 24019-2423.
- Wu D, Hayek MG, Meydani S. Vitamin E and macrophage cyclooxygenase regulation in the aged. J Nutr 2001; 131: 382S-388S.
- Atmaca G. Antioxidant effects of sulfur-containing amino acids. Yonsei Med J 2004; 45: 776-788.
- Jafari B, Ouyang B, Li LF, Hales CA, Quinn DA. Intracellular glutathione in stretch-induced cytokine release from alveolar type-2 like cells. Respirology 2004; 9: 43-53.
- Poole LB, Schoneich C. Introduction: what we do and do not know regarding redox processes of thiols in signaling pathways. Free Radic Biol Med 2015; 80: 145-147.
- Klatt P, Molina EP, De Lacoba MG, et al. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J 1999; 13: 1481-1490.
- Hill BG, Bhatnagar A. Protein S-glutathiolation: redox-sensitive regulation of protein function. J Mol Cell Cardiol 2012; 52: 559-567.
- Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem 2000; 267: 4928-4944.
- Mokry J, Urbanova A, Medvedova I, et al. Effects of tadalafil (PDE5 inhibitor) and roflumilast (PDE4 inhibitor) on airway reactivity and markers of inflammation in ovalbumin-induced airway hyperresponsiveness in guinea pigs. J Physiol Pharmacol 2017; 68: 721-730.
- Mikolka P, Kopincova J, Tomcikova Mikusiakova L, et al. Effects of surfactant/budesonide therapy on oxidative modifications in the lung in experimental meconium-induced lung injury. J Physiol Pharmacol 2016; 67: 57-65.
- Mokra D, Drgova A, Mokry J, Antosova M, Durdik P, Calkovska A. N-acetylcysteine effectively diminished meconium-induced oxidative stress in adult rabbits. J Physiol Pharmacol 2015; 66: 101-110.
- Sun S, Zhang H, Xue B, et al. Protective effect of glutathione against lipopolysaccharide-induced inflammation and mortality in rats. Inflamm Res 2006; 55: 504-510.
- Mitsopoulos P, Omri A, Alipour M, Vermeulen N, Smith MG, Suntres ZE. Effectiveness of liposomal-N-acetylcysteine against LPS-induced lung injuries in rodents. Int J Pharm 2008; 363: 106-111.
- Dannenberg AJ, Altorki NK, Boyle JO, et al. Cyclooxygenase 2: a pharmacological target for the prevention of cancer. Lancet Oncol 2001; 2: 544-551.
- Shao J, Sheng H, Inoue H, et al. Regulation of constitutive cyclooxygenase-2 expression in colon carcinoma cells. J Biol Chem 2000; 275: 33951-33956.
- Kosaka T, Miyata A, Ihara H, et al. Characterization of the human gene (PTGS2) encoding prostaglandin-endoperoxide synthase 2. Eur J Biochem 1994; 221: 889-897.
- Reddy ST, Wadleigh DJ, Herschman HR. Transcriptional regulation of the cyclooxygenase-2 gene in activated mast cells. J Biol Chem 2000; 275: 3107-3113.
A c c e p t e d : August 30, 2018