Since the GLU and NO pathways engaged in the phenomenon of the posthyperoxic hypoxic ventilatory enhancement may intertwine, discerning the functional role of either is difficult. In the present study we addressed this issue in an anesthetized rat model by comparing the effects on the phenomenon of separate blockades of either pathway with those of concurrent blockade of them both. We found that the posthyperoxic hypoxic ventilatory enhancement is indeed similarly, but only partially, dampened by either a competitive glutamate blocker, 2-amino-5-phosphonopentanoic acid (AP5), or nNOS blocker, 7-nitroindazol (7NI). A combination of the two blockers further diminishes, but does not abolish, the hypoxic ventilatory enhancement. Thus, although the GLU-NO system accounts for an appreciable part of the posthyperoxic hypoxic ventilatory enhancement, other as yet unclear mechanisms contribute as well.
Animals, neuromodulators, vehicles, doses, and the volume and route of administration
Twenty six adult Wistar rats of either sex, weighing 276.5 ±10.0 g (range 202-340 g) were used for the study. The rats were housed in a light (12 h day/12 h night) and temperature controlled (23°C) room. The animals were divided into four experimental groups that differed with regard to the chemical agent injected. The number of animals in each group was different. The groups were the following: control or 0.9% NaCl-treated (n=4), AP5-treated (n=6), 7NI-treated (n=9), and AP5+7NI-treated (n=5). In addition, in two rats injections, as outlined below, of the vehicle dimethylsulfoxide (DMSO) were performed. The latter injections had no appreciable effects on the ventilatory variables studied and their results were not included in any of the experimental group reported herein. AP5 and 7NI, obtained from Sigma-RBI (Sigma-Aldrich, Poznañ, Poland), were dissolved in 0.9% NaCl and in DMSO, and injected in a dose of 10 mg/kg and 50 mg/kg, respectively. All injections were intraperitoneal in a volume of 0.2 ml per rat. The doses and route of administration of both neurotransmitter blockers have previously been validated (15, 16). Each animal was used for one substance injection once, as the substances were mostly long acting and also to avoid undue side effects. The only exception was the mixed group in which AP5 and 7NI were given from two different abdominal punctures 5 min apart. All procedures were performed in accordance with the accepted standard principles in the care and use of animals. The study protocol was approved by a local Ethics Committee (permits no. 136 and 188).
Surgical and recording procedures
Animals were anesthetized with alpha-chloralose and urethane (35 and 800 mg/kg, ip, respectively), tracheostomized, and spontaneously breathing through a two-way valve. The vagus nerves were isolated and transected bilaterally in the neck. Respiratory output profiles were recorded using diaphragmatic EMG activity. Two stainless steel unipolar electrodes were inserted into the costal diaphragm. The EMG signal, recorded differentially to ground, was amplified and band pass filtered (50 Hz-5000 Hz). The output signals were rectified and integrated with a time constant of 70 ms to obtain moving time averages of EMG activity (Digitimer-Neurolog System, Welwyn Garden City, UK). End-tidal fractional concentration of CO2 (FETCO2) was monitored at the trachea with an infra-red Engström Eliza analyzer (Gambro Engström AB, Sweden) and arterial blood pressure was continuously measured with a strain gauge and an electromanometer (MCK4011S, Femed, Zabrze, Poland).
All these recordings were displayed on a strip-chart hot-stylus polygraph (Honeywell-Omnilight 8M36, NEC San-ei Instruments, Tokyo, Japan). Arterial blood samples were withdrawn for measurements of blood gas content and pH during changes of inspired gas mixtures with a Compact 2 Blood Gas Analyzer (AVL List, Graz, Austria). The preparation included cannulations of the femoral artery and vein. Rectal temperature was maintained at ~38°C with a heating pad.
Experimental protocol
A schematic of the experimental protocol is presented in Fig. 1. After a period of stabilization of the respiratory profile during normoxia, the animal was subjected to hypoxia (Hypoxia I). After recovery, a given chemical agent was injected and the effects of injection were followed for 25 min. Then another hypoxic test (Hypoxia II) was performed, followed by a 15 min period of 100% O2 breathing and a repeat hypoxic test (Hypoxia III). All hypoxic tests were of a steady-state type, with 12% O2 in N2, and took 2.5 min. The recovery from hypoxia was achieved within 1-2 min. The end-tidal CO2 was allowed to run free during all gas changes.
 |
| Fig. 1. Diagram showing a schematic of sequential steps of the experimental protocol. |
Reaction of arterial blood pressure and respiration to pinching of a hind paw was used as an indicator of the depth of anesthesia. An extra dose of the anesthetics (5% of the initial dose) was injected intravenously, if there was an evident withdrawal reflex or blood pressure or respiratory instability. The frequency of such injections did not exceed 1-2 during the experimental period.
Measurements and data analysis
Measurements were done breath-by breath and all data were collected off-line from the strip-chart recording. The following variables were quantified from the moving time average of the EMG activity trace: the peak diaphragm amplitude (A, arbitrary units), taken as a surrogate of phrenic nerve motor activity running down to the diaphragm and therefore of tidal volume, the breathing frequency (f, breaths/min), taken as 60/duration of the EMG activity cycle, from the inspiratory onset of activity to its next onset, and the minute diaphragmatic EMG output (MDO), an index of minute ventilation, taken as the product of EMG amplitude and frequency.
The mean value of each variable was computed in the period of 3 full respiratory cycles at baseline, every 30 s during hypoxic exposures, and before and after injections of chemical agents. Since respiratory values usually show inter-animal variation that depends on a number of poorly controlled biological and technical factors, such as the sensitivity to anesthesia, the exact position of recording electrodes in a muscle or the gain of signal amplifiers, data were standardized by expressing them as the percent change from a baseline value, taken as 100%. The group percentage means ±SE were calculated. The time course of ventilatory variables during the three hypoxic conditions: control or preinjection hypoxia (Hypoxia I) postinjection or preoxygen hypoxia (Hypoxia II), and postoxygen hypoxia (Hypoxia III) was compared with a multivariate analysis of variance (MANOVA), followed by the Scheffe or LSD test, as indicated, in the case of a significant interaction. The effects of injections of chemical agents alone were compared with a two-tailed paired t-test. A P<0.05 was considered to denote significant changes for all comparisons. Commercial Statsoft software was used for statistical analyses.
Figure 2 demonstrates the time courses of hypoxic ventilatory profiles in all the experimental conditions and groups, i.e., the baseline condition before any pharmacologic maneuver, 25 min after injection of a given agent, and then after a 15-min O2 breathing period; each in the NaCl, AP5, 7NI, and AP5+7NI groups. For each group the ventilatory profile encompasses the minute diaphragm output (upper row), the amplitude of integrated diaphragm activity (middle row), an index of tidal volume, and the frequency of diaphragm volleys (bottom row), an index of respiratory rate.
Control hypoxic ventilatory response
The control ventilatory response to hypoxia (Hypoxia I), taken before implementation of a pharmacologic maneuver, may be characterized on the basis of the NaCl-treated group (Fig. 2A). The response was a biphasic response. The mean minute diaphragm output peaked at 0.5 min, increasing by 40.7 ±4.6% over the baseline level, and then started declining. The decline leveled off at 1.0-1.5 min, with the diaphragm output remaining 24% above baseline. Thus, ventilation reverted by ~40% from its peak increase. Thereafter, the ventilatory decrease accelerated and neared the baseline at 2.5 min, the test end. This profile of response to hypoxia was due both to the significant increases in f and A (P<0.05).
Ventilatory effects of administration of chemical agents
The mean percentage changes in ventilatory variables recorded 25 min after administration of chemical agents are displayed in Table 1. On the whole, changes were unremarkable with fluctuations around the preinjection baseline level. A second exposure to hypoxia following administration of chemical agents (Hypoxia II) caused variable effects depending on the agent used. In the NaCl group no differences were noted in the ventilatory variables compared with the preinjection hypoxia (Fig. 2A). Both AP5 and 7NI blockers caused a qualitatively similar change. The change was one of a depression of the minute diaphragmatic output (P<0.005) (Fig. 1B, C). The depression was due primarily to a significant decrease in f (P<0.04) with inappreciable changes in the peak diaphragmatic amplitude. The effect on the hypoxic ventilatory response of concurrent administration of the two blockers was not additive. The minute diaphragmatic output appeared to decrease, particularly in the stimulatory part of the hypoxic response, but the decrease was not significant (Fig. 2D).
| Table 1. Changes in ventilatory variables 25 min after administration of chemical agents. |
 |
| Values are a mean ±SE percent of the preinjection baseline level. MDO, minute diaphragmatic output; A, peak amplitude of the moving time average of EMG measurement; f, breathing frequency. No significant changes were noted. |
Posthyperoxic hypoxic ventilatory enhancement
The phenomenon of the posthyperoxic hypoxic ventilatory enhancement is best exemplified by the response in the NaCl-treated group. Hypoxic exposure performed after a 15 min period of O2 breathing (Hypoxia III) shifted the minute diaphragmatic output curve upward (Fig. 2A). The stimulatory/depressant profile was retained. The peak stimulation at 0.5 min was by 125.2 ±17.1% over baseline as compared with 41.0 ±6.7% in the preoxygen hypoxic exposure, a 3-fold increase in response. Since the postoxygen ventilatory decrease began from a higher level, ventilation was still 61.6 ±21.0% over the baseline level at 2.5 min from the onset of hypoxia. Ventilatory enhancement was driven by both volume and frequency components. There was a significant interaction between the ventilatory preoxygen and postoxygen profiles for the minute ventilation, volume, and frequency responses (P<0.05).
After administration of AP5 alone the posthyperoxic hypoxic ventilatory enhancement was partially dampened (Fig. 2B). The mean minute diaphragmatic output increase was more than halved compared with that in the control group (Table 2). The dampening was due more to driving down the volume than frequency respiration. 7NI alone acted in like manner (Fig. 1C). Neither blocker influenced the biphasic pattern of the response. Although the posthyperoxic hypoxic ventilatory enhancement was lessened by either blocker given alone, there still remained a significant interaction between the preoxygen and postoxygen profiles of all ventilatory variables (Fig. 2B, C) (P<0.04).
| Table 2. Differences in the increase of minute diaphragmatic output between the preoxygen and postoxygen hypoxic exposures after administration of the chemical agents used in the study. |
| Values are
mean ±SE percentage differences ( |
Concurrent administration of AP5 and 7NI further attenuated the posthyperoxic hypoxic ventilatory enhancement, but the effect was less than additive, leaving behind an appreciable portion of the enhancement (Fig. 2D, Table 2). The attenuation concerned foremost the volume respiration. The remaining increase in ventilation was due mainly to the persisting frequency effect. Significant interactions between the preoxygen and postoxygen minute diaphragmatic output and frequency persisted (P<0.0002) (Fig. 1D). The basic pattern of the response remained biphasic. Moreover, the ventilatory decrease at 2.5 min was ~30% off the peak increase, which is similar to that in the initial hypoxic test in the control group.
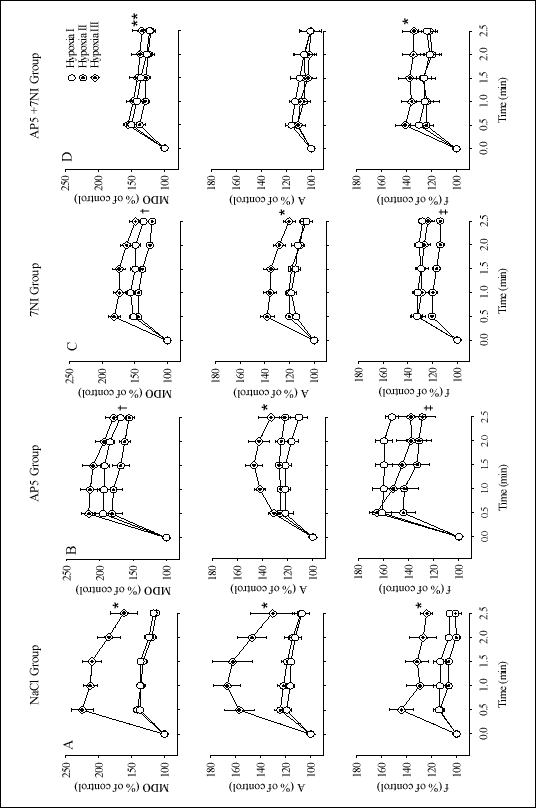 |
| Fig. 2. Respiratory profile in the experimental groups studied during consecutive hypoxic tests. A - control NaCl group, B - AP5 group, C - 7NI group, and D - AP5+7NI group. Symbols: open circles - control initial hypoxia (Hypoxia I), target circles - hypoxia after administration of a given chemical agent (Hypoxia II), target rhombs - hypoxia after prior oxygen breathing (Hypoxia III). MDO - minute diaphragm EMG output, A - peak diaphragm signal amplitude, f - frequency of diaphragm respiratory volleys. Symbols are mean ±SE percentage changes from control. A lack of the SE bar indicates the SE which is smaller than the symbol size. *Hypoxia III different from the remaining two at P<0.001, each hypoxia different from each other at P<0.005, Hypoxia II different from the remaining two at P<0.04, **Hypoxia III different from Hypoxia II at P<0.0002. Each set of variables was tested with multivariate analysis of variance for repeated measures (MANOVA). |
Arterial blood gas content and arterial blood pressure changes
Changes in the arterial blood gas content were fairly similar in each of the hypoxic exposures, irrespective of the chemical agent administered (Table 3). Hypoxia caused a decrease in PaO2 from the normoxic level of about 94-83 mmHg down to 51-45 mmHg. Since CO2 was allowed to run free, hypoxic hyperventilation was associated with hypocapnia and an alkaline shift in pHa. The majority of these changes were significant in each hypoxic run (P
| Table 3. Arterial blood gas content and pH in the gas conditions studied in each experimental group. |
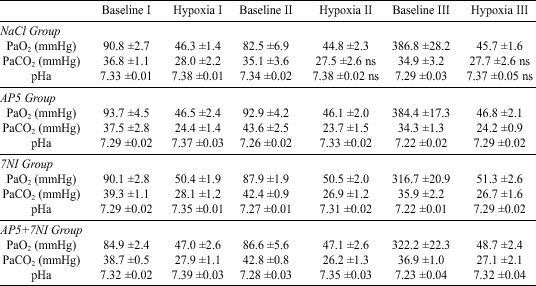 |
| Values are
means ±SE. Baseline I is the basic control, Baseline II is the control
level after injection of chemical agents, Baseline III is at the end of
exposure to hyperoxia. Differences between respective baseline and hypoxic
values in each hypoxic run were assessed with a paired t-test.
These difference, when unmarked, were significant at P |
Arterial blood pressure changes during consecutive exposures to hypoxia are shown in Table 4. Both systolic and diastolic pressures decreased at the peak of hypoxic ventilatory stimulation. The decrease was in a range of 15-20 mmHg and was significant in the majority of cases, although blood pressure remained within reasonable limits. There was a more pronounced decrease of arterial blood pressure at the nadir of hypoxic ventilation. Blood pressure rebounded during the recovery periods, so that there was no difference between the consecutive baseline levels. The pattern of arterial blood pressure changes was similar during every exposure to hypoxia in all experimental groups.
| Table 4. Arterial blood pressure changes during consecutive exposures to hypoxia in the experimental groups differentiated by chemical agents injected. |
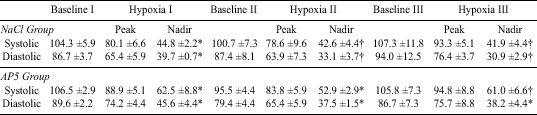 |
| Values are means ±SE of mmHg. Baseline I is the basic control, Baseline II is the control level after injection of chemical agents, Baseline III is at the end of exposure to hyperoxia. Each hypoxic run along with its preceding baseline was analyzed statistically with one-way ANOVA. (*) denotes that each blood pressure point within a given hypoxic run, including its baseline level, is different from one another at P<0.05; () denotes the hypoxic nadir points different from the remaining two points in a given hypoxic run at P<0.05. |
This study focused on two neurotransmitter mechanisms, glutamatergic and nitrergic, both of the known central stimulatory action on ventilation (5, 6, 17), which have previously been quoted as being possibly involved in the posthyperoxic hypoxic ventilatory enhancement (2, 3). These mechanisms were dealt with by use of specific pharmacologic blockers before studying the effects on ventilation of gas changes. We found that either AP5, an NMDA receptor blocker, or 7NI, an nNOS blocker, attenuated, to a closely similar degree, the posthyperoxic hypoxic ventilatory enhancement. The enhancement was further suppressed, but still measurable, after concurrent administration of the two blockers. These findings support the role of the glutamatergic and nitrergic pathways in the posthyperoxic ventilatory enhancement found in other studies (2, 3), but point to multiple, and still not fully resolved, underlying neurotransmitter mechanisms.
Enhancement of the posthyperoxic hypoxic ventilatory response has previously been reported in conscious humans (3) and rats (2). In the former study the plausible involvement of the excitatory NMDA receptor neurotransmission was suggested on the basis of increased glutamine, a precursor of GLU, concentration in the blood and in the latter study was associated with NO action, since the enhancing effect was entirely abolished by 7NI. In contrast, in the present study we report only a partial diminution by 7NI of the posthyperoxic hypoxic ventilatory enhancement. We cannot be certain about the cause of the discrepancy, but a different experimental model may account for it. We used anesthetized, vagotomized rats and no enrichment of CO2 in inspired gas mixtures. The hypoxic ventilatory profile also was different, with ventilatory depression appearing within a couple of minutes, at a time when no changes in ventilation are yet seen in conscious rats (2).
The experimental model of this study allowed to exclude some potential mechanisms that might be responsible for the posthyperoxic hypoxic ventilatory enhancement. One such mechanism might be wakefulness during which hypoxia alters cortical activity (18) and central respiratory pattern generation (19). Anesthesia severs cortical pathways descending down to the respiratory brain stem areas. Thus cortical influences could not interact with respiration and its responses to hypoxia. Anesthesia used in this study, albeit potentially suppressant for ventilation, has a relatively mild and balanced effect on various, rather than any single, neurotransmitter systems (20) and should not factor much in ventilatory modifications observed. Likewise, local NO-related processes in the lungs modulating neuronal circuits controlling respiration via the vagus nerve (21) or the stimulatory effect on pulmonary stretch receptors of developing hypocapnia (22) would not modify breathing pattern in vagotmized animals. In fact, hypocapnia induced by hypoxic hyperventilation could blunt the magnitude of the nascending hypoxic ventilation, as it is a strong inhibitory stimulus for the hypoxic ventilatory response (23).
Both GLU and NO neurotransmitter systems are present and engaged in local transneuronal communication in the brainstem respiratory areas (24, 25). Hypoxic excitation of carotid body chemoreceptor afferent discharge releases GLU in the solitary tract nucleus (5, 24), which through the activation of NMDA receptors is thought to underlie excitatory synaptic inputs to respiratory neurons (26). Hypoxia also seems to have a direct, carotid body independent excitatory effect on respiratory neurons. It induces release of GLU and activates respiratory neuronal discharges in in vitro preparations devoid of carotid chemoreceptor input (27, 28). Activation of neuronal NMDA receptors is closely linked to the metabolism of NO, as the accompanying intracellular calcium signal activates NOS associated with the postsynaptic density-95 protein to produce NO (8, 29). Postsynaptically released NO can act as a retrograde messenger to further the presynaptic release of GLU (11, 12). GLU may also affect respiratory neuronal transmission via nonNMDA receptors (30). The role of this possibility in the hypoxic ventilatory enhancement remains to be explored by using other pharmacologic tools.
Since the two neurotransmitter systems are interrelated, the resolution of the issue of which system predominates in the posthyperoxic hypoxic ventilatory enhancement is not easy. Blockade of either NMDA glutamate receptors or NO would suppress the NO-mediated actions. An independent effect mediated by NMDA receptors alone would be liable to potentiate suppression of the posthyperoxic hypoxic ventilatory enhancement, when blockers of both systems are applied simultaneously. Such potentiation, to some extent, was observed in the present study (Fig. 2D, Table 2). We believe, however, that the study findings have not proved the explicit role of either NMDA or NO neurotransmitter system in the phenomenon. Sustainment of the posthyperoxic hypoxic ventilatory enhancement after the combined blockade of both systems suggests the involvement of multiple mechanisms and may be an example of the brain stem's ability to modify its respiratory function under a changing environment.
The study findings show that the biphasic profile of the hypoxic ventilatory response was retained after blockade of either the glutamatergic or nitrergic or both systems together. Moreover, the magnitude of a decrease from the ventilatory peak level remained proportionate to that in the basic control condition. These results do not support an active involvement of GLU or NO in upholding ventilation during developing central depression as suggested, particularly for NO, in some other studies (31). In accordance with other studies we found a significant degree of hypotension during the ventilatory depressant phase (17). Changes in blood pressure may influence respiration through the carotid baroreceptor reflex. These changes were, however, similar in all hypoxic tests in all the experimental groups of this study. Besides, prevention of the late hypoxic drop in blood pressure does not alter the ventilatory profile or magnitude of a hypoxic response (17). Therefore, it seems unlikely that blood pressure changes could factor in the results obtained.
One limitation of the present study is that we used diaphragmatic muscle activity as an index of ventilation. The possibility arises that gas changes could lead to recruitment of muscle fibers without actual changes in respiratory drive. Muscle activity would then not accurately reflect the descending neural drive. However, short-term hypoxia in anesthetized rats, as opposed to conscious animals, has a definite inhibitory effect on diaphragm muscle activity (32, 33). Little is known about the role of GLU in diaphragmatic muscle contraction under hypoxic conditions. On the other hand, endogenous NO increases in the hypoxic diaphragm muscle and seems important for the optimal muscle function (34). An intact diaphragm should then better reflect physiologic conditions and could blunt, rather than overestimate, the hypoxic ventilatory enhancement after blockade of the nitrergic pathway.
In summary, the results of this study suggest that the posthyperoxic hypoxic ventilatory enhancement is a probable result of multiple neurotransmitter mechanisms. The role of the posthyperoxic hypoxic ventilatory enhancement in hypoxic homeostasis is unclear. Insofar as the hypoxic ventilatory response functions to maintain tissue oxygenation during hypoxemia, the phenomenon has potential clinical implications. Brief hyperoxic exposures could increase hypoxic reactivity and consequently be advantageous for tissue oxygenation, which could lead to the deliberate use of such exposures in hypoxic pathologies. Given the significant effects that antecedent oxygen has on hypoxic ventilation, further research on the underlying mechanisms is clearly of interest.
Acknowledgments: This study was funded by the statutory budget of the Polish Academy of Sciences Medical Research Center. E. Kolesnikova was supported by an intergovernmental research exchange program between the Polish Academy of Sciences and the National Academy of Sciences of Ukraine. The authors are grateful to Ms. E. Wielechowska for excellent technical assistance.
- Marczak M, Kolesnikova E E, Pokorski M. Hypoxic ventilatory profile in the anesthetized rat. J Physiol Pharmacol 2004; 55 Suppl. 3: 89-94.
- Gozal D. Potentiation of hypoxic ventilatory response by hyperoxia in the conscious rat: putative role of nitric oxide. J Appl Physiol 1998; 85: 129-132.
- Honda Y, Tani H, Masuda A et al. Effect of prior O2 breathing on ventilatory response to sustained isocapnic hypoxia in adult humans. J Appl Physiol 1996; 81: 1627-1632.
- Pokorski M, Marczak M, Jenajczyk U. Augmentation of hypoxic respiration after brief hyperoxia in the anesthetized cat: Putative function of GABAA neurotransmission. J Biomed Sci 2004; 11: 322-330.
- Mizusawa A, Ogawa H, Kikuchi Y et al. In vivo release of glutamate in nucleus tractus solitarii of the rat during hypoxia. J Physiol (Lond) 1994; 478: 55-66.
- Prabhakar NR, Cherniack NS, Haxhiu MA. Inhibitory and excitatory effects of nitric oxide on respiratory responses to hypoxia. In Ventral Brainstem Mechanisms and Control of Respiration and Blood Pressure. OC Trouth, RM Millis, HF Kiwull-Schöne, ME Schläfke (eds). New York, Marcel Dekker, 1995, vol. 82, pp. 393-404.
- East SJ, Garthwaite J. NMDA receptor activation in rat hippocampus induces cyclic GMP formation through the L-arginine-nitric oxide pathway. Neurosci Lett 1991; 123: 17-19.
- Garthwaite J, Charles SL, Chess-Williams R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988; 336: 385-388.
- Schuman EM, Madison DV. Nitric oxide and synaptic function. Annu Rev Neurosci 1994; 17: 153-183.
- Ogawa H, Mizusawa A, Kikuchi Y, Hida W, Miki H, Shirato K. Nitric oxide as a retrograde messenger in the nucleus tractus solitarii of rats during hypoxia. J Physiol (Lond) 1995; 486: 495-504.
- Stanton PK, Winterer J, Bailey CP et al. Long-term depression of presynaptic release from the readily releasable vesicle pool induced by NMDA receptor-dependent retrograde nitric oxide. J Neurosci 2003; 23: 5936-5944.
- Jurado S, Sanchez-Prieto J, Torres M. Differential expression of NO-sensitive guanylyl cyclase subunits during the development of rat cerebellar granule cells: regulation via N-methyl-D-aspartate receptors. J Cell Sci 2003; 116: 3165-3175.
- Volke V, Wegener G, Vasar E. Augmentation of the NO-cGMP cascade induces anxiogenic-like effect in mice. J Physiol Pharmacol 2003; 54: 653-660.
- Ignarro L.J. Nitric oxide as a unique signaling molecule in the vascular system: A historical overview. J Physiol Pharmacol 2002; 53: 503-514.
- Kline DD, Overholt JL, Prabhakar NR. Mutant mice deficient in NOS-1 exhibit attenuated long-term facilitation and short-term potentiation in breathing. J Physiol (Lond) 2002; 539: 309-315.
- Smith JB, Ogonowski AA. Behavioral effects of NMDA receptor agonists and antagonists in combination with nitric oxide-related compounds. Eur J Pharmacol 2003; 471: 121-128.
- Richter DW, Schmidt-Garcon P, Pierrefiche O, Bischoff AM, Lalley PM. Neurotransmitters and neuromodulators controlling the hypoxic respiratory response in anaesthetized cats. J Physiol (Lond) 1999; 514: 567-578.
- Pokorski M, Trojecka A, Marczak M, Wierzbicka A, Jernajczyk W. Cortical activity during hypoxic hyperventilation. J Physiol Pharmacol 2003; 54 Suppl. 1: 29-34.
- Lovering AT, Dunin-Barkowski WL, Vidruk EH, Orem JM. Ventilatory response of the cat to hypoxia in sleep and wakefulness. J Appl Physiol 2003; 95: 545-554.
- Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg 2002; 94: 313-318.
- Iben S.C., Dreshaj IA, Farver CF, Haxhiu MA, Martin RJ. Role of endogenous nitric oxide in hyperoxia-induced airway hyperreactivity in maturing rats. J Appl Physiol 2000; 89: 1205-1212.
- Davies A, Dixon M, Callanan D, Huszczuk A, Widdicombe JG, Wise JC. Lung reflexes in rabbits during pulmonary stretch receptor block by sulphur dioxide. Respir Physiol 1978; 34: 83-101.
- Jounieaux V, Parreira VF, Aubert G, Dury M, Delguste P, Rodenstein DO. Effects of hypocapnic hyperventilation on the response to hypoxia in normal subjects receiving intermittent positive-pressure ventilation. Chest 2002; 121: 1141-1148.
- Vardhan A, Kachroo A, Sapru H.N. Excitatory amino acid receptors in commissural nucleus of the NTS mediate carotid chemoreceptor responses. Am J Physiol 1993; 264: R41-R50.
- Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience 1992; 46: 755-784.
- Ohtake PJ, Torres JE, Gozal YM, Graff GR, Gozal D. NMDA receptors mediate peripheral chemoreceptor afferent input in the conscious rat. J Appl Physiol 1998; 84: 853-861.
- Ramirez JM, Quellmalz UJA, Wilken B, Richter DW. The hypoxic response of neurons within the in vitro mammalian respiratory network. J Physiol (Lond) 1998; 507: 571-581.
- Richter DW, Bischoff AM, Anders K, Bellingham M, Windhorst U. Response of the medullary respiratory network of the cat to hypoxia. J Physiol (Lond) 1991; 443: 231-256.
- Sheng M, Pak DT. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol 2000; 62: 755-778.
- Pierrefiche O, Foutz AS, Champagnat J, Denavit-Saubie M. NMDA and non-NMDA receptors may play distinct roles in timing mechanisms and transmission in the feline respiratory network. J Physiol (Lond) 1994; 474: 509-523.
- Gozal D, Gozal E, Torres JE, Gozal Y, Nuckton TJ, Hornby PJ. Nitric oxide modulates ventilatory responses to hypoxia in the developing rat. Am J Respir Crit Care Med 1997; 155: 1755-1762.
- Bonora M, Vizek M. Ventilation, EELV and diaphragmatic activity in rats during the early phase of normobaric hypoxia. Respir Physiol 2001; 128: 131-45.
- Pierce JD, Clancy RL. Effects of hypoxia on diaphragm activity in anesthetized rats. J Perianesth Nurs 2001; 16: 181-186.
- Zhu X, Heunks LM, Machiels HA, Ennen L, Dekhuijzen PN. Effects of modulation of nitric oxide on rat diaphragm isotonic contractility during hypoxia. J Appl Physiol 2003; 94: 612-620.