We recently developed a computational model that simulates cardiac metabolism under normal and ischemic conditions (8). Unlike previous models of cardiac metabolism, which focused on glycolysis and/or the citric acid cycle and considered glucose as the only energy source (9-11), our model incorporated acetyl-CoA formation from the oxidation of both carbohydrates (glucose and lactate) and fatty acids. Our model also incorporated several key control metabolites (ADP, ATP, NADH, NAD+, acetyl-CoA, and free-CoA) into the regulation of pyruvate oxidation, glycolysis, lactate production, the citric acid cycle, and oxidative phosphorylation.
In our previous paper on the development of the model we found good agreement when we compared model-simulated results with previously published data from pigs, dogs and humans, however prospective validation studies have not been performed. Thus, in this study, our goal was to test the robustness of our model of myocardial metabolism (8) by evaluating the agreement between model responses and experimental data obtained in our lab under similar conditions. Accordingly, an abrupt (step) and a gradual (ramp) reduction in flow of similar magnitude were implemented and used as model input. The ramp reduction was elicited over a 30-min period. Once flow reductions reached 60%, ischemia was maintained constant for 60 minutes in both groups. Results of model simulations from the two flow reduction patterns were compared to data from in vivo swine experiments. Previous studies show that a gradual onset of myocardial ischemia is associated with lessened metabolic derangements when compared to those caused by a sudden onset of ischemia (1; 7). We hypothesized that the responses to these flow-reduction profiles will display different transients (concentration and flux rates) but similar steady-state values, and that our model-simulated responses will predict the results of the animal experiments.
Model Simulations
Minor modifications to our previously developed model (8) were made so that under normal flow conditions the concentration and flux values matched our experimental values collected during the nonischemic steady state baseline period (Tables 2-5). Simulations were then performed for the two different flow inputs (step and ramp reductions in flow). The metabolic species and reactions included in the model are depicted in Figure 1. As an initial-value problem, the parameter values (including initial fluxes (
 |
| Figure 1. Schematic depiction of the metabolic reactions incorporated in the computational model. |
| Table 1 Initial Concentrations |
 |
| * Denotes data from present study. Abbreviations: GL, glucose; FA, fatty acids; GP, glucose 6-phosphate; GY, glycogen; TG, triglyceride; PY, pyruvate; LA, lactate; AC, acetyl-CoA; FC, free CoA; CoA Pool, total CoA pool; NAD+, oxidized nicotinamide adenine dinucleotide; PC, phosphocreatine; NADH, reduced nicotinamide adenine dinucleotide. |
| Table 2. Blood-Tissue Partition Coefficients |
 |
| * Denotes data from present study. |
| Table 3. Initial Fluxes (µmol g wet wt-1 min-1) |
 |
| * Denotes data from present study. |
| Table 4. Km Values (mmol g wet wt-1) |
 |
| Table 5. Rate Coefficients (min-1) |
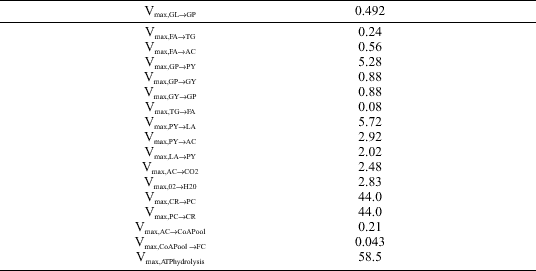 |
Animal Preparation
Experiments were performed on 16 domestic pigs (mean weight, 37.1 ± 1.1 kg). These studies were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication Number 85-23) and the Institutional Animal Care and Use Committee at Case Western Reserve University.
We used a previously developed open-chest model that allows for simultaneous measurements of myocardial substrate utilization (glucose, lactate, and fatty acids), metabolite concentrations in arterial and coronary venous blood, and tissue substrate concentrations (12-15). Following an overnight fast, animals were sedated with 6 mg/kg of Telazol (im), given isoflurane (5%) by mouth, intubated via tracheotomy, maintained with isoflurane (0.75%-1.25%) and ketamine (4 mg kg-1 hr-1), and ventilated to maintain blood gases in the normal range (PO2 > 100 mmHg, PCO2 35-45 mmHg, and pH 7.35-7.45) (14). The heart was exposed via a midline sternotomy with a left-side rib resection and suspended in a pericardial sling. A femoral vein was cannulated through which heparin was infused to prevent clotting (200 U/kg bolus, followed by 150 U/kg-hr i.v), and a 20% triglyceride emulsion (Intralipid 20%, 0.3 ml/kg-hr iv) were infused to raise circulating free fatty acids (FFA) to human levels (0.6 mM). A femoral artery was cannulated and used to prime the perfusion line using a roller pump. The left anterior descending coronary artery (LAD) was cannulated above the first diagonal branch and perfused with blood supplied from the femoral artery via the extracoporal perfusion circuit. The flow was adjusted to give an interventricular venous hemoglobin saturation of 35-40% (14; 16; 17). Arterial blood samples were obtained from the perfusion line and venous samples from the anterior interventricular vein. The isotopic tracers (14C-glucose, 13C-lactate, 3H-oleate), heparin, and triglyceride emulsion were infused into a femoral vein. Heart rate, aortic pressure, left ventricular pressure, and systolic thickening were continuously recorded using online data acquisition software (BioPac Acknowledge). Left ventricular contractile function was monitored by measuring left ventricular pressure with a 7-Fr Milar Mikrotip transducer catheter and anterior wall shortening using sonimicrometer crystals (Triton Technologies, San Diego, CA) placed in the mid myocardium (16; 18).
Experimental Protocol
Our experiments consisted of two groups: the Step Group underwent a rapid (~15 seconds) 60% step reduction in LAD blood flow, while in the Ramp Group the LAD flow was reduced linearly by 60% over a 30 min period (Figure 2). After completion of the surgical preparation, tracer was initiated with an intravenous bolus injection of 13C-lacate (80 mg), 14C-glucose (20 µCi) followed by a constant infusion of 13C-lactate (93.3 mg/hr), [U-14C]glucose (13.3 µCi/hr), and [9,10-3H]oleate (40 µCi/hr) at a rate of 3 ml/hr (Figure 2). After 45 min of tracer infusion, the first initial state blood samples were drawn. As shown in Figure 2, time = 0 was set to the onset of a 60% flow reduction in the LAD. In the Step Group the tracer infusion was initiated at -60 minutes, with aerobic control values take at -15 and -2 minutes. In the Ramp Group the tracer infusion started at -90 min, and aerobic control samples were taken at -45 and -32 min, then at -30 min the LAD flow was reduced linearly by 60% over 30 mins, and blood samples were drawn at -25, -20, -15, -10, and -5 mins. The two groups had the same sampling pattern during the 60 min of constant ischemia where blood samples were taken at 1, 3, 6, 10, 15, 20, 30, 40, 50, and 60 min. Tissue biopsy samples were obtained using a 14-gauge biopsy needle between the two aerobic control blood samples, and at 7, 25, and 60 min of ischemic period. At the conclusion of the experiment, a terminal punch biopsy was obtained from both the LAD bed and the circumflex bed (~3 g). All biopsies were immediately frozen (3-5 s) using metal blocks pre-cooled in liquid nitrogen and stored at -80°C for later analysis. The cardiovascular measurements were recorded immediately before the collection of each set of arterial and venous blood samples. Heart rate, left ventricular pressure (LVP), the peak positive and negative first derivatives of LVP (dP/dt), and segment length were continuously recorded using an online data acquisition system (Crystal Biotech model CBI8000 with Biopaq software).
 |
Fig. 2. Study protocols. (A) Protocol for step reduction in flow. (B) Protocol for ramp reduction in blood flow. Blood plasma samples, CO2 vials, and saturated salicylic acid (SSA) samples were collected at six different blood sample time points. |
Analytic Methods
Arterial and venous pH, PCO2, and PO2 were measured using a blood gas analyzer (NOVA Profile Stat 3, NOVA Biomedical; Waltham, MA) and oxygen saturation and hemoglobin were measured on a hemoximeter (Avoximeter; San Antonio, TX). Blood samples used to measure glucose, lactate, 14C-glucose, and 14C-lactate concentrations were immediately deproteinized in ice-cold 1 M perchloric acid (1:2 vol/vol). They were then analyzed for glucose and lactate using enzymatic-spectrophotometric assays (12; 13; 15). The specific radioactivity of [14C]glucose was measured on deproteinized blood samples using ion-exchange resin chromatography and blood 14CO2 concentration was assessed by trapping it in hyamine hydroxide, as previously described (19-21). Plasma fatty acid and 3H-oleate concentrations were measured as previously described (22; 23). 3H2O was measured by determining the difference in dpm/mL of water distilled from plasma using a Hickman still (Kontes Glass, Custom Shop) (24). The enrichment of lactate with 1-13C-lactate was measured in plasma deproteinized with salicylic acid using GC-MS analysis of the tert-butyldimethylsilyl (TBDMS) derivative as previously described (25).
The tissue biopsies taken during the initial state and after 7 min, 25 min, and 60 min of the 60% reduction in flow were used to measure the tissue levels of ATP, ADP, glycogen, and lactate. Tissue levels of ATP and ADP were measured luminometrically by methods previously described (26). Tissue glycogen was measured with perchloric acid extracts using the amyloglucosidase method by methods previously described (27). Tissue lactate measurements were completed with an enzymatic spectrofluorometric assay, using modified technique similar to that of the blood lactate assay. The 3-gram punch biopsy taken at the conclusion of each experiment was used to measure NAD+ and triglyceride levels. Tissue was extracted in ice-cold chloroform/methanol (2:1 vol/vol) and triglyceride levels were measured using an enzymatic spectrophotometric assay (Triglyceride E kit, Wako Chemicals, Richmond, VA). NAD+ was measured spectrophotometrically using a previously described method (28).
Calculations
The net uptakes (µmol g wet weight-1 min-1) for glucose, lactate, and free fatty acids were calculated based on the product of the arterial and coronary venous substrate difference and the normalized myocardial blood flow (ml g wet weight-1 min-1). The rates of exogenous glucose and fatty acid oxidation were calculated as the product of the release of either 14CO2 or 3H2O (dpm/ml) and normalized myocardial blood flow, divided by the arterial specific radioactivity of glucose or free fatty acids (dpm/µmol) (23). Simultaneous lactate uptake and release were calculated from the 13C-lactate enrichment and blood lactate concentrations as previously described (12; 20). The rates of glucose and fatty acid uptake and oxidation, and the rates of lactate uptake and production were used in determining the initial flux values (Table 3). Myocardial blood flow was measured from the calibrated pump flow of the perfusion circuit and normalized by dividing by the weight of the heart being perfused by the LAD (17).
The LVP-segment length loop area was calculated off-line from approximately 30 consecutive heartbeats by using software developed in Matlab. This was used as an index of the external work of the anterior free wall of the left ventricle and was expressed as a fraction of the initial state values. The LVP-segment length loop area multiplied by the heart rate was used as an index of the external power of the anterior wall.
Statistical Analysis
Triglyceride, and NAD+ concentrations were compared between the LAD and circumflex (CFX, control) bed and between step and ramp groups using a two-way analysis of variance. Uptakes of glucose, lactate, and free fatty acid; all hemodynamic parameters; regional anterior wall work and power indices; and glycogen and ATP concentration were compared using a two-way repeated measure analysis of variance, using a Student-Newman-Keuls test for post hoc comparisons. All tests for significance were performed at the 0.05 level of significance and all values are reported as means ± SE.
Simulation Results and Experimental Data
Ramp ischemia was induced by decreasing flow from 100% to 40% (0.76 ± 0.04 ml g-1 min-1 to 0.31 ± 0.002 ml g-1 min-1) of normal coronary blood flow in a linear manner over 30 min while step ischemia was induced by decreasing the flow the same magnitude (0.73 ± 0.025 ml g-1 min-1 to 0.29 ± 0.001 ml g-1 min-1) over 1 min. To test the robustness of our computational model with updated initial conditions and parameter values, we compared the concentrations and flux rates that were measured experimentally with the corresponding model-simulated results.
With the introduction of ischemia, there was an immediate decrease in myocardial oxygen consumption (MVO2) (Figure 3 A) and a proportionate decrease in ATP (Figure 3 B). The model simulations for both the step and ramp show a 50% decrease in MVO2 while experimentally the decreases were 54% ± 2% and 58% ± 2%, respectively (Table 6). These results show that the two different flow reduction profiles result in similar steady-state decreases in MVO2. The decreases in ATP for step were 40% and 41% for experimental and simulated, respectively, while those for ramp ischemia were 69% and 42%.
| Table 6. Hemodynamic values from in vivo swine experiments |
 |
| Values are means ± SE with 7 pigs in the ramp group and 9 pigs in the step group. HR, heart rate; dP/dt, first derivative of pressure; LVP, left ventricular pressure; MVO2, myocardial O2 consumption. Work and power are expressed as the fraction of the baseline value. * P < 0.05 compared to baseline. |
 |
| Figure 3. Myocardial oxygen consumption (MVO2) (A) and ATP concentration changes (B). Dashed lines represent model-simulated responses from ramp induction of ischemia while solid lines represent responses from step induction of ischemia. The open circles represent experimental data from the step group with the filled circles represent experimental data from the ramp group. * p<0.05 between the step vs. ramp groups. |
Another consequence of ischemia is an acceleration of glycolysis that results in increased utilization of glycogen, accumulation of lactate in the heart, and a switch from net lactate uptake to net lactate release by the heart. In the step group, glycogen content decreased -after 60 minutes of ischemia- by 83% ± 4% in the animal experiments and by 76% in the simulations (Figure 4 A). In the ramp group, glycogen content decreased by 83% ± 3% in the experiments while the model predicted an 88% decrease (Figure 4 A). Even though the steady-state glycogen concentration values were similar in all four cases (step vs. ramp and experiments vs. simulations) after 60 minutes of ischemia, the transient behavior was different when experiments were compared to computer simulations. Experimentally, glycogen concentration decreased in a hyperbolic or exponential manner while the model predicted a linear decrease in glycogen concentration regardless of flow-reduction pattern. Thus, the experimental rate of glycogen utilization increased more rapidly initially and tapered off quicker than that predicted by the computer model (Figure 4 A).
 |
| Figure 4. Glycogen (A) and lactate (B) concentration changes. Dashed lines represent model-simulated responses from ramp induction of ischemia while solid lines represent responses from step induction of ischemia. The open circles represent experimental data from the step group with the filled circles represent experimental data from the ramp group. |
Lactate accumulation and the switch from net uptake to net release of lactate by the heart results from the increased rate of glycolysis observed during ischemia. The model simulated results for tissue lactate fit within the standard error bars of the measured concentrations, and increased two to three fold above initial values in both groups (Figure 4 B). As was the case with glycogen breakdown, the model-simulated time course of lactate accumulation to a step reduction in blood flow was slower than seen experimentally. The model simulated results showed a delay in the switch from net lactate uptake to net release, and a greater rate of lactate release from 10 to 60 minutes of ischemia compared to the experimental data (Figure 5). While both the simulated and experimental results showed decreased glycogen concentration, accumulation of lactate, and net lactate release with ischemia, the onset of the rate of increase of glycogenolysis and the switch to lactate efflux occurred sooner and more rapidly in the experiments than in the simulations. Thus, the experimental rate of glycogen utilization and lactate production increased more rapidly initially and tapered off quicker than that predicted by the computer model.
 |
| Figure 5. Net lactate uptake by the heart. The positive values represent net lactate uptake while the negative values show net lactate release by the heart. The peak lactate release by the heart was significantly different between the step and ramp groups (p<0.05). |
Many changes induced by ischemia, including the increased rate of glycolysis and decreased rates of oxygen consumption and electron transport chain flux, result in a decrease in the concentration of NAD+ and an increase in the redox state (NADH/NAD+). The myocardial NAD+ contents were 0.41±0.02 and 0.39±0.01 µmols/g in the nonischemic CFX perfusion territory for the step and ramp groups, respectively; and they were reduced by 22% ± 4% and 25% ± 6% in the ischemic LAD territory. The model simulation results predicted decreases were 40% and 44% for step and ramp, respectively, which followed a time course similar to MVO2. The experimental results and the model-simulated results both show a decrease in the concentration of NAD+ and the model predicts a seven-fold increase in the redox ratio.
Comparison of Step and Ramp
Values for the measured hemodynamic variables are presented in Table 6. There were no significant differences between the step and ramp groups regarding any of these values. Peak left ventricular pressure (LVP) for both groups combined was significantly higher at baseline than at any time point during the 60 min of 60% reduction in blood flow. However LVP values were not significantly different from each other beyond 6 min (Table 6). Work and power at 6 min were significantly different from those at 60 min. Myocardial oxygen consumption was significantly different between baseline values and values during the 60% reduction in blood flow.
Peak lactate release was greater in the step group (0.90 ± 0.12 µmol/g/min) than in the ramp group (0.69 ± 0.10 µmol/g/min), P < 0.05. However, no difference was seen between the groups in tissue lactate concentration, lactate production, and steady-state lactate release at the end of the 60 min of 60% reduction in blood flow. The tissue ATP concentration was lower in the ramp group (P < 0.05). There was no difference in glucose oxidation, fatty acid oxidation, glycogen concentration, or any of the other measured species concentrations or fluxes.
We previously presented model simulations for 60 min at a 60% reduction in blood flow and validated the simulations with retrospective experimental data (8). In the present study, we prospectively compared model-simulated and experimental results from two different flow-reduction profiles, showing the robustness and limitations of the model. The two different induction patterns of 60% ischemia resulted in the same decrease in steady-state MVO2 and in similar steady-state values for metabolite concentrations and fluxes. Model simulations in response to step and ramp blood flow reductions agreed well with some of the experimental data (e.g. lactate concentration, final glycogen concentration) while some of the results had different changes in magnitude and time courses (e.g. MVO2, lactate release, glycogen depletion).
We have developed the first computational model of myocardial energy metabolism that incorporates the key carbon substrates of human myocardial metabolism. In this study we tested the model with two different inductions of ischemia. For both flow reductions used, our model-simulated results agreed relatively well with the experimental data we collected from the corresponding in vivo animal studies and other previously published data. The time courses for the decreases in MVO2, ATP, NAD+, and glycogen differed between the step and ramp groups although their measured and model-simulated steady-state values were similar. The same relationships held for the accumulation of lactate and the switch from net lactate uptake to net release.
We selected the two different flow-reduction profiles in order to show the ability of the model, to accurately predict changes occurring from both flow-reductions using a single set of parameter values. The step flow-reduction profile was selected because it is the most commonly used method of flow-reduction in experimental studies of ischemia, and it results in a rapid reduction in MVO2, oxidative phosphorylation, and activation of glycolysis. In addition, previous studies showed that the ramp flow reduction profile results in less activation of glycolysis (1; 7}. As seen in Figure 3 A, the results agreed with our hypothesis that the two methods of blood flow reduction result in similar decreases in MVO2 although with different time courses. The experimental results (55 and 58% decreases in MVO2 for step and ramp, respectively) showed a larger decrease than predicted by the model (50%). The initial experimental MVO2 values (2.6 and 2.8 µmol/g wet wt for step and ramp, respectively) from our current study were substantially lower than previously published experimental values, which ranged from 7.5 to 2.5 µmol/g wet wt/min, with an average of 4.8 µmol/g wet wt/min and a median value of 4.7 µmol/g wet wt/min (2; 5-7; 13; 17; 29-33). Three previous studies involving a 60% reduction in blood flow resulted in 46, 49, and 50% decreases in MVO2 due to ischemia (5; 6; 30; 31). Our model-simulated results, with a 50% reduction in MVO2, are comparable with these previous results.
Another difference between our experimental data and published data from other in vivo pig studies is the baseline blood flow values. The range of normal blood flow values reported ranged from 0.70 to 1.43 ml/g/min (1; 2; 5-7; 13; 17; 30-35) and our experimental blood flow was 0.73 and 0.76 ml/g/min for step and ramp groups, respectively. Although our measured experimental values for MVO2 and blood flow are lower than some published values with similar preps, these current values agree with those recently published using the same preparation (29). Furthermore, during these experiments the swine were not ischemic or stressed during the initial state, as evidenced by the lack of net lactate production, the relatively low heart rates, and stable LV pressures.
The concentration of NAD+ is expected to decrease with an increase in NADH concentration during ischemia. Unfortunately, there is limited data available on the concentrations of NAD+ and NADH due to the difficulty of measuring them. Our model-simulated results reflect these changes and we attempted to measure them both using the terminal punch biopsies collected during the experiments. The measured decreases in NAD+ were less than those predicted by the model and there was no difference between the step and ramp groups. In order to set the initial concentration value for NADH in the model, we used a baseline ratio for NADH to NAD+ from previously published values (0.11) (Table 1) (36; 37) and our measured NAD+ values to calculate the corresponding NADH concentration. Although our measurements of NAD+ provide useful insight into the possible changes in the pyridine nucleotide pool with the introduction of ischemia, the lack of other experimental data in the area makes it difficult to draw clear conclusions from our data.
While the steady-state experimental values for tissue glycogen and lactate content at 60 minutes of ischemia agreed reasonably well with simulated results, the model-simulated time courses during the transition to 60% ischemia lagged behind for both metabolites. Specifically, the model did not predict the rapid burst in glycogen depletion and lactate production during the initial minutes of ischemia, nor the decline from ~15 to 60 min. In this case, experimental data suggests the model components that need to be revised by incorporating more biochemical detail or control mechanisms (e.g., reactions or controllers). This could lead to a better correspondence between model simulations and experimental results during the dynamic phase of the response. However, since muscle biopsies to measure glycogen content in the myocardium are difficult to take frequently, other methods of model validation need to be implemented to provide enough in vivo data to describe the time course of glycogen depletion. The sluggishness of some of the responses likely occurs because known controllers of glycogenolysis and glycolysis are missing in the current model. The addition of inorganic phosphate (Pi), cytosolic Ca2+ and the adenine nucleotides (AMP, adenosine, IMP, inosine), which control key regulatory enzymes in these pathways, should help speed up these time courses. They are not included in the model due to lack of reliable data and analytical measurements applicable to our in vivo experimental preparation. In order to accurately model the responses of Pi, Ca2+ and adenine metabolites it would be necessary to increase the complexity of the reactions included the model and to obtain some realistic experimental values. In addition, our model does not separate the cytosolic compartment from the mitochondria; addition of a cytosolic compartment would increase the sensitivity of glycolysis to regulatory metabolites in the cytosol. Cortassa et al recently developed an integrated thermokinetic model describing control of cardiac mitochondrial bioenergetics, and demonstrated the importance of mitochondrial matrix Ca2+ in matching energy supply with demand in cardiac myocytes (38). While this important model is very useful for understanding cardiac mitochondrial function, it does not include glycolysis or fatty acid oxidation, and it cannot be directly applied to in vivo conditions. Clearly, future model development and subsequent validation studies should aim to incorporate these key metabolites and multiple cell compartments.
In summary, we present a computational model of human-like myocardial energy metabolism that incorporates both fatty acid and carbohydrate metabolism. We tested the model with two different inductions of 60% ischemia, a ramp reduction (completed over 30 min) and a step reduction (completed over 1 min). The results agreed with our hypotheses that the two methods of blood flow reduction result in similar steady-state metabolite values, although the time courses are different. The steady-state experimental values for tissue glycogen and lactate content at 60 minutes of ischemia agreed well with simulated results, however the model-simulated time courses during the transition to 60% ischemia lagged behind for both substrates. In conclusion, this study demonstrates the utility of computer models for predicting experimental outcomes in studies of metabolic regulation under physiological and pathological conditions. Future models, however, need to incorporate more complex control mechanisms to more accurately predict dynamic changes.
Acknowledgements: This work was supported by the National Institutes of Health (GM066309), NASA-NSBRI (IHF00205), and the American Heart Association (9660355V). The authors wish to thank Naveen Sharma, Bridgette Christopher, and Dr. Nelson Chavez for their assistance.
- Arai AE, Grauer SE, Anselone CG, Pantely GA and Bristow JD. Metabolic adaptation to a gradual reduction in myocardial blood flow. Circulation 1995; 92: 244-252.
- Arai AE, Pantely GA, Anselone CG, Bristow J and Bristow JD. Active downregulation of myocardial energy requirements during prolonged moderate ischemia in swine. Circ Res 1991; 69: 1458-1469.
- Pantely GA, Malone SA, Rhen WS, Anselone CG, Arai A, Bristow J and Bristow JD. Regeneration of myocardial phosphocreatine in pigs despite continued moderate ischemia. Circ Res 1990; 67: 1481-1493.
- McNulty PH, Sinusas AJ, Shi CQ, Dione D, Young LH, Cline GC and Shulman GI. Glucose metabolism distal to a critical coronary stenosis in a canine model of low-flow myocardial ischemia. J Clin Invest 1996; 98: 62-69.
- Stanley WC, Hall JL, Smith KR, Cartee GD, Hacker TA and Wisneski JA. Myocardial glucose transporters and glycolytic metabolism during ischemia in hyperglycemic diabetic swine. Metabolism 1994; 43: 61-69.
- Stanley WC, Hall JL, Stone CK, Hacker TA. Acute myocardial ischemia causes a transmural gradient in glucose extraction but not glucose uptake. Am J Physiol 1992; 262: H91-H96.
- Ito BR. Gradual onset of myocardial ischemia results in reduced myocardial infarction. Association with reduced contractile function and metabolic downregulation. Circulation 1995; 91: 2058-2070.
- Salem JE, Saidel GM, Stanley WC, Cabrera ME. Mechanistic model of myocardial energy metabolism under normal and ischemic conditions. Ann Biomed Eng 2002; 30: 202-216.
- Achs MJ, Garfinkel D. Computer simulation of energy metabolism in anoxic perfused rat heart. Am J Physiol 1977; 232: R164-R174.
- Ch'en FF, Vaughan-Jones RD, Clarke K, Noble D. Modelling myocardial ischaemia and reperfusion. Prog Biophys Mol Biol 1998; 69: 515-538.
- Kashiwaya Y, Sato K, Tsuchiya N, Thomas S, Fell DA, Veech RL, Passonneau JV. Control of glucose utilization in working perfused rat heart. J Biol Chem 1994; 269: 25502-25514.
- Hall JL, Stanley WC, Lopaschuk GD, Wisneski JA, Pizzurro RD, Hamilton CD, McCormack JG. Impaired pyruvate oxidation but normal glucose uptake in diabetic pig heart during dobutamine-induced work. Am J Physiol 1996; 271: H2320-H2329.
- Hall JL, Lopaschuk GD, Barr A, Bringas J, Pizzurro RD, Stanley WC. Increased cardiac fatty acid uptake with dobutamine infusion in swine is accompanied by a decrease in malonyl CoA levels. Cardiovasc Res 1996; 32: 879-885.
- Stanley WC, Hernandez LA, Spires D, Bringas J, Wallace S, McCormack JG. Pyruvate dehydrogenase activity and malonyl CoA levels in normal and ischemic swine myocardium: effects of dichloroacetate. J Mol Cell Cardiol 1996; 28: 905-914.
- Hall JL, Henderson J, Hernandez LA, Kellerman LA, Stanley WC. Hyperglycemia results in an increase in myocardial interstitial glucose and glucose uptake during ischemia. Metabolism 1996; 45: 542-549.
- Mazer CD, Cason BA, Stanley WC, Shnier CB, Wisneski JA, Hickey RF. Dichloroacetate stimulates carbohydrate metabolism but does not improve systolic function in ischemic pig heart. Am J Physiol 1995; 268: H879-H885.
- Panchal AR, Comte B, Huang H, Kerwin T, Darvish A, Des RC, Brunengraber H, Stanley WC. Partitioning of pyruvate between oxidation and anaplerosis in swine hearts. Am J Physiol Heart Circ Physiol 2000; 279: H2390-H2398.
- Hall JL, Sexton WL, Stanley WC. Exercise training attenuates the reduction in myocardial GLUT-4 in diabetic rats. J Appl Physiol 1995; 78: 76-81.
- Wisneski JA, Gertz EW, Neese RA, Gruenke LD, Morris DL, Craig JC. Metabolic fate of extracted glucose in normal human myocardium. J Clin Invest 1985; 76: 1819-1827.
- Wisneski JA, Stanley WC, Neese RA, Gertz EW. Effects of acute hyperglycemia on myocardial glycolytic activity in humans. J Clin Invest 1990; 85: 1648-1656.
- Gertz EW, Wisneski JA, Stanley WC, Neese RA. Myocardial substrate utilization during exercise in humans. Dual carbon- labeled carbohydrate isotope experiments. J Clin Invest 1988; 82: 2017-2025.
- Chandler MP, Huang H, McElfresh TA, Stanley WC. Increased nonoxidative glycolysis despite continued fatty acid uptake during demand-induced myocardial ischemia. Am J Physiol. 2002; 282:H1871-878.
- Wisneski JA, Gertz EW, Neese RA, Mayr M. Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J Clin Invest 1987; 79: 359-366.
- Myears DW, Sobel BE, Bergmann SR. Substrate use in ischemic and reperfused canine myocardium: quantitative considerations. Am J Physiol 1987; 253: H107-H114.
- Comte B, Vincent G, Bouchard B, Jette M, Cordeau S, Rosiers CD. A 13C mass isotopomer study of anaplerotic pyruvate carboxylation in perfused rat hearts. J Biol Chem 1997; 272: 26125-26131.
- Lowry OH, Passonneau JV. A Flexible System of Enzymatic Analysis. New York: Academic Press, 1972.
- Passonneau JV, Lauderdale VR. A comparison of three methods of glycogen measurement in tissues. Anal Biochem 1974; 60: 405-412.
- Methods of enzymatic analysis. New York: Academic press 1985.
- Chavez PN, Stanley WC, McElfresh TA, Huang H, Sterk JP, Chandler MP. Effect of hyperglycemia and fatty acid oxidation inhibition during aerobic conditions and demand-induced ischemia. Am J Physiol Heart Circ Physiol 2003; 284: H1521-H1527.
- Hacker TA, Hall JL, Stone CK, Stanley WC. Alanine, glutamate, and ammonia exchanges in acutely ischemic swine myocardium. Basic Res Cardiol 1992; 87: 184-192.
- Hacker TA, Renstrom B, Paulson D, Liedtke AJ, Stanley WC. Ischemia produces an increase in ammonia output in swine myocardium. Cardioscience 1994; 5: 255-260.
- Fedele FA, Gewirtz H, Capone RJ, Sharaf B, Most AS. Metabolic response to prolonged reduction of myocardial blood flow distal to a severe coronary artery stenosis. Circulation 1988; 78: 729-735.
- Zhu P, Lu L, Xu Y, Greyson C, Schwartz GG. Glucose-insulin-potassium preserves systolic and diastolic function in ischemia and reperfusion in pigs. Am J Physiol Heart Circ Physiol 2000; 278: H595-H603.
- Chavez PN, Stanley WC, McElfresh TA, Huang H, Sterk JP, Chandler MP. Effect of hyperglycemia and fatty acid oxidation inhibition during aerobic conditions and demand-induced ischemia. Am J Physiol Heart Circ Physiol 2003; 284: H1521-H1527.
- Guth BD, Wisneski JA, Neese RA, White FC, Heusch G, Mazer CD, Gertz EW. Myocardial lactate release during ischemia in swine. Relation to regional blood flow. Circulation 1990; 81: 1948-1958.
- Ferrari R, Cargnoni A, Bernocchi P, Pasini E, Curello S, Ceconi C, Ruigrok TJ. Metabolic adaptation during a sequence of no-flow and low-flow ischemia. A possible trigger for hibernation. Circulation 1996; 94: 2587-2596.
- Ceconi C, Bernocchi P, Boraso A, Cargnoni A, Pepi P, Curello S, Ferrari R. New insights on myocardial pyridine nucleotides and thiol redox state in ischemia and reperfusion damage. Cardiovasc Res 2000; 47: 586-594.
- Cortassa S, Aon MA, Marban E, Winslow RL, O'Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J 2003; 84: 2734-2755.