BENEFICIAL ANTI-INFLAMMATORY EFFECT OF ANGIOTENSIN-CONVERTING ENZYME INHIBITOR AND ANGIOTENSIN RECEPTOR BLOCKER IN THE TREATMENT OF DEXTRAN SULFATE SODIUM-INDUCED COLITIS IN MICE
2Faculty of Veterinary Medicine, Department of Basic Veterinary Sciences, University of Helsinki, Helsinki, Finland
INTRODUCTION
The renin-angiotensin system (RAS) is a critical regulator of blood pressure but it has several other functions both systemically and locally. For example, RAS activation promotes inflammation and fibrosis in many tissues, including the intestine (1, 2). The majority of RAS’s blood-pressure elevating and pro-inflammatory properties are mediated by angiotensin II (Ang II) and its receptor, angiotensin II receptor subtype 1 (AT1R). Ang II is produced from angiotensin I mainly by the activity of angiotensin-converting enzyme (ACE). RAS participates in the regulation of gastrointestinal inflammation (1, 3) and has been linked to human inflammatory bowel diseases (IBD) (4-7). The expression of angiotensinogen is reduced in active disease sites in ileal Crohn’s disease (6) but certain angiotensinogen (AGT) and ACE gene variants are more prevalent in IBD patients (5, 8). The levels of angiotensin I and II are increased in the colons of Crohn’s disease patients (4), which could contribute to fibrosis in IBD via Ang II-mediated induction of transforming growth factor beta1 (TGF-β1) (2). The pro-inflammatory ACE - Ang II - AT1R arm is generally referred to as the classical RAS. Components of an alternative RAS, ACE2 - angiotensin 1-7 (Ang 1-7) - Mas receptor, are considered anti-inflammatory, and their levels are elevated in the blood of IBD patients (9).
The chemically-induced inflammation in DSS colitis is claimed to resemble human ulcerative colitis (UC) morphologically and symptomatically and offers interesting insights to IBD pathogenesis, nevertheless also with marked differences (10). Pharmacological inhibition of classical RAS is capable of alleviating even severe colitis in experimental animal models. Several drugs which were originally developed as antihypertensives, including ACE inhibitors, e.g. enalapril (11), enalaprilat (12-15), captopril (16, 17) and lisinopril (17) as well as angiotensin receptor blockers (ARBs), valsartan (18), candesartan (19, 20), losartan and its analogs (15, 20-24), olmesartan (25) and telmisartan (26) have been demonstrated to prevent and alleviate dextran sodium sulfate (DSS) and trinitrobenzene sulfonic acid (TNBS) -induced colitis in mice and rats. In addition, knockout of angiotensinogen (21) or angiotensin II receptor 1 (AT1R) (27) has been demonstrated to protect mice from colitis, whereas mice overexpressing renin are more susceptible to TNBS-induced colitis (24). Furthermore, Ang 1-7 administration has exerted beneficial effects on DSS-induced colitis (28) and in experimental model of gastric ulcers (29).
Despite the evidence emerging from preclinical studies highlighting the benefits of ACE inhibitors and ARBs in experimental colitis, the use of either drug as a treatment for IBD has not been investigated to any significant extent. There are rather few studies describing human data and none of them are randomized controlled trials. In one study, IBD patients on ARB therapy exhibited a lower expression of several pro-inflammatory cytokines and chemokines in their intestinal mucosa compared to those who did not receive ARBs (24). In contrast, in another retrospective study, presented as a conference abstract, Jacobs et al. (30) noted that in IBD patients, ACE inhibitor and ARB use was associated with higher all-cause hospitalization and higher corticosteroid use. In addition to inflammation, IBD can lead to severe fibrosis, and therefore a reduction of angiotensin II signalling could offer additional benefits for IBD patients by reducing scarring and the formation of strictures (2, 31). This proposal is supported by preclinical studies in which both ACE inhibitors and ARBs were able to prevent colitis-induced extracellular matrix accumulation and fibrosis in rats (16, 22). In addition, intestinal epithelial cell apoptosis has been indicated in IBD (32, 33), and ACE inhibitors and ARBs have been reported to reduce apoptosis rate in intestinal epithelium in preclinical colitis studies (11, 12). Combination therapy of ACE inhibitors and ARBs can potentially have synergistic effects and strengthen the anti-inflammatory and tissue-protective properties of these medications in alleviation of intestinal inflammation. Pharmacological and biological treatments for IBD have their own shortcomings, i.e. adverse effects and the development of drug resistance, leading to a declining effect and relapse (34, 35). Therefore, preclinical studies aimed at the modulation of intestinal RAS remain an important study target while paving the way to clinical studies.
We have previously reported that the ACE ectodomain is shed from the intestine during DSS-induced colitis (36). Furthermore, the ACE inhibitor, captopril, reduced the gene expression of angiotensinogen, Ace and Cyp11b1, the gene coding for the rate-limiting enzyme of corticosterone synthesis, in colon (37), and intestinal corticosterone synthesis was increased by Ang II (36). However, we could not detect any amelioration of colitis after oral captopril treatment (37). Interestingly, we have found that mesenchymal stem cell therapy reduces protein levels of pro-inflammatory ACE in colon of mice in the DSS model (Salmenkari et al., unpublished data).
Intestinal RAS clearly plays a part in human and experimental colitis. In this preclinical study, our aim was to compare the efficacy of orally given ACE inhibitor, enalapril, the ARB, losartan, and their combination in alleviating DSS-induced colitis and local production of anti-inflammatory corticosterone production in mice. We measured the gene expression of several RAS components and ACE protein and corticosterone synthesis in the colons of the mice.
MATERIALS AND METHODS
Animals
The study was approved by National Animal Experimentation Committee of Finland (ESAVI/114/04.10.07/2015) according to EC Directive 86/609/ECC and Finnish Experimental Animal Act 62/2006.
A total of 40 Balb/C male mice were obtained from Envigo (Horst, Netherlands) at the age of 7 weeks. The mice were housed in individually-ventilated cages in specific-pathogen free laboratory conditions with 12-h light/dark cycle. The animals were provided with standard rodent feed, 2018 Teklad Global 18% Protein Rodent Diet (Harlan Laboratories, Indianapolis, IN, USA) and drinking water ad libitum.
Administration of medications and induction of colitis
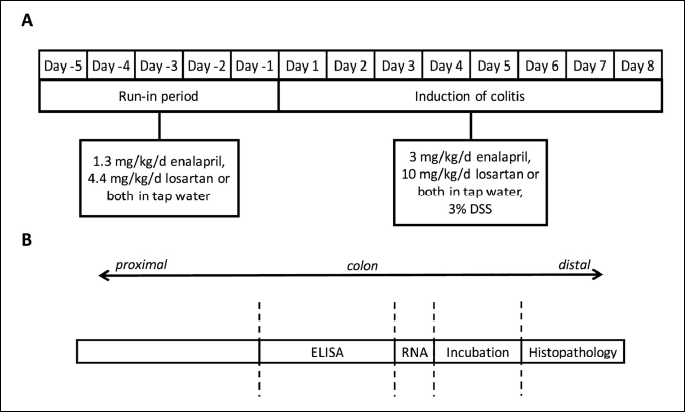
The mice were assigned into five experimental groups (n = 8 in each group) with similar starting weights (23.5 ± 0.1 g). At the beginning of the experiment, the mice in the intervention groups were given orally in drinking water either 11 mg/l enalapril (PHR1289, Sigma Aldrich, St. Louis, MO, USA), 37 mg/l losartan (PHR1602, Sigma Aldrich), corresponding to daily doses of approximately 1.3 mg/kg/d of enalapril and 4.4 mg/kg/d of losartan, respectively, or a combination of both drugs for 5 days to acquaint the animals to the medications. Subsequently, 3% (w/vol) 40 kDa DSS (TdB Consultancy, Uppsala, Sweden) was added to the drinking water of the three intervention groups and a DSS-control group for 7 days, and the dosing of medications in the intervention groups were concurrently increased to 25 mg/l enalapril and 83 mg/l losartan, corresponding to daily doses of approximately 3 mg/kg/d enalapril and/or 10 mg/kg/d losartan, respectively. These doses are based on effective doses reported in other publications (11, 23). The study design is presented in Fig. 1A. The healthy controls received water throughout the experiment. The animals were weighed daily throughout the experiment. The daily fluid intake was measured, and the intake was not affected by addition of either the drugs or DSS. At the end of the experiment, the animals were sacrificed by exsanguination under isoflurane anaesthesia.
Macroscopic evaluation of colitis
The macroscopic scoring criteria was adapted from Holma et al., and Melgar et al., (38, 39). The colons of mice were excised, and the lengths were measured. The colons were opened longitudinally and the stool consistency was evaluated on a scale from 0 to 4 (0 = normal, 1 = moist, 2 = loose, 3 = liquid, 4 = none) and the presence of blood was scored on a scale from 0 to 3 (0 = none, 1 = streaks, 2 = clearly visible, 3 = heavy bleeding). The colons were cleaned of intestinal contents and edema was scored on a scale of 0 to 3 (0 = normal, 1 = mild, 2 = moderate and 3 = marked). These scores were combined as a total macroscopic score (scale 0 – 10). The sample collection schematic is presented in Fig. 1B.
Histopathologic evaluation of colitis
Pieces of the most distal part of colon were fixed in 10% neutral-buffered formalin (Sigma Aldrich) for 24 hours, embedded longitudinally into paraffin blocks, sectioned at 4 µm thickness, and stained with hematoxylin and eosin (H&E). Histopathological changes were assessed in a partly blinded manner; the pathologist (JL) was aware of the group of each animal, but unaware of the group identities and treatments. Briefly, surface epithelial injury, crypt epithelial injury, and crypt dilatation and distortion were graded on a scale of 0 – 3 each and combined into an injury score (scale 0 – 9). The presence of granulocytes was graded on a scale of 0 – 3, the presence of lymphocytes and macrophages on a scale of 0 – 4, and the number and extent of granulomatous inflammatory foci (granulomas) on a scale of 0 – 2 and combined into an inflammation score (scale 0 – 9). Finally, the injury and inflammation scores were combined as the histopathology scores (scale 0 – 18). The grading was simplified from a grading system used in a study examining chemotherapy-induced gastrointestinal toxicity in rats (40) and employed lesion definitions generally used in toxicological pathology (41). Criteria for grading are detailed in Table 1.
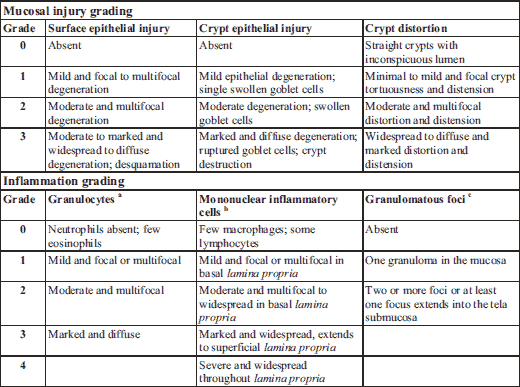
b Macrophages, lymphocytes and plasma cells (very few) were graded together as mononuclear inflammatory cells.
c Granulomatous foci (granulomas) consisted of a dense aggregation of macrophages encircled basally and laterally by lymphocytes. The macrophages were superficially admixed with variable number of neutrophils infiltrating into the surface epithelium. Some foci were ulcerated.
Ex-vivo incubation samples of colon
Ex-vivo incubation samples were prepared as previously described (37). Briefly, pieces of distal colon, adjacent to the histological samples, were incubated in a pre-oxygenated Krebs buffer (119 mmol/l NaCl, 25 mmol/l NaHCO3, 15 mmol/l KCl, 11 mmol/l glucose, 1.6 mmol/l CaCl2, 1.2 mmol/l KH2PO4, 1.2 mmol/l MgSO4) for 90 min at 37°C with gentle agitation. The samples were centrifuged at 13,300 g for 3 min. The supernatant was used for ACE protein (#DY1513 R&D System, Minnesota, MO, USA) and corticosterone (#501320 Cayman Chemical, Michigan, MI, USA) assays, and the tissue pieces for protein quantification (Pierce™ BCA Protein Assay Kit, Thermo Scientific).
Angiotensin-converting enzyme protein quantification from colonic tissue
Tissue pieces of mid-colon were homogenized in 100 mM Tris - 150 mM NaCl, pH 8.3 using a Precellys 24 cryobead homogenizer (Bertin Technologies, Montigny le Bretonneux, France). The samples were centrifuged at +4°C for 20 min and ACE protein and total protein were quantified from the supernatant (#DY1513 R&D System and Pierce™ BCA Protein Assay Kit, Thermo Scientific).
Reverse-transcription quantitative PCR
RNA was extracted from mid-distal colon using NucleoSpin RNA kit (#740955, Macherey Nagel, Duren, Germany), and reverse transcribed to cDNA using iScript™ cDNA Synthesis Kit (#1708891, BioRad, Hercules, CA, USA). RT-qPCR was run using LightCycler® 480 SYBR Green I Master (#04707516001, Roche Diagnostics Corp., Indianapolis, IN, USA). The primers were ordered from Sigma-Aldrich (St- Louis, MO, USA).
The primer sequences used for the study are:
Rplp0 F: 5’-TAACCCTGAAGTGCTCGACA-3’,
R: 5’-GGTACCCGATCTGCAGACA-3’;
S18 F: 5’-AACGAACGAGACTCTGGCAT-3’,
R: 5’-ACGCCACTTGTCCCTCTAAG-3’;
Eef2 F: 5’-TGTCAGTCATCGCCCATGTG-3’,
R: 5’-CATCCTTGCGAGTGTCAGTGA-3’;
IL-1b F: 5’-CTCCAGCCAAGCTTCCTTGT-3’,
R: 5’-TCATCACTGTCAAAAGGTGGCA-3’;
Tnf-a F: 5’-GCCTCTTCTCATTCCTGCTTG-3’,
R: 5’- CTGATGAGAGGGAGGCCATT-3’ (42);
Ace F: 5’-GCTGGAGGGTCTTTGATGGA-3’,
R: 5’-AGTCACCTTGGGATCTTGGC-3’;
Agt F: 5’-CTTCCAAGGAACGATGAGAGGTT-3’,
R: 5’-ACAGACACCGAGATGCTGTT-3’;
Cyp11b1 F: 5’-GGAACCCACCATCAGTAAGGA-3’,
R: 5’-TCTTCCCTCACGCATGACAA-3’;
Lrh-1 F: 5’-GCATGGGAAGGAAGGGACAA-3’,
R: 5’-CGCTGATCGAACTGAAGGGA-3’.
Target gene expression was normalized to the geometric mean of Rplp0, S18 and Eef2 expression.
Statistical analysis
RT-qPCR data is presented in graphs as individual values and geometric mean, and in the text, as percent increase or decrease of the geometric means. All other parametric data is presented as mean ± SD in the text and individual values and mean in the graphs. Non-parametric data is presented as median and interquartile range (IQR) in the tables. Statistical analyses were conducted with SPSS versions 22 or 23. The differences between groups were analyzed using either one-way ANOVA with Tukey’s test or Kruskal-Wallis and Mann-Whitney U test. P-values lower than 0.05 were considered statistically significant.
RESULTS
Macroscopic disease activity is reduced by losartan
DSS induced a relatively mild colitis during the 7-day DSS administration. During that time, the body weight of the mice remained close to the initial weight in all groups and by the end of the trial, the weight had declined only slightly in the colitis groups (Fig. 2A). DSS induced diarrhea, swelling of the colon and colonic bleeding in all colitis groups, which were measured as macroscopic scores (Table 2). All colitis groups had elevated macroscopic scores (P < 0.01 for all groups) when compared with the healthy mice. The macroscopic scores were lower in the losartan group compared with the DSS-control group (P < 0.05) but otherwise the colitis groups did not statistically significantly differ from each other (Table 2).
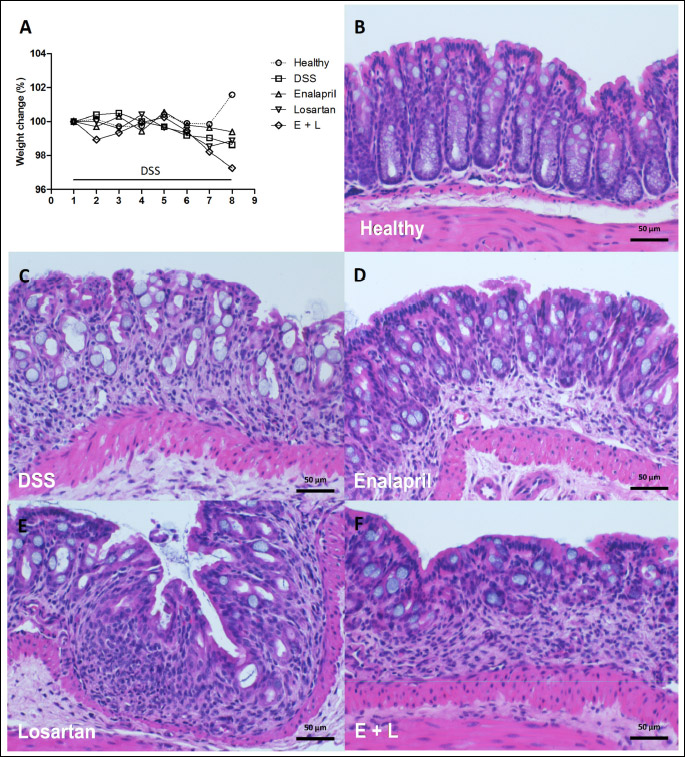
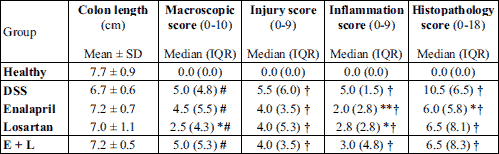
Alleviation of histopathologic colitis by enalapril and losartan
The histopathological changes induced by DSS were relatively mild (Fig. 2B-2F, Table 2). Injury, inflammation and histopathology scores were higher in all colitis groups (P < 0.001 for all groups) when compared with healthy controls (Table 2). Both enalapril (P = 0.007) and losartan (P = 0.038) alone, but not their combination, reduced the inflammatory infiltrate i.e. the inflammation score in colon when compared with the DSS control group (Table 2). The lower inflammation scores were mainly due to reduced number of neutrophils both in enalapril- and losartan-treated mice, and by a lower number of granulomatous foci in enalapril-treated mice. The treatments had no statistically significant impact on the injury scores. Overall, the reduction in histopathology score remained significant in the enalapril group when compared with DSS controls (P = 0.021) but the reduction attributable to either losartan or the combination of enalapril and losartan was not statistically significant. Furthermore, the differences between enalapril and losartan or the combination group were not statistically significant.
Enalapril reduces colonic IL-1b following dextran sodium sulfate colitis
In the next part of the experiment, we evaluated whether the medications influenced any of the molecular markers of inflammation by measuring the levels of Il-1β and Tnf-α mRNA in the colons of the mice (Fig. 3A). DSS increased the expression of IL-1β mRNA by 173% compared with healthy controls (almost statistically significant, P = 0.060). The expression was also increased by 149% in the losartan and by 254% in the combination group (P = 0.040 and 0.002, respectively) compared with healthy controls. In comparison with the DSS controls, enalapril, but not losartan or the combination treatment, reduced IL-1β expression by 72% (P = 0.020). The level of IL-1β expression was similar between DSS controls, losartan and the combination groups, but the expression was lower in enalapril group than in the losartan (P = 0.012) or the combination groups (P = 0.001). The expression pattern of Tnf-α was similar to IL-1β, but there were no statistically significant differences between the groups (Fig. 3B). The geometric mean was 376% higher in the DSS group than in the healthy group and 37% and 40% lower in the enalapril and the combination groups when compared with the DSS-control group, respectively. The expression was 37% higher in the losartan group than the DSS controls.
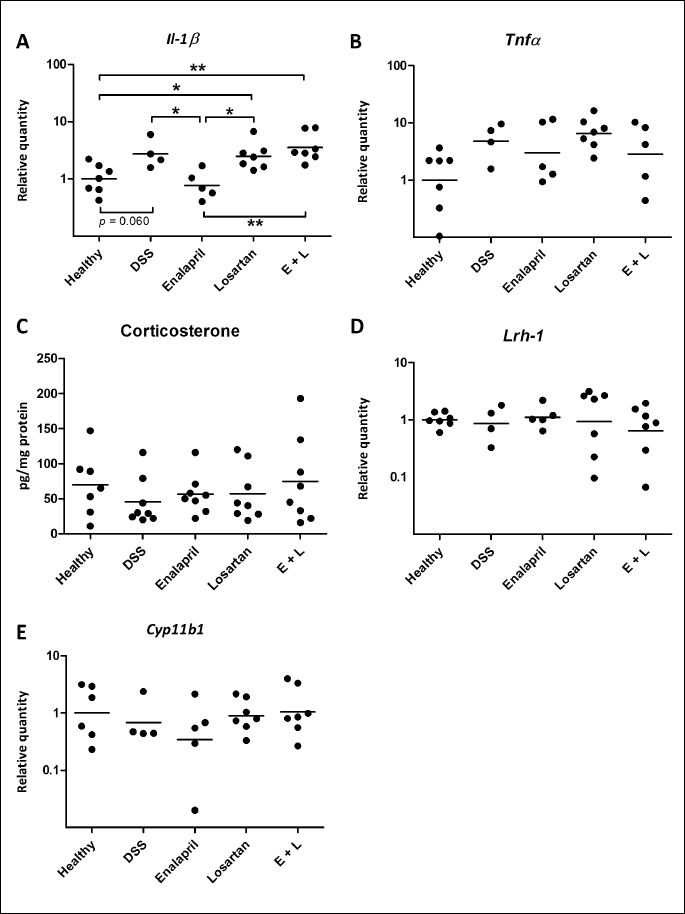
Anti-inflammatory corticosterone production was not affected by colitis or interventions in distal colon
In order to investigate whether ACE inhibition or AT1R blockade affects colonic corticosterone synthesis pathway, we measured corticosterone production and the gene expression of the transcription factor liver receptor homolog-1 (Lrh-1), which drives intestinal glucocorticoid synthesis, and the rate-limiting enzyme of corticosterone synthesis Cyp11b1 in the colons of the mice (Fig. 3C-3E). Corticosterone production in distal colon was measured from intestinal incubation supernatants and the expression levels of Lrh-1 and Cyp11b1 were measured from the adjacent mid-distal colon segment (Fig. 1B). Neither DSS colitis nor the medications exerted any influence on corticosterone secretion (Fig. 3C) or Lrh-1 (Fig. 3D) or Cyp11b1 (Fig. 3E) gene expression.
Dextran sodium sulfate induces angiotensin-converting enzyme shedding in distal colon
ACE protein was quantified from incubation supernatants of distal colon (Figs. 1B and 4A) and tissue segments of mid-colon (Figs. 1B and 4B) in order to examine whether the medications had influenced the levels of ACE in colon while Ace gene expression was quantified in mid-distal colon (Figs. 1B and 4C). The ACE-ectodomain shedding was increased in all colitis groups (9.8 ± 4.7 ng/mg protein, P = 0.032 in DSS-control, 13.1 ± 4.6 ng/mg, P < 0.001 in enalapril, 12.1 ± 3.0 ng/mg, P = 0.001 in losartan and 11.3 ± 2.5 ng/mg, P = 0.005 in the combination group samples) compared with healthy controls (3.8 ± 1.5 ng/mg), and it was not altered by any of the interventions (Fig. 4A). In contrast, Ace expression and tissue-bound ACE levels were not modulated by DSS or by any of the treatments (Fig. 4B, 4C). Furthermore, the gene expression of angiotensinogen did not differ between the groups (Fig. 4D). Additionally, we measured ACE shedding in distal ileum, but this was not affected by DSS or the medications (data not shown).
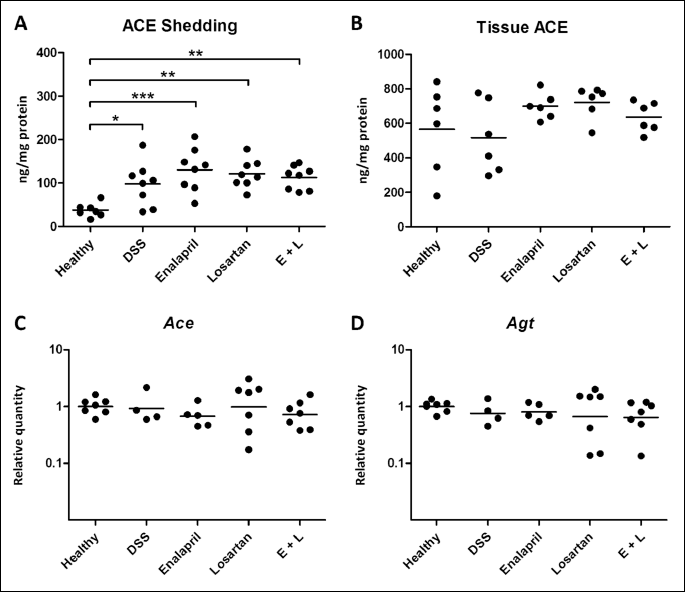
Abbreviations: DSS, dextran sodium sulfate, E + L, enalapril + losartan combination group.
DISCUSSION
Modulation of RAS activity has been beneficial in the treatment of experimental colitis (11-23, 25, 26). Therefore, we compared two common drugs, the ACE inhibitor, enalapril, and the ARB, losartan, and their combination in alleviation of DSS-induced colitis in mice. Several ACE inhibitors and ARBs and different administration regimens have been evaluated in earlier studies, but as far as we are aware, previously they have not been mutually compared or examined in combination in the treatment of colitis. We observed an amelioration of colitis by both ACE inhibition and ARB alone but not by their combination. Our findings support the beneficial role of ACE inhibitors and ARBs in murine intestinal inflammation and reveal some potential differences in their disease-alleviating outcomes but indicate there are no added benefits in combining these drugs, in fact, there may be disadvantages.
The histopathological changes in DSS colitis in the acute phase are described to encompass goblet cell depletion, erosion and ulceration as well as infiltration of granulocytes into the lamina propria and submucosa (10, 43, 44). Our results demonstrate that both pharmacological ACE inhibition as well as angiotensin receptor blockade can reduce the severity of colitis by reducing colonic inflammatory cell infiltration. More specifically, both drug classes reduced the number of infiltrating neutrophils and enalapril reduced the number of granulomas. One possible explanation is that there is a reduction in leukocyte recruitment. Ang II promotes leukocyte homing into many inflamed tissues including the intestine, which occurs via AT1R signalling followed by a subsequent increase in cell adhesion molecules (19, 45). Protein expression of MAdCAM-1, an important adhesion molecule in the pathogenesis of human IBD, was shown to be inhibited in AT1R knockout mice during DSS-induced colitis (19).
In acute disease model studies, drugs are commonly administered by injections or oral gavage because of the possibility of exact dosing of compounds to each animal and to avoid any possible risk of interaction with for example DSS. Multiple oral gavage treatments, however, can cause weight loss, esophageal tearing, or lowering of white blood cell counts of the animals (46), which would be problematic and confounding in colitis studies. Administration in the drinking fluid has the benefit of stable intake in case of drugs with short half-life like losartan. Therefore, we chose to investigate the effect of the drugs when given orally in drinking water. Nevertheless, the stability of the drugs in fluid and interactions with DSS is of concern and must be tested with every individual compound. Losartan has been previously administered in drinking water (23, 24) in TNBS model. Valsartan, another angiotensin receptor blocker with a similar chemical formula as losartan, has been employed together with DSS in drinking water by Santiago et al. (18). Enalapril was used successfully in drinking water in a diabetic kidney model by Sasaki et al. (47). In this study, we can conclude that both drugs had an anti-inflammatory effect when administered together with DSS.
The comparison of ACE inhibition and AT1R blockade made it possible to reveal the differences between the drugs in how they alleviate the histopathological, macroscopic and biochemical symptoms of colitis, as well as the apparent self-regulatory role of intestinal RAS observed in our earlier studies (37). On the other hand, we chose an experimental design with only a single dose, which does not allow a full pharmacological comparison of the therapeutic effect between ACE inhibition and AT1R blockade. Other groups have experimented on the dosages of both enalapril and losartan. Oral doses of 1 and 5 mg/kg/d of enalapril were tested by Lee et al., (11), and 5 mg/kg dose was more effective in the treatment of colitis. Oral doses of 7 (22), 10 (23) and 15 mg/kg/d (24) losartan were all effective against TNBS colitis. In our study, 3 mg/kg/d enalapril and 10 mg/kg of losartan represent effective but submaximal doses for mice, which could have therefore had synergistic effects in treatment of colitis. Nevertheless, enalapril treatment resulted in a lowering of the overall histopathologic scores and IL-1β expression, whereas administration of losartan reduced the macroscopic and histopathologic inflammation scores. This data demonstrates that both enalapril and losartan can alleviate colitis but suggest that the outcomes and obviously the are not identical. ACE inhibition exerted a stronger anti-inflammatory effect, but only ARB reduced the macroscopic manifestations (stool consistency, colonic edema and bleeding) of DSS colitis. Neither of the drug was able to prevent mucosal injury. A recent study found that losartan reduces the angiotensin II-dependent contractibility of both small and large intestine ex vivo (48). This could in part explain the beneficial effect of losartan on the macroscopic scores (especially stool consistency) in our study. It is possible that the dose of enalapril was more potent in alleviating DSS colitis than that of losartan, but losartan might have the additional benefit of reducing smooth muscle contractibility.
Both drug classes inhibit the classical proinflammatory RAS pathway, but due to the complexity of the RAS network the drugs might cause a shift in production of other angiotensin metabolites than Ang II, such as Ang 1-7 (3). ACE inhibition reduces Ang II formation, and to some degree, the breakdown of the anti-inflammatory Ang 1-7 (49). Ang II mainly acts via AT1R, although AT2R is constitutively expressed in the intestine, albeit to a lesser degree (50). AT1R signalling is among other things pro-inflammatory, and pro-apoptotic, as well as induces contractions of the smooth muscle cells both in the intestinal and vascular wall (1, 51). AT2R and the Mas receptor, the receptor for Ang 1-7, act in opposite or counteract the effects of AT1R signalling (1). Small angiotensinogen-derived peptides (e.g. from Ang 1-5 to Ang 1-2) have recently been found to activate Mas receptor (52). The complex network of peptides and receptors might be the underlying cause of the small differences between ACE inhibitors and ARBs.
Angiotensin receptor blockers have been reported to lower the colonic Il-1β and Tnf-α expression in several TNBS-colitis studies (21, 23, 25). In the DSS model, 10-fold higher doses than in our study, administered per rectum reduced proinflammatory cytokine expression (20), but we found no other mention of losartan’s or other ARB’s effects on colonic Il-1β in the DSS model. Some studies report ARBs decrease TNF-α levels in DSS model (18, 25). The relatively low dose of losartan might explain the lack of Il-1β reduction in our study.
We believed that simultaneous inhibition of ACE and AT1R blockade would lead to a more potent inhibition of AT1R signalling, and thus more efficient alleviation of colitis. However, the benefits of the drugs were lost when they were combined, suggesting that either the combination introduces additional adverse effects due to excessive RAS inhibition or, more unlikely, the drugs have some unknown interaction which cancels out the beneficial effects. Although we chose doses that were lower than the biggest effective doses for colitis found in literature (11, 24), reducing doses in combination therapy might have yielded better results similar to single medications. ACE inhibitors and ARBs are important antihypertensive drugs. We did not measure the blood pressure of the mice, but according to literature the chosen doses of enalapril and losartan are unlikely to cause hypotension for normotensive mice (53, 54). The combination of an ACE inhibitor and an ARB has not been studied in the context of experimental colitis or human IBD, but several clinical trials have been conducted regarding post-myocardial infarction therapy and diabetic nephropathy (55-58). Several studies reported that the combination therapy increased the occurrence of potentially serious adverse effects (56-58), including hyperkalemia and kidney injury, and only one reported that combination treatment reduced the risk of cardiovascular events (55). These results advocate caution for use in other disease areas like gastrointestinal medicine. However, several factors might distinguish the actions of either enalapril or losartan in different tissues, and therefore the results of cardiovascular studies might not directly apply in gastrointestinal tract. The degree and duration of ACE inhibition can differ between tissues, as described using lisinopril (59). Activation of AT1R might have different outcomes in the intestine than in the vessels due to selective G-protein coupling (60). In addition, some ACE inhibitors also inhibit other peptidases of the gut to some extent (61, 62), and together this could lead to reduced effect during combination treatment. However, our results show that combination therapy is unlikely to confer additional benefits in the treatment of colitis.
We have previously reported that the ACE inhibitor captopril lowered the gene expression of RAS components, Ace and angiotensinogen, and the rate-limiting enzyme in corticosterone synthesis, Cyp11b1 (37). In this study, we wanted to investigate whether a similar effect could be obtained with another ACE inhibitor, enalapril, or with an ARB. However, neither enalapril nor losartan exerted a similar effect on RAS modulation as captopril in acute/subacute DSS-induced colitis and the treatments did not affect the expression levels of these genes. There could be several reasons for these discrepancies; differences in the severity of colitis, differences in the effective dose of ACE inhibitors, or structural differences between the ACE inhibitors, e.g. captopril’s sulfhydryl-moiety. There was a relatively mild colitis applied in this study, i.e. the animals experienced minimal weight loss, moderate macroscopic and histopathology scores and there was a lack of induction of corticosterone synthesis. Corticosterone is an anti-inflammatory hormone which is produced primarily in the adrenal gland but also locally throughout the intestine (63). In the intestine, glucocorticoid synthesis is regulated by the transcription factor, LRH-1 (64). During acute intestinal inflammation, TNF-α activates LRH-1 to promote the transcription of the genes of glucocorticoid synthesis e.g. the rate-limiting enzyme Cyp11b1 but the signal becomes suppressed during chronic inflammation (65-67). Here, despite the evident increase in the levels of pro-inflammatory IL-1β and the disease scores provoked by DSS, the colitis might have been too mild to induce corticosterone synthesis. Similarly, the gene expression levels of Lrh-1 and Cyp11b1 were not elevated by the DSS treatment.
ACE protein is shed from vascular endothelium, and probably also the intestinal epithelium, by ADAM9 protease (68). In addition to the vascular endothelium, ACE is prominently expressed in the intestine and in macrophages. We have determined that DSS induces ACE shedding in jejunum and to some extent in distal colon, but the shedding is not strongly induced in the proximal parts of colon (36, 37). In line with these results, ACE shedding was prominently increased by DSS in distal parts of colon, where the DSS-induced damage is most distinct. In distal ileum, the basal ACE shedding was not affected by DSS. Therefore, ACE shedding seems to take place in certain parts of the intestine in the presence of inflammatory stimuli. We hypothesized that the role of ACE shedding could be to regulate the local Ang II concentration either by downregulating cellular ACE or by directing soluble ACE to sites of inflammation. It would be interesting to determine whether the shedding increases in other colitis models as well or is it rather an indicator of the toxic damage evoked by DSS.
In conclusion, this study strengthens the evidence for the benefits of using ACE inhibitors or ARBs in the treatment of experimental colitis, but based on our results, combination therapy of ACE inhibitors and ARBs does not seem advisable. Although both ACE inhibition and angiotensin receptor blockade alleviate experimental colitis, their benefits seem to be partially dissimilar. Mechanistic studies of the anti-colitogenic effects of these two drug classes, and the possible involvement of the extended RAS, should be conducted to determine the most suitable treatment approach for gastrointestinal diseases. Clinical, and to some degree epidemiological, studies of ACE inhibitor and ARB use in IBD patients will be needed to translate the findings from experimental models into human therapeutics. In addition, it would be interesting to investigate the potential of ACE inhibitors and ARBs in the alleviation of established colitis.
Abbreviations: ACE, angiotensin-converting enzyme; ADAM9, A disintegrin and metalloproteinase domain-containing protein 9; Ang II, angiotensin II; Ang 1-7, = angiotensin 1-7; AGT, angiotensinogen; ARBs, angiotensin receptor blockers; AT1R, angiotensin receptor subtype 1; AT2R, angiotensin receptor subtype 2; DSS, dextran sodium sulfate; IBD, inflammatory bowel disease; IL-1β, interleukin 1beta; LRH-1, liver receptor homolog-1; RAS, renin-angiotensin system; TGF-β1, transforming growth factor beta1; TNBS, trinitrobenzene sulfonic acid; TNF-α, tumor necrosis factor alpha; UC, ulcerative colitis.
Author contributions: H. Salmenkari, H. Vapaatalo and R. Korpela designed the study plan; H. Salmenkari, J. Linden and L. Pasanen performed the experiments; H. Salmenkari analyzed the data; H. Salmenkari, J. Linden, H. Vapaatalo and R. Korpela wrote the paper.
Acknowledgements: We thank Helena Taskinen at Tissue Preparation and Histochemistry Unit, Medicum, Department of Anatomy, University of Helsinki for preparation of the H&E-stained tissue slides. We are grateful to Hanna Launonen at Medicum, Department of Pharmacology, University of Helsinki for her skilful technical assistance during this study. We sincerely thank Dr. Ewen MacDonald for the grammar and style revision. The study was funded by Juhani Aho Medical Research Foundation, Finland, and Finska Lakaresallskapet, Einar och Karin Stroems Stiftelse, Finland.
Conflict of interests: None declared.
REFERENCES
- Garg M, Angus PW, Burrell LM, Herath C, Gibson PR, Lubel JS. Review article: the pathophysiological roles of the renin-angiotensin system in the gastrointestinal tract. Aliment Pharmacol Ther 2012; 35: 414-428.
- Szabo H, Fiorino G, Spinelli A, et al. Review article: anti-fibrotic agents for the treatment of Crohn’s disease - lessons learnt from other diseases. Aliment Pharmacol Ther 2010; 31: 189-201.
- Brzozowski T. Role of renin-angiotensin system and metabolites of angiotensin in the mechanism of gastric mucosal protection. Curr Opin Pharmacol 2014; 19: 90-98.
- Jaszewski R, Tolia V, Ehrinpreis MN, et al. Increased colonic mucosal angiotensin I and II concentrations in Crohn’s colitis. Gastroenterology 1990; 98: 1543-1548.
- Hume GE, Fowler EV, Lincoln D, et al. Angiotensinogen and transforming growth factor beta1: novel genes in the pathogenesis of Crohn’s disease. J Med Genet 2006; 43: e51. Doi: 10.1136/jmg.2005.040477
- Hume GE, Doecke JD, Huang N, et al. Altered expression of angiotensinogen and mediators of angiogenesis in ileal Crohn’s disease. J Gastrointestin Liver Dis 2016; 25: 39-48.
- Hume GE, Radford-Smith GL. ACE inhibitors and angiotensin II receptor antagonists in Crohn’s disease management. Expert Rev Gastroenterol Hepatol 2008; 2: 645-651.
- Saibeni S, Spina L, Virgilio T, et al. Angiotensin-converting enzyme insertion/deletion gene polymorphism in inflammatory bowel diseases. Eur J Gastroenterol Hepatol 2007; 19: 976-981.
- Garg M, Burrell LM, Velkoska E, et al. Upregulation of circulating components of the alternative renin-angiotensin system in inflammatory bowel disease: a pilot study. J Renin Angiotensin Aldosterone Syst 2015; 16: 559-569.
- Eichele DD, Kharbanda KK. Dextran sodium sulfate colitis murine model: an indispensable tool for advancing our understanding of inflammatory bowel diseases pathogenesis. World J Gastroenterol 2017; 23: 6016-6029.
- Lee C, Chun J, Hwang SW, Kang SJ, Im JP, Kim JS. Enalapril inhibits nuclear factor-kappaB signaling in intestinal epithelial cells and peritoneal macrophages and attenuates experimental colitis in mice. Life Sci 2014; 95: 29-39.
- Spencer AU, Yang H, Haxhija EQ, Wildhaber BE, Greenson JK, Teitelbaum DH. Reduced severity of a mouse colitis model with angiotensin converting enzyme inhibition. Dig Dis Sci 2007; 52: 1060-1070.
- Koga H, Yang H, Adler J, Zimmermann EM, Teitelbaum DH. Transanal delivery of angiotensin converting enzyme inhibitor prevents colonic fibrosis in a mouse colitis model: development of a unique mode of treatment. Surgery 2008; 144: 259-268.
- Sueyoshi R, Ignatoski KM, Daignault S, Okawada M, Teitelbaum DH. Angiotensin converting enzyme-inhibitor reduces colitis severity in an IL-10 knockout model. Dig Dis Sci 2013; 58: 3165-3177.
- Okawada M, Wilson MW, Larsen SD, Lipka E, Hillfinger J, Teitelbaum DH. Blockade of the renin-angiotensin system prevents acute and immunologically relevant colitis in murine models. Pediatr Surg Int 2016; 32: 1103-1114.
- Wengrower D, Zanninelli G, Pappo O, et al. Prevention of fibrosis in experimental colitis by captopril: the role of tgf-beta1. Inflamm Bowel Dis 2004; 10: 536-545.
- Jahovic N, Ercan F, Gedik N, Yuksel M, Sener G, Alican I. The effect of angiotensin-converting enzyme inhibitors on experimental colitis in rats. Regul Pept 2005; 130: 67-74.
- Santiago OI, Rivera E, Ferder L, Appleyard CB. An angiotensin II receptor antagonist reduces inflammatory parameters in two models of colitis. Regul Pept 2008; 146: 250-259.
- Mizushima T, Sasaki M, Ando T, et al. Blockage of angiotensin II type 1 receptor regulates TNF-alpha-induced MAdCAM-1 expression via inhibition of NF-kappaB translocation to the nucleus and ameliorates colitis. Am J Physiol Gastrointest Liver Physiol 2010; 298: G255-G266.
- Okawada M, Koga H, Larsen SD, et al. Use of enterally delivered angiotensin II type Ia receptor antagonists to reduce the severity of colitis. Dig Dis Sci 2011; 56: 2553-2565.
- Inokuchi Y, Morohashi T, Kawana I, Nagashima Y, Kihara M, Umemura S. Amelioration of 2,4,6-trinitrobenzene sulphonic acid induced colitis in angiotensinogen gene knockout mice. Gut 2005; 54: 349-356.
- Wengrower D, Zanninelli G, Latella G, et al. Losartan reduces trinitrobenzene sulphonic acid-induced colorectal fibrosis in rats. Can J Gastroenterol 2012; 26: 33-39.
- Liu TJ, Shi YY, Wang EB, Zhu T, Zhao Q. AT1R blocker losartan attenuates intestinal epithelial cell apoptosis in a mouse model of Crohn’s disease. Mol Med Rep 2016; 13: 1156-1162.
- Shi Y, Liu T, He L, et al. Activation of the renin-angiotensin system promotes colitis development. Sci Rep 2016; 6: 27552. doi: 10.1038/srep27552
- Nagib MM, Tadros MG, ElSayed MI, Khalifa AE. Anti-inflammatory and anti-oxidant activities of olmesartan medoxomil ameliorate experimental colitis in rats. Toxicol Appl Pharmacol 2013; 271: 106-113.
- Arab HH, Al-Shorbagy MY, Abdallah DM, Nassar NN. Telmisartan attenuates colon inflammation, oxidative perturbations and apoptosis in a rat model of experimental inflammatory bowel disease. PLoS One 2014; 9: e97193. doi: 10.1371/journal.pone.0097193
- Katada K, Yoshida N, Suzuki T, et al. Dextran sulfate sodium-induced acute colonic inflammation in angiotensin II type 1a receptor deficient mice. Inflamm Res 2008; 57: 84-91.
- Khajah MA, Fateel MM, Ananthalakshmi KV, Luqmani YA. Anti-inflammatory action of angiotensin 1-7 in experimental colitis. PLoS One 2016; 11: e0150861. doi: 10.1371/ journal.pone.0150861
- Pawlik MW, Kwiecien S, Ptak-Belowska A, et al. The renin-angiotensin system and its vasoactive metabolite angiotensin-(1-7) in the mechanism of the healing of preexisting gastric ulcers. The involvement of Mas receptors, nitric oxide, prostaglandins and proinflammatory cytokines. J Physiol Pharmacol 2016; 67: 75-91.
- Jacobs J, Gulotta G, Liao C, Li YC, Bossonnette M, Pekow J. Angiotensin II blockade is associated with worse clinical outcomes in IBD patients. Am J Gastroenterol 2015; 110 (Suppl. 1): S766-S7847.
- Latella G, Sferra R, Speca S, Vetuschi A, Gaudio E. Can we prevent, reduce or reverse intestinal fibrosis in IBD? Eur Rev Med Pharmacol Sci 2013; 17: 1283-1304.
- Zeissig S, Bojarski C, Buergel N, et al. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut 2004; 53: 1295-1302.
- Blander JM. Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. FEBS J 2016; 283: 2720-2730.
- Quetglas EG, Mujagic Z, Wigge S, et al. Update on pathogenesis and predictors of response of therapeutic strategies used in inflammatory bowel disease. World J Gastroenterol 2015; 21: 12519-12543.
- Sales-Campos H, Basso PJ, Alves VB, et al. Classical and recent advances in the treatment of inflammatory bowel diseases. Braz J Med Biol Res 2015; 48: 96-107.
- Salmenkari H, Issakainen T, Vapaatalo H, Korpela R. Local corticosterone production and angiotensin-I converting enzyme shedding in a mouse model of intestinal inflammation. World J Gastroenterol 2015; 21: 10072-10079.
- Salmenkari H, Holappa M, Forsgard RA, Korpela R, Vapaatalo H. Orally administered angiotensin-converting enzyme-inhibitors captopril and isoleucine-proline-proline have distinct effects on local renin-angiotensin system and corticosterone synthesis in dextran sulfate sodium-induced colitis in mice. J Physiol Pharmacol 2017; 68: 355-362.
- Holma R, Salmenpera P, Riutta A, Virtanen I, Korpela R, Vapaatalo H. Acute effects of the cys-leukotriene-1 receptor antagonist, montelukast, on experimental colitis in rats. Eur J Pharmacol 2001; 429: 309-318.
- Melgar S, Karlsson A, Michaelsson E. Acute colitis induced by dextran sulfate sodium progresses to chronicity in C57BL/6 but not in BALB/c mice: correlation between symptoms and inflammation. Am J Physiol Gastrointest Liver Physiol 2005; 288: G1328-G1338.
- Forsgard RA, Korpela R, Holma R, et al. Intestinal permeability to iohexol as an in vivo marker of chemotherapy-induced gastrointestinal toxicity in Sprague-Dawley rats. Cancer Chemother Pharmacol 2016; 78: 863-874.
- Nolte T, Brander-Weber P, Dangler C, et al. Nonproliferative and proliferative lesions of the gastrointestinal tract, pancreas and salivary glands of the rat and mouse. J Toxicol Pathol 2016; 29 (Suppl. 1):1S-125S.
- Yamakawa I, Kojima H, Terashima T, et al. Inactivation of TNF-alpha ameliorates diabetic neuropathy in mice. Am J Physiol Endocrinol Metab 2011; 301: E844-E852.
- Schepp-Berglind J, Atkinson C, Elvington M, Qiao F, Mannon P, Tomlinson S. Complement-dependent injury and protection in a murine model of acute dextran sulfate sodium-induced colitis. J Immunol 2012; 188: 6309-6318.
- Chassaing B, Aitken JD, Malleshappa M, Vijay-Kumar M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol 2014; 104: Unit 15.25. doi: 10.1002/0471142735.im1525s104
- Suzuki Y, Ruiz-Ortega M, Lorenzo O, Ruperez M, Esteban V, Egido J. Inflammation and angiotensin II. Int J Biochem Cell Biol 2003; 35: 881-900.
- Jones CP, Boyd KL, Wallace JM. Evaluation of mice undergoing serial oral gavage while awake or anesthetized. J Am Assoc Lab Anim Sci 2016; 55: 805-810.
- Sasaki M, Uehara S, Ohta H, et al. Losartan ameliorates progression of glomerular structural changes in diabetic KKAy mice. Life Sci 2004; 75: 869-880.
- Patten GS, Abeywardena MY. Effects of antihypertensive agents on intestinal contractility in the spontaneously hypertensive rat: angiotensin receptor system downregulation by losartan. J Pharmacol Exp Ther 2017; 360: 260-266.
- Chappell MC, Pirro NT, Sykes A, Ferrario CM. Metabolism of angiotensin-(1-7) by angiotensin-converting enzyme. Hypertension 1998; 31: 362-367.
- Sechi LA, Valentin JP, Griffin CA, Schambelan M. Autoradiographic characterization of angiotensin II receptor subtypes in rat intestine. Am J Physiol 1993; 265: G21-G27.
- Fandriks L. The renin-angiotensin system and the gastrointestinal mucosa. Acta Physiol (Oxf) 2011; 201: 157-167.
- Moraes PL, Kangussu LM, da Silva LG, Castro CH, Santos RAS, Ferreira AJ. Cardiovascular effects of small peptides of the renin angiotensin system. Physiol Rep 2017; 5: e13505. doi: 10.14814/phy2.13505
- Harding P, Stonier C, Aber GM. Dose-dependent effects of angiotensin converting enzyme (ACE) inhibitors on glomerular prostanoid production by normotensive rats. Br J Pharmacol 1993; 108: 327-330.
- Diop-Frimpong B, Chauhan VP, Krane S, Boucher Y, Jain RK. Losartan inhibits collagen I synthesis and improves the distribution and efficacy of nanotherapeutics in tumors. Proc Natl Acad Sci USA 2011; 108: 2909-2914.
- McMurray JJ, Ostergren J, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiotensin-converting-enzyme inhibitors: the CHARM-Added trial. Lancet 2003; 362: 767-771.
- Pfeffer MA, McMurray JJ, Velazquez EJ, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 2003; 349: 1893-1906.
- Phillips CO, Kashani A, Ko DK, Francis G, Krumholz HM. Adverse effects of combination angiotensin II receptor blockers plus angiotensin-converting enzyme inhibitors for left ventricular dysfunction: a quantitative review of data from randomized clinical trials. Arch Intern Med 2007; 167: 1930-1936.
- Fried LF, Emanuele N, Zhang JH, et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N Engl J Med 2013; 369: 1892-1903.
- Sakaguchi K, Chai SY, Jackson B, Johnston CI, Mendelsohn FA. Inhibition of tissue angiotensin converting enzyme. Quantitation by autoradiography. Hypertension 1988; 11: 230-238.
- Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol 2006; 20: 953-970.
- Reinhardt D, Sigusch HH, Hensse J, Tyagi SC, Korfer R, Figulla HR. Cardiac remodelling in end stage heart failure: upregulation of matrix metalloproteinase (MMP) irrespective of the underlying disease, and evidence for a direct inhibitory effect of ACE inhibitors on MMP. Heart 2002; 88: 525-530.
- Efsen E, Saermark T, Hansen A, Bruun E, Brynskov J. Ramiprilate inhibits functional matrix metalloproteinase activity in Crohn’s disease fistulas. Basic Clin Pharmacol Toxicol 2011; 109: 208-216.
- Cima I, Corazza N, Dick B, et al. Intestinal epithelial cells synthesize glucocorticoids and regulate T cell activation. J Exp Med 2004; 200: 1635-1646.
- Mueller M, Cima I, Noti M, et al. The nuclear receptor LRH-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J Exp Med 2006; 203: 2057-2062.
- Noti M, Corazza N, Mueller C, Berger B, Brunner T. TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med 2010; 207: 1057-1066.
- Noti M, Corazza N, Tuffin G, Schoonjans K, Brunner T. Lipopolysaccharide induces intestinal glucocorticoid synthesis in a TNFalpha-dependent manner. FASEB J 2010; 24: 1340-1346.
- Huang SC, Lee CT, Chung BC. Tumor necrosis factor suppresses NR5A2 activity and intestinal glucocorticoid synthesis to sustain chronic colitis. Sci Signal 2014; 7: ra20. doi: 10.1126/scisignal.2004786
- English WR, Corvol P, Murphy G. LPS activates ADAM9 dependent shedding of ACE from endothelial cells. Biochem Biophys Res Commun 2012; 421: 70-75.
A c c e p t e d : August 30, 2018