Peptic ulcer is generally considered as a
result of the imbalance between the gastroduodenal mucosal defense mechanisms
and damaging factors breaching in the mucosa and extending through the
muscularis
mucosae into the submucosa and deeper. The recognition by Schwartz (1) in
the first decade of the twentieth century that the formation of gastroduodenal
ulcers is caused by the aggressive action of acid (his
famous dictum
was, "no acid-no ulcer") and that the decrease of gastric acid favors the repair
of gastric lesions, changed the surgical way of thinking and opened new avenues
of anti-ulcer strategies. The discovery of cytoprotective effects of prostaglandins
(PG) on gastric mucosa in experimental animals (2) raised a hope that these
PG may be an ideal remedy for ulcer prevention and healing, but subsequent clinical
trials failed to support their clinical usefulness in ulcer therapy, mainly
due to side effects. With the identification of the histamine-2 receptor subtype
and the development by Black and his associates (3) of agents specifically capable
of blocking acid secretion by antagonism of this receptor revolutionized the
management of peptic ulcer disease and virtually obliterated surgery as a therapeutic
option for peptic ulcer disease, except that in the case of an emergency and
complications. With discovery of drugs that inhibit H
+,K
+ATPase,
the proton pump of the parietal cell (4), the most effective inhibitors of gastric
acid secretion so called proton pump inhibitors (PPI) are available and widely
used in ulcer therapy (5).
Although gastric acid and pepsin are requisites for ulcer formation (1) and
appropriate acid suppression is required for optimal ulcer healing (6, 7), the
most notable aggressive factor in pathogenesis of peptic ulcerations appears
to be
Helicobacter pylori (
H. pylori) (8-10). Most of peptic ulcers have
been associated either with gastric
H. pylori infection or with ingestion of
nonsteroidal anti-inflammatory drugs (NSAID). Numerous studies showed that peptic
ulcer recurred infrequently when either
H. pylori infection or NSAID use is
eliminated (10). With the recognition of important role of
H. pylori in pathogenesis
of peptic ulcer, its healing and recurrence, the antisecretory therapy has been
combined with antimicrobial treatment in order to accelerate ulcer healing,
to reduce ulcer complications and to prevent ulcer recurrence (10).
Following the discovery of cytoprotective activity of PG, stable PGE analogues
were obtained, suggesting that they could be useful in the treatment of peptic
ulcer, particularly that they were found to be effective gastric acid inhibitors
in humans (11). Indeed, several clinical trials (12-14) documented that these
analogues were effective in accelerating healing rate of gastroduodenal ulcers
not only accompanying NSAID therapy, when the deficiency of endogenous PG exists
(15), but also in NSAID-independent peptic ulcerations (12-14). It was found
that PGE
1 stable analog misoprostol, significantly
lowered the frequency of gastroduodenal ulcers occurring in patients with long
term therapy of NSAID (15). This analogue was, however, effective in enhancing
peptic ulcer healing mainly by gastric acid inhibition than by cytoprotective
activity, indicating that cytoprotection, exerting so dramatic preventive action
against acute gastric lesions in experimental animals (2), plays no part in
healing of chronic peptic ulcer that involves mucosal repair (16). Although
in patients at high risk for recurrent gastric ulcer, the use of cotherapy with
misoprostol was found to be almost equally affective as PPI such as lanzoprazole
or omeprazole (17, 18), exogenous PGE or its stable analogues are not widely
used in peptic ulcer therapy because of their diarrhogenic and abortifaciant
effects. This, "unfulfilled promise" (16), regarding the clinical usefulness
of prostaglandin in peptic ulcer therapy, does not exclude the possibility that
endogenous PG generated by cyclooxygenase (COX)-1 or COX-2 in the ulcer area
are implicated in the pathogenesis and healing of peptic ulcerations.
The purpose of this article is to overview the mechanisms of ulcer healing in experimental model of acetic acid-induced chronic gastric ulceration in rats, especially the role of endogenous PG generated by COX-1 and COX-2.
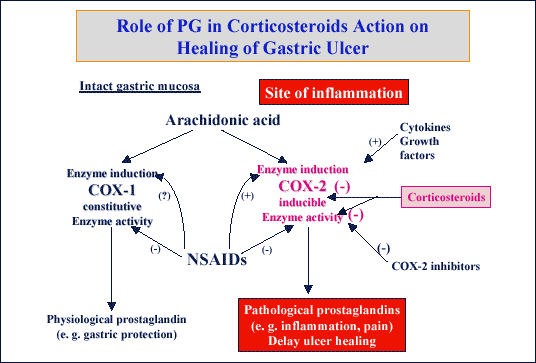
Role of exogenous and endogenous PG in healing of peptic ulcers
Healing of peptic ulcer is an active and complex process including the reconstruction of the mucosa by formation of granulation tissue at the ulcer base, formation of new vessels (angiogenesis) and re-establishment of glandular architecture (19). PG generated especially at an ulcer margin by COX-2, appear to play a crucial role in ulcer healing through triggering the cell proliferation, promotion of angiogenesis and restoration of mucosal integrity. Unlike COX-1, which is constitutively expressed in intact gastric mucosa to produce PG that regulate mucosal blood flow and epithelial secretion of mucus and bicarbonate, PGs from COX-2 influence epithelial proliferation and endothelial-leukocyte adherence. COX-2 has been shown to be induced in ulcerated and inflamed gastric mucosa (20-24).
In this study, we used an experimental ulcer model obtained by serosal application
of 100% acetic acid on the area of 28 mm
2 for
25 s according to our modified method (25) (
Fig. 1). Histologically,
such acute ulcer develops immediately after serosal application of acetic acid
on mid portion of the stomach and involves the entire mucosa and submucosa to
become chronic within 2-3 days. It heals spontaneously depending on initial
size within 2-4 weeks without perforation or penetration to surrounding organs.
After recovery from surgery, the animals start normal chow diet next day after
ulcer induction and can be treated either with vehicle (saline) or various substances
such as PG, PPI, COX-inhibitor, growth factors or gut hormones. The animals
were then lightly anesthetized with ether after 3, 7, 10 or 14 days upon ulcer
induction, the abdomen was opened and the gastric mucosal blood flow at the
ulcer margin was determined using H
2-gas clearance
technique. The stomach was then opened and the area of gastric ulcers was determined
using planimetry. In addition, the large (50 mg) of biopsy samples were taken
from the ulcer margin and intact mucosa and immediately frozen in liquid nitrogen
for further studies of gene or protein expression of COX-1, COX-2. The blood
samples were also taken for the assessment of plasma levels of gastrin, melatonin
and cytokines as described before (20, 26). Each experimental group included
6-10 animals that were fasted about 24 h before the anesthesia. The studies
were approved by Institutional Ethic Committee of the Jagiellonian College of
Medicine, Cracow, Poland.
 |
| Fig. 1.
Production of gastric ulcer by serosal application of acetic acid in rats
treated daily with prostaglandins (PG), proton pump inhibitors, COX-1
or COX-2 inhibitors, growth factors, gut hormones or melatonin without
and with pretreatment with neurotoxic dose of capsaicin. At the end of
experiments the animals were anesthetized and the area of gastric ulcers
were measured by planimetry, mucosal blood flow was measured by H2
gas clearance and biopsy samples were taken for the determination of mucosal
generation of PGE2 and expression of
COX-1 or COX-2. |
As shown on
Fig 2, in rats with chronic acetic acid-induced gastric ulcer
the mRNA expression for COX-1 was similar in the intact mucosa and at ulcer
margin with gastritis as well as in the ulcer base. It did not change significantly
also following healing of gastric ulcer. In contrast, the expression for COX-2
markedly increased both in the margin of ulcer as well as in ulcer itself and
disappeared following the ulcer healing. PGE
2
generation rose significantly at the ulcer margin and the ulcer base as compared
to the intact mucosa to decline after ulcer healing. These results indicate
that the induction of gastric ulcer and accompanying gastritis induce dramatic
rise in expression of COX-2 as reported previously (20).
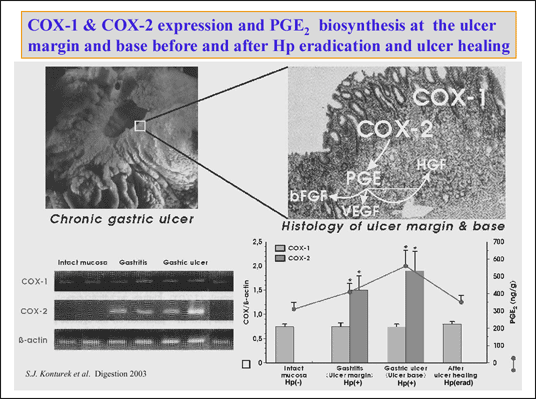 |
| Fig. 2.
Macrosopic and microscopic pictures of chronic gastric ulcer in rats induced
by serosal application of acetic acid (upper panel). COX-1 and COX-2 expression
and mucosal generation of PGE2 in H.
pylori negative intact gastric mucosa, at the ulcer margin H. pylori
postive with gastritis, at the H. pylori positive ulcer base and
in the area of healed H. pylori eradicated ulcer. COX-2 generated
PGE2 stimulates the expression of growth
factors in the mucosa. |
It is of interest that PG generated from COX-1 tonically suppress COX-2 activity
in the GI tract. COX-2 is rapidly up-regulated after COX-1 inhibition, when
the mucosa is exposed to potentially damaging agents or when the mucosal injury
or ulceration occurs. Recent studies showed that PGE
2
release by fibroblasts at the ulcer margin expressing COX-2 is accompanied by
the release of growth factors in the ulcer area that may contribute to mucosal
repair and angiogenesis (26) (
Fig. 3).
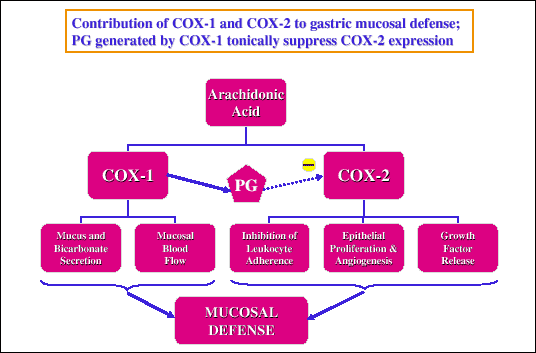 |
| Fig. 3.
Contribution of COX-1 and COX-2 to various aspects of gastric mucosal
defense. PGE2 generated by COX-1 tonically
suppresses COX-2 activity. |
The question remains what is the effect of exogenous PG on ulcer healing and
COX-1 and COX-2 expression in the ulcer area. We demonstrated before (25) that
the small non-antisecretory dose of 16,16 dimethyl PGE
2
(dmPGE
2) failed to affect the healing and these
results have not been included. In this study we used larger dose of dmPGE
2,
which in experiments with chronic gastric fistula rats caused significant inhibition
of gastric acid secretion. Such larger dose of this PGE
2
analogue (50 µg/kg/d) was found in the present study to be as effective in the
acceleration of ulcer healing as omeprazole used in equipotent gastric inhibitory
dose (40 mg/kg/d). In vehicle-treated rats, the ulcer area gradually decreased,
the reduction in ulcer area being significant at day 7 and 10 to disappear almost
completely at day 14 (
Fig. 4). Thus, we can conclude that exogenous PGE
analogue, applied in larger dose, was equally effective in acceleration of ulcer
healing as omeprazole administered in equipotent gastric inhibitory dose.
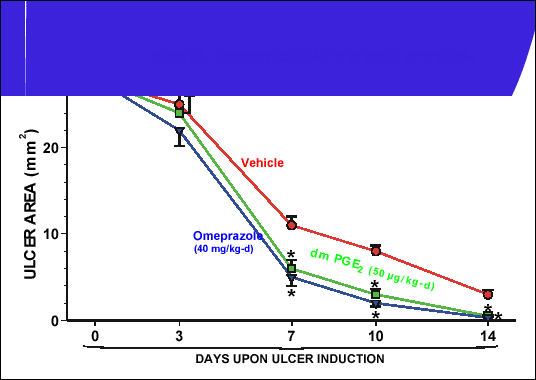 |
| Fig. 4.
Healing of gastric ulcers in rats treated with intragastric vehicle (saline),
dmPGE2 (50 µg/kg/d) or omeprazol 40 mg/kg/d.
Mean ±SEM of 6 experiments on 6 rats. Asterisk indicates significant decrease
below the value recorded in vehicle-treated animals (unpublished results). |
The question remained whether the ulcer healing effect of exogenous PGE analogue
and omeprazole, representing PPI, is only due to the inhibition of gastric acid
secretion or whether these agents also affect COX-PG system in the ulcerated
mucosa.
Fig. 5 shows that administration of exogenoms PGE
2
analogue or omeprarole in gastric inhibitory dose caused significant increase
in mucosal generation of PGE
2, especially in
the first days of drug application. As shown on
Fig.6, COX-1 showed similar
expression in the intact mucosa and at ulcer margin in rats without or with
administration of dmPGE
2 or omeprazole. In contrast,
COX-2, which showed only negligible expression in the intact mucosa was pronounced
at the ulcer area even of vehicle-treated controls, but treatment with dmPGE
2
or omeprazole resulted in further significant elevation of COX-2 expression
in the ulcerated mucosa (
Fig. 7). Thus, the excessive generation of PGE
2
in dmPGE
2- or omeprazole-treated rats originated
from the upregulation of COX-2 at the ulcer margin by the tested agents.
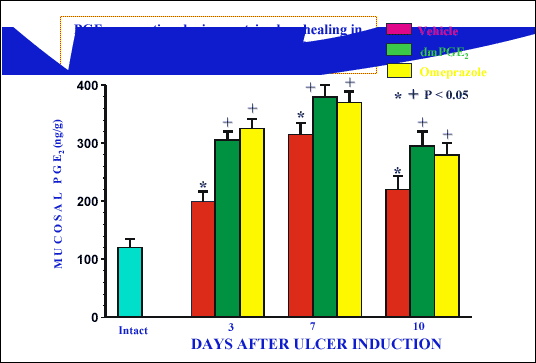 |
| Fig. 5.
Mucosal PGE2 generation during ulcer
healing in rats treated with dmPGE2 (50
µg/kg/d), omeprazole (40 mg/kg/d) or vehicle (unpublished results). |
 |
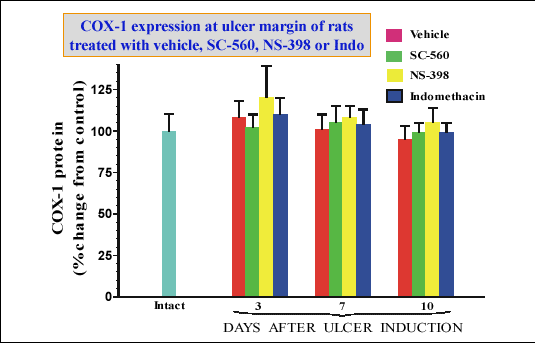
| Fig. 6.
COX-1 expression in intact gastric mucosa and at ulcer margin of rats
treated with vehicle, dmPGE2 or omeprazole
at day 3, 7 and 10 upon ulcer induction (unpublished results). |
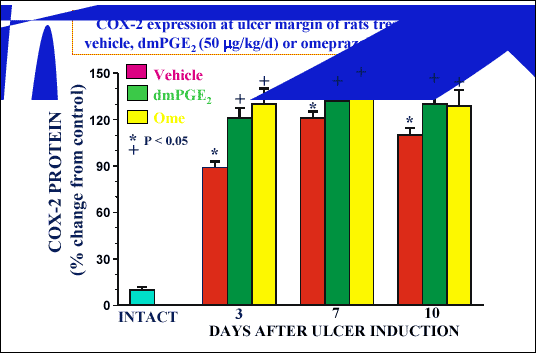 |
| Fig. 7.
COX-2 expression in intact gastric mucosa and at ulcer margin of rats
treated with vehicle, dmPGE2 or omeprazole
at day 3, 7 and 10 upon ulcer induction. Asterisk indicates significant
increase above the value recorded in gastric mucosa of intact rats. Cross
indicates significant increase above the value recorded in vehicle-treated
rats (unpublished results). |
The mechanism of COX-2 upregulation at an ulcer margin, particularly following
treatment with dmPGE
2 or omeprazole is unknown,
but we suspect that this could be attributed, at least in part, to the hypergastrinemia
that was found to be accompanying the production of gastric ulceration itself
and the administration of gastric inhibitory dose of dmPGE
2
or omeprazole (
Fig. 8). The upregulation of COX-2 by gastrin, released
in higher amounts following administration of gastric inhibitors such as lanzoprazole,
has been suggested before by Tsuji
et al. (28), who reported that the
protective effects of this PPI, against ethanol-induced gastric damage could
be attributed to the upregulation of COX-2 due to the action of gastrin released
in excessive amounts because the blockade of specific gastrin receptors abolished
lanzoprazole-induced enhancement of PGE
2-generation
and the upregulation of COX-2. The acceleration of ulcer healing combined with
upregulation of COX-2 and elevated generation of PGE
2
in our tests with dmPGE
2 could also be attributed
to hypergastrinemia resulting from gastric inhibition by this PGE
2-analogue
as reported previously(11-13). An alternative explanation could be that exogenous
stable PGE analogue by itself could directly increase the COX-2 expression and
activity in similar fashion to that exerted by this PGE analogue in prostate
cancer cells (29) but this possibility requires confirmation in the model of
gastric ulceration (
Fig. 7).
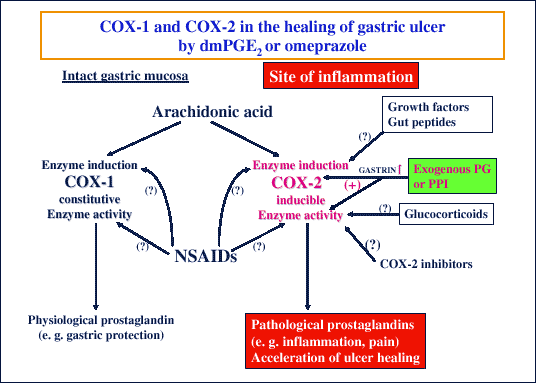 |
| Fig. 8.
Schematic presentation of the arachidonic metabolism via COX-1
and COX-2 under physiological and pathological conditions such as gastric
ulcerations treated with exogenous dmPGE2
or proton pump inhibitor (PPI) such as omeprazole. Enhanced release of
plasma gastrin is possibly responsible for the upregulation of COX-2 at
the ulcer margin and accelerated ulcer healing. |
In summary, the ulcer healing efficacy of omeprazole (and probably other PPI)
and exogenous potent PGE analogues could be attributed not only to their gastric
acid inhibitory action but also to the upregulation of COX-2 in the ulcer area
(
Fig. 8).
If endogenous PGE
2 generated by the upregulated
COX-2 at the ulcer margin, contributes to ulcer healing it is expected that
the inhibition of COX-1 and/or COX-2 should delay ulcer healing as shown by
other studies (30) including our own (20). This delay in ulcer healing by NSAID
has been associated with the inhibition of endothelial cell proliferation and
the reduction in angiogenesis at the ulcer site. In the present report we confirmed
that specific inhibitor of either COX-1 (SC-56) or COX-2 (NS-398) as well as
nonspecific inhibitor of both COX-1 and COX-2 (indomethacin) delayed ulcer healing
(
Fig. 9). This delay in ulcer healing by COX-inhibitors was accompanied
by expected strong reduction in PGE
2 generation,
especially in ulcerated gastric mucosa normally exhibiting more pronounced release
of PGE. The inhibition of PGE
2 generation was
more impressive after the application of indomethacin than of specific COX-2
inhibitor (NS-398) because the former agent is known to inhibit non-specifically
both COX-1 and COX-2 activity, and, therefore, is more effective inhibitor of
PGE
2 generation than NS-398 (
Fig. 10).
This remains in agreement with studies of Wallace and Devchand (26), who proposed
that selective inhibition of only COX-1 or only COX-2 activity results in rather
small mucosal damage, but suppression of both isoforms of COX, as achieved with
indomethacin, causes significantly more pronounced mucosal damage. As endogenous
PGE
2 release was more suppressed by indomethacin
than by NS-398, it is obvious that the COX-2 expression was more enhanced with
indomethacin than with NS-398.
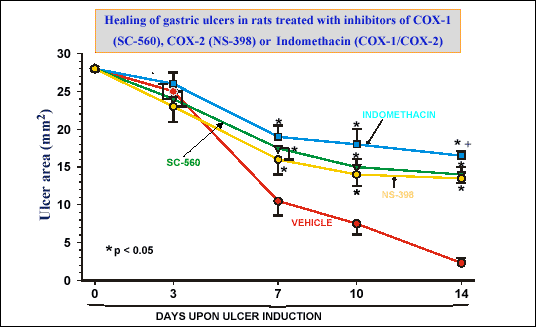 |
| Fig. 9.
Mean area of gastric ulcers measured at day 0, 3, 7, 10 and 14 upon ulcer
induction in vehicle-treated control rats and those treated with indomethacin,
SC-560 or NS-398. Mean ± SEM of 6 experiments on 6 rats. Asterisk indicates
significant increase, above the value in vehicle-treated rats (unpublished
data) |
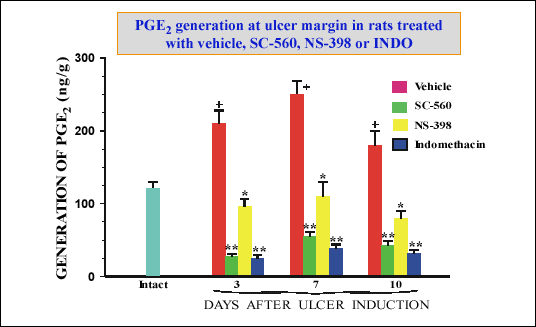 |
| Fig. 10.
PGE2 generation in the ulcerated and
non-ulcerated gastric mucosa of rats treated with vehicle, SC-560, NS-398
or indomethacin. Mean ± SEM of 6 experiments on 6 rats. Single asterisk
indicates significant decrease, below the value in vehicle-treated rats.
Double asterisks indicate significant decrease below the value obtained
after inhibition of COX-2 by NS398. (unpublished data) |
Davies
et al (31) were first to observed significant upregulation of
COX-2 in the rats following the administration of aspirin and suggested that
diminished mucosal generation of PGE
2 by this
NSAID was responsible for triggering this upregulation of COX-2. In this study
we found that marked reduction in PGE
2 generation
due to inhibition of COX-1 and COX-2 activity was not accompanied by any change
in expression of COX-1 (
Fig. 11). In contrast, the COX-2 expression was
significantly upregulated in tests with indomethacin but not with NS-398 that
produced smaller fall in PGE
2 generation. (
Fig.
12). It is of interest that even small doses of exogenous dmPGE
2
given to rats treated with COX-1 or COX-2 inhibitors restored completely the
healing of gastric ulcers and increased mucoal blood flow (
Fig. 13).
These observations lead to hypothesis that inhibition of COX-1 activity is associated
with delay of ulcer healing and that decrease in local PGE
2
release is combined with an increase in COX-2 expression in the ulcerated mucosa
(
Fig. 14). Thus, inhibition of COX-1 and COX-2 activity by nonselective
inhibitors such as indomethacin reduces COX activity and elevates the expression
of COX-2 at the ulcer area, while specific COX-2 inhibitor does not affect the
expression of this COX isoform (
Fig. 14).
 |
| Fig. 11.
COX-1 expression in intact gastric mucosa and at ulcer margin of rats
treated with vehicle, SC-560, NS-398 or indomethacin or at day 3, 7 and
10 upon ulcer induction (unpublished results). |
 |
| Fig. 12.
COX-2 expression in intact gastric mucosa and at ulcer margin of rats
treated with vehicle, indomethacin or SC-560 or NS-398 at day 3, 7 a dn10
upon ulcer induction. Asterisk indicates significant increase above the
value recorded in gastric mucosa of vehicle-treated rats. Cross indicates
significant increase above the value recorded in vehicle-treated rats
(unpublished results). |
 |
| Fig. 13.
It is of interest that minute amount of exogenous dmPGE2
(5 µg/kg/d) administered to rats receiving vehicle, SC-560, NS-398 or
indomethacin abolished the delay in ulcer-healing and restored gastric
blood flow caused by pretreatment with COX-1 or COX-2-inhibitor or by
indomethacin. Asterisk iondicates significant change as compared to vehicle-treated
rats, Cross indicates significant change as compared to the value obtained
in SC-560, NS-398 or indoemthacin administration. (unpublished results) |
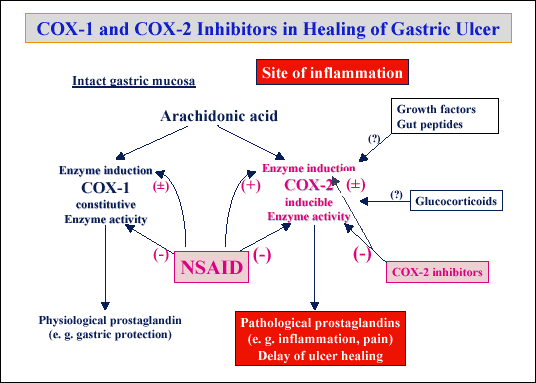 |
| Fig. 14.
Schematic presentation of the arachidonic metabolism via COX-1
and COX-2 under physiological and pathological conditions such as gastric
ulcerations treated with nonspecific NSAID or specific COX-2 inhibitors; |
Effects of corticosteroids on peptic ulcer healing
The ulcerogenic effect of corticosteroids in the stomach is controversial. While some investigators suggested that there is no association between corticosteroids therapy and ulcerogenesis, others emphasized an increased risk of peptic ulcer and its complications (33-37) or reported that peptic ulcer is rather rare complications of corticosteroid therapy (34) and prophylaxis should be considered only in patients with increased risk factors such as concurrent NSAID therapy or previous history of peptic ulceration (35,36). Animal experiments with gastric ulcers showed that hydrocortisone (37), dexamethasone (38) or prednisolone (39) delayed gastric ulcer healing and this healing could be improved by the addition of exogenous PGE (37, 39).
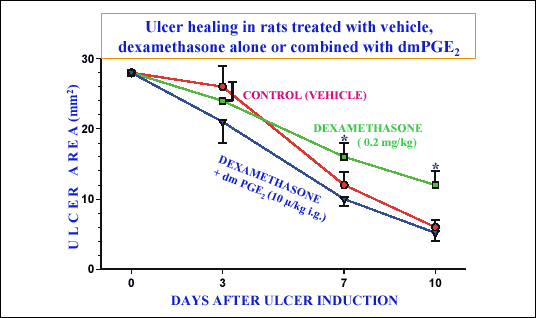 |
| Fig. 15.
Mean area of gastric ulcers measured at day 0, 3, 7 and 10 upon ulcer
induction in vehicle-treated control rats and those treated with vehicle,
dexamethasone (0.2 mg/kg) alone and dexamethasone combined with dmPGE2
(10 µg/kd) Mean ± SEM of 6 experiments on 6 rats. Asterisk indicates significant
increase, above the values in vehicle-treated rats. (unpublished results) |
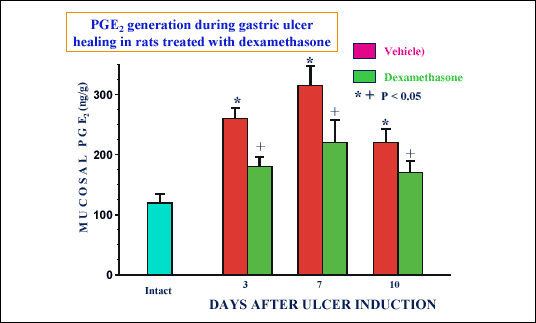 |
| Fig. 16.
Mucosal generation of PGE2 in intact
rats and in those with gastric ulceration treated with vehicle and dexamethasone
(0.2 g/kg/d) at day 3, 7 and 10 upon ulcer induction. Mean ± SEM of 6
experiments on 6 rats. Asterisk indicates significant increase above the
value recorded in intact mucosa. Cross indicates significant decrease
below the value recorded at ulcer margin in vehicle-treated rats (unpublished
results). |
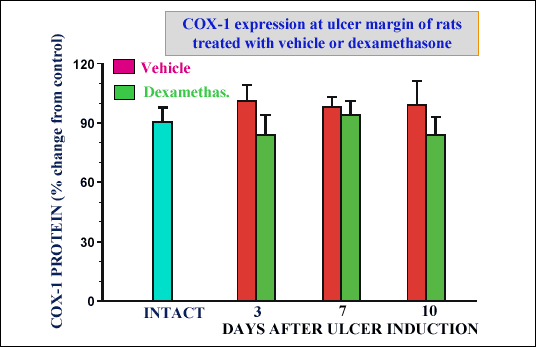
In this preliminary study we used non-ulcerogenic dose of dexamethasone (38)
and confirmed that such dose delayed gastric ulcer healing rate after 7 and
10 days of treatment (
Fig. 15). Addition of dmPGE at a small dose (10µg/kg/d),
that by itself failed to affect ulcer healing rate when given alone (data not
shown), reversed the delay of healing caused by dexamethosone. We confirmed
that dexamethasone reduced the generation of PGE
2
(
Fig. 16) and decreased both expression and activity of COX-2 but not
COX-1 at the ulcer area (
Figs 17 and
18). We can conclude, therefore,
that corticosteroids such as dexamethasone at the dose used delayed ulcer healing
most likely due to the inhibition of both expression and activity of COX-2 (
Fig.
19). Our results coincide with previous reports showing that dexamethasone
at non-ulerogenic dose (38) causes an inhibition of both COX-1 and COX-2 activity
and this is required for induction by this corticosteroid of gastric mucosal
damage (40). The mechanism by which dexamethasone induced reduction in COX-2
expression and attenuated PGE
2 formation could
delay ulcer healing is not obvious, but it could be related to the decrease
in the proliferation of gastric epithelial cells (41) and angiogenesis (42),
that are normally enhanced by COX-2-derived PGE
2
and associated with induction of hepatocyte growth factor expression (41). Depletion
of mucosal PGE
2 by dexamethasone seems to play
a key role in delay of ulcer healing as supplementation with dmPGE
2
returned the ulcer healing rate back to normal level observed in vehicle-treated
animals. In conclusion, ulcer production by acetic acid activate the repair
system in the gastric mucosa including epithelial cell proliferation and angiogenesis
at ulcer margin that are mediated by COX-2-PGE
2
and hepatocyte growth factor production. The interference of dexamethazone in
COX-2-PGE
2 system appears to deter the above
repair mechanisms leading to worsening of the ulcer healing process (
Fig.
19).
 |
| Fig. 17.
COX-2 expression in the mucosa of intact rats and at the ulcer margin
of rats treated with vehicle or dexamethasone. Mean ± SEM of 6 experiments
on 6 rats. Asterisk indicates significant increase above the value recorded
in intact mucosa. Cross indicates significant decrease below the value
recorded in vehicle-treated rats (unpublished results). |
 |
| Fig. 18.
COX-1 expression in intact gastric mucosa and that at ulcer margin of
rats treated with vehicle or dexamethasone at day 3, 7, and 10 upon ulcer
induction. Mean ± SEM of 6 experiments on 6 rats (unpublished results) |
 |
| Fig. 19.
Schematic presentation of the inhibitory action of corticosteroids on
COX-2 induction and COX-2 activity in gastric mucosa of rats with gastric
ulcer. |
Effects of growth factors and gut hormones on ulcer healing
It is well-known that ulcer healing involves local expression of various growth
factors in the ulcer area such as epidermal growth factor (EGF), transforming
growth factor (TGF
alpha), hepatocyte growth
factor (HGF) and basic fibroblast growth factor (bFGF) as well as gastrin (27).
According to our observations besides COX-PG system, the most effective in the
acceleration of ulcer healing are growth promoting factors such as EGF, HGF,
TGF
alpha and bFGF (
Fig. 20). These growth
factors were found to be expressed at the ulcer margin during ulcer healing
and could contribute to the healing process (43). It is of interest that expression
of these growth factors, coincides with the inhibition of gastric acid secretion
and increased mucosal blood flow at the ulcer margin as well as hypergastrinemia
(44). Administration of gastrin, that increases gastric acid secretion, also
accelerates ulcer healing (see
Fig. 20), indicating that the healing
effect of this hormone is unrelated to its gastric acid stimulation. Furthermore,
gastrin receptors (CCK
2-receptors) were found
to be expressed in the regenerative mucosal ulcer margin as demonstrated by
RT-PCR and autoradiography (45), reinforcing the concept that in addition to
growth factors, gastrin also contributes to the mucosal cell proliferation at
the ulcer margin (46, 47). It is of interest that the acceleration of ulcer
healing by growth factors or gastrin can be attenuated by local application
of antibodies against these growth factors or gastrin (27), emphasizing the
specificity of their ulcer healing promotion through stimulation of mucosal
growth and angiogenesis at the ulcer margin. It may be important to stress that
treatment with growth factors does not affect the gene expression of COX-1,
but elevates COX-2 expression in the ulcerated mucosa (
Fig. 21). As acceleration
of ulcer healing by growth factors can be delayed by the administration of COX-1
and COX-2 inhibitors such as indomethacin and is accompanied by the upregulation
of COX-2 at the ulcer margin, it is reasonable to conclude that COX-2 derived
PG mediate the acceleration of ulcer healing by various growth factors expressed
at the ulcer margin (
Fig. 22).
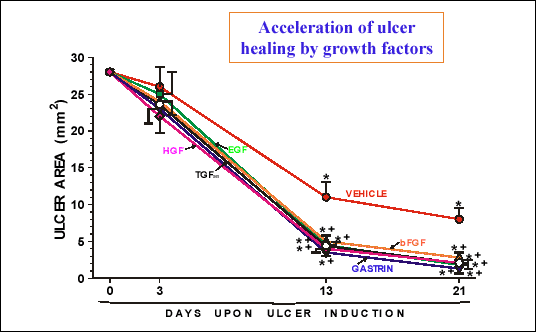 |
| Fig. 20.
Mean area of gastric ulcers in rats with daily treatment with various
growth factors and gastrin at a dose of 10 µg/kg/d injected i.p. Asterisk
indicates significant decrease below the value obtained at day 0. Cross
indicates significant decrease below the value obtained in rats treated
with vehicle. |
 |
| Fig. 21.
Gene expression of COX-1 and COX-2 (presented as ratio of COX to beta-actin)
in the intact gastric mucosa and at the gastric ulcer margin in rats treated
for 13 days with EGF, HGF and bFGF. Asterisk indicates significant increase
above the value recorded in intact mucosa. Cross indicates significant
increase above the value obtained in vehicle-treated mucosa. |
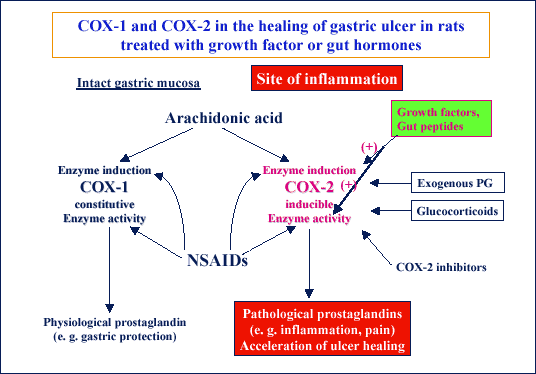 |
| Fig. 22.
Schematic presentation of the mechanism of stimulatory action of growth
factors and certain gut hormones such as gastrin on the induction of expression
and activity of COX-2 that contribute to the stimulation of ulcer healing
effects of these substances. |
The list of ulcer healing factors includes several others factors of gastrointestinal
origin such as cholecystokinin (CCK), gastrin releasing peptide (GRP), ghrelin,
leptin, somatostatin and insulin (50-53). As shown on
Fig. 23, the ulcer
healing activity of various gut hormones is accompanied by the stimulation of
mucosal growth (except somatostatin) and depends, in most instances, on the
mucosal generation of PG due to elevation of COX-2 expression (
Fig. 23).
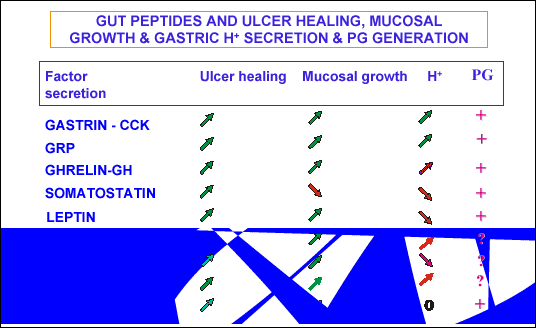 |
| Fig. 23.
List of gut hormones affecting the ulcer healing, mucosal growth, gastric
acid secretion and prostaglandin generation in the gastric mucosa |
The ulcer healing action of various gut hormones controlling food intake such as CCK, leptin or ghrelin is independent on gastric acid secretion but involves the activation of brain-gut axis
via stimulation of peripheral sensors and afferent nerves. This is supported by the finding that inactivation of sensory nerves with neurotoxic dose of capsaicin attenuated the ulcer healing effects of these appetite-regulating gut hormones (49-53).
Melatonin that was thought to originate primarily from the pineal glands, but
recently it has been detected in large amounts in the digestive organs, such
as stomach, gut and the pancreas (54, 55). Although its gastroprotective activity
has been attributed to scavenging of reactive oxygen species and increase of
antioxidative enzymes (56), we found that this indole, as well as its substrate
L-tryptophan, accelerates ulcer healing (
Fig. 24), at least in part,
by activation of COX-PG system because it was accompanied by the increase in
gastric mucosal generation of PGE
2 and the inhibition
of COX-1/COX-2 system by indomethacin delayed the ulcer healing promoted by
melatonin (
Fig. 25). Furthermore, the deactivation of sensory nerves
with neurotoxic dose of capsaicin reversed the healing acceleration by metalonin
and L-tryprophan and supplementation with calcitonin-gene related peptide (CGRP),
a neuropeptide that is deficient in such sensory deactivated animals, restored
the healing effects of melatonin and its precursor (
Fig. 26). Its is,
therefore, reasonable to assume that melatonin enhances ulcer healing through
the activation of brain-gut axis and stimulation of afferent sensory nerves
(54-58).
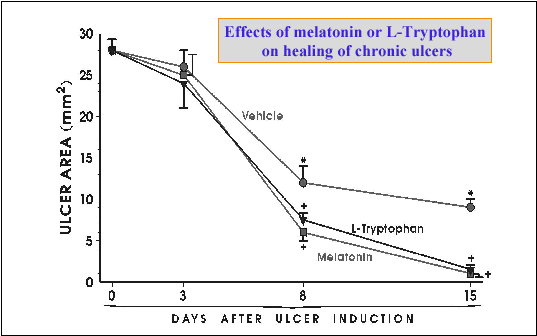 |
| Fig. 24.
Time sequence of healing rate of preexisting ulcers by melatonin and its
precursor, L-tryptophan. Asterisk indicates significant difference compared
to initial value at day 0. Cross indicates significant difference compared
to vehicle control. (unpublished results) |
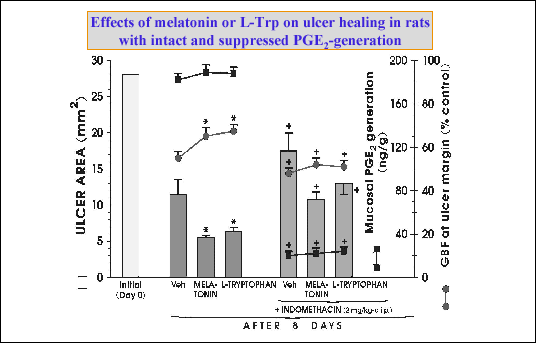 |
| Fig. 25.
Effects on melatonin and its substrate, L-tryptophan, on the area of gastric
ulcers, mucosal blood flow (GBF) and mucosal generation of PGE2
after 8 days upon ulcer production in rats without an with treatment with
indomethacin. Mean ± SEM of 6 experiments on 6 rats. Asterisk indicates
significant decrease below the value in vehicle-treated rats. Cross indicates
significant change as compared to the values recorded in rats without
pretreatment with indomethacin. (unpublished results) |
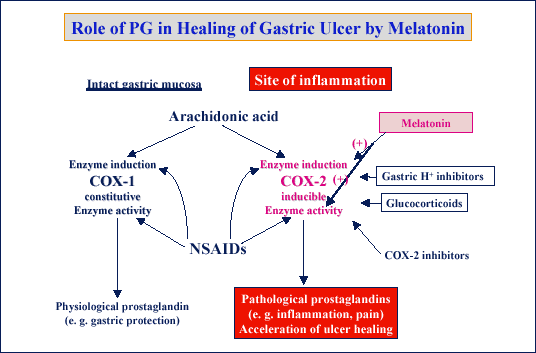 |
| Fig. 26.
Role of COX-1 and COX-2 expression in melatonin induced acceleration of
ulcer healing. |
As indicated in the introduction,
H. pylori infection and NSAID are known
to delay ulcer healing in humans but it is not clear how these two factors interact
on the healing process of experimental ulcer. Using our acetic acid model in
rats with or without
H. pylori infection, we found that infection significantly
delays ulcer healing as compared with vehicle control (
Fig.27). Similar
effects are exerted by the application of aspirin (50 mg/kg/d) or rofecoxib
(10 ng/kg/d). With the combination of
H. pylori infection and addition
of aspirin or rofecoxib, the ulcer area was significantly reduced as compared
to that obtained with aspirin or rofecoxib alone without
H. pylori infection.
The increased ulcerogenicity of aspirin or rofecoxib in rats without
H. pylori
infection could be simple attributed to the inhibiton of PG generation with
subsequent decrease in mucus alkaline secretion, attenuation of mucosal blood
flow and neutrophil adherence to vascular epithelium. The reduced ulcerogenicity
of aspirin and rofecoxib in
H. pylori-infected rats could result from
activation of the inflammatory cascade, release of various cytokines and most
important from the increased expression of COX-2 and enhanced generation of
PGE
2 by the presence of
H. pylori in
the ulcer area but this requires further documentation. Clinical implication
of this finding would be that the eradication of
H. pylori in NSAID ingesting
patients should not be recommended except when ulcer complications occur, but
this is the controversial issue requiring further studies.
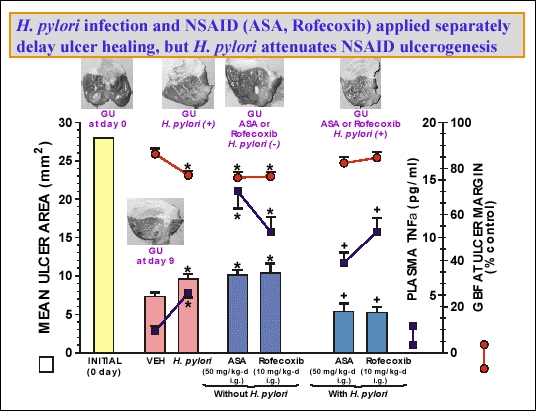 |
| Fig. 27.
Mean ulcer area, gastric blood flow (GBF) at the ulcer margin and plasma
level of tumor necrosis factor (TNFalpha)
in rats given saline (control) or inoculated with H. pylori without
or with administration for 9 days of aspirin, rofecoxib. Asterisk indicates
significant change as compare to the value recorded in in vehicle-treated
rats. Cross indicates significant change as compared to the value obtained
in rats treated with aspirin or rofecoxib but not inoculated with H.
pylori. (unpublished results) |
In conclusion, PG of E series are involved in mucosal repair and healing at least in part, due to increased expression and activity of COX-2 in the ulcer area. Exogenous PGE enhance ulcer healing only at higher gastric inhibitory dose and exerts similar stimulatory action on COX-2 activity and its expression at ulcer margin to that of proton pump inhibitors. Both nonspecific NSAID and specific COX-2 inhibitor reduce the activity of COX-1 and COX-2, while enhancing the expression of COX-2. Corticosteroids inhibit both the expression and activity of COX-2 and this is probably the major mechanism of their ulcerogenic effects. Growth factors and certain gut peptides such as gastrin and CCK or melatonin accelerate ulcer healing due to stimulation of mucosal cell proliferation and, at least in part, expression and activity of COX-2-PG system in the ulcer area as well as activation of brain gut-axis. The infection of gastric mucosa with
H. pylori while increasing by itself the ulcerogenesis, appears to reduce the ulcerogenicity of NSAID in our experimental model possibly due to increase in the expression and activity of COX and release of PG in the ulcer area.
REFERENCES
- Schwarz K. Uber penetrierende Magen- and Jejunalgeschwure, 1910.
- Robert A, Nezamis JE, Lancaster C et al. Cytoprotection by prostaglandins in rat. Prevention of gastric necrosis produced by alcohol, HCl, hypertonic NaCl, and thermal injury. Gastroenterology 1979; 77: 433-443.
- Black JW, Duncan AM, Durant CJ, Ganellin CR, Parsons EM. Definition and antagonism of histamine H2 receptors. Nature 1972, 236, 385-390.
- Sachs G. Physiology of the parietal cell and therapeutic implications. Pharmacotherapy 2003; 23: 68S-73S.
- Anersson T. Pharmacokinetics, metabolism and interactions of acid pump inhibitors. Focus on omeprazole, lanzoprazole and pantoprazole. Clin Pharmacokinet 1996; 31: 9-28.
- Jones DB, Howden CW, Burget DW, Kerr GD, Hunt RH. Acid suppression in duodenal ulcer: a metaanalysis to define optimal dosing with antisecretory drugs. Gut 1987; 28: 1120-1127.
- Hunt RH, Cederberg C, Dent J, et al. Optimizing acid suppression for treatment of acid-related diseases. Dig Dis Sci 1995; 40: 24S-49S.
- Marshall BJ. Helicobacter pylori. Am J Gastroenterol 1994; 89: (Suppl. 1), S116-128.
- Watanabe T, Higuschi K, Tominaga K, Fujiwara Y, Arakawa T. Peptic ulcer recurrences after successful eradication of Helicobacter pylori - clinical characteristics and managements. Nippon Rinsho 2004; 62: 495-498.
- Kurata JH, Nogawa AN. Meta-analysis of risk factors for peptic ulcers; Nonsteroidal anti-inflammatory drugs, Helicobacter pylori and smoking. J Clin Gastroenterol 1997; 24: 2-17.
- Konturek SJ, Kwiecien N, Swierczek J, Oleksy J, Sito E, Robert A. A comparison of methylayed prostaglandin E analogues given orally in the inhibition of gastric responses to pentagastrin and peptone meal in man. Gastroenterology 1976; 70: 683-687.
- Gibinski K, Rybicka J, Mikos E, Nowak A. Double-blind clinical trial on gastroduodenal ulcer healing with prostaglandin analogues. Gut 1977: 18: 636-639
- Rybicka J, Gibinski K. Methylated prostaglanndin E2 analogues for healing of gastroduodenal ulcers. Scand J Gastroenterol 1987; 13:155-159.
- Poynard T., Pignon JP. Acute treatment of duodenal ulcer analysis of 293 randomized clinical trials. Paris, John Libbey Eurotext, 1989, p. 7.
- Graham DY, White RH, Moreland LW et al. Duodenal and gastric ulcer prevention with misoprostol in arthritis patients taking NSAID. Misoprostol study group. Ann Intern Med 1993; 19: 257-262.
- Hawkey CJ, Walt RP. Proostaglandins for peptic ulcer. A promise unfulfilled. Lancet 1986; 2: 1084-1087.
- Goldstein JL, Huang B, Amer F, Christopulos NG. Ulcer recurrence in high-risk patients receiving nonsteroidal antiinflammatory drugs plus low-dose aspirin: results of a post HOC subanalysis. Clin Ther 2004; 26: 1637-1643.
- Graham DY. Critical effect of Helicobacter pylori infection on the effectiveness of omeprazole for prevention of gastric or duodenal ulcer in chronic NSAID users. Helicobacter 2002; 7: 1-8.
- Perini RF, Ma L., Wallace J. Mucosal repair and COX-2 inhibition. Curr Pharm Design. 2003; 9: 2207-2211.
- Brzozowski T, Konturek PC, Konturek SJ et al. Classic NSAIDand selective cyclooxygenase (COX)-1 and COX-2 inhibition in healing of chronic gastric ulcers. Microsc Res Tech 2001; 53: 343-353.
- Mizuno H, Sakamoto C, Matsuda K et al. Induction of cyclooxygenase 2 in gastric mucosal lesions and its inhibition by the specific antagonist delays healing in mice. Gastroenterology 1997; 112: 387-397.
- Takahashi S, Shigeta J, Inoue H et al. Localization of cyclooxygenase-2 and regulation of its mRNA expression in gastric ulcers in rats. Am J Pharmacol Gastrointest Liver Physiol 1998; 275: G1137-G1145.
- Tatsuguchi A, Sakamoto C, Wada K et al. Localization of cyclooxygenase 1 and cyclooxygenase 2 in Helicobacter pylori related gastritis and gastric ulcer tissues in humans. Gut 2000; 46: 782-789.
- Mizuno H, Sakamoto C, Matsuda K, Wajda K et al. Induction of cyclooxygenase-2 in gastric mucosal lesions and its inhibition by specific antagonist delays healing in mice. Gastroenterology 1997; 112: 387-397.
- Konturek SJ, Stachura J, Radecki T, Drozdowicz D, Brzozowski T. Cytoprotective and ulcer healing properties of prostaglandin E2 in rats. Digestion 1987; 38: 103-113.
- Wallace JL, Devchand PR. Emerging roles for cyclooxygenase-2 in the gastrointestinal mucosal defense. Br J Pharmacol 2005; 145; 275-282.
- Brzozowski T, Konturek PC, Konturek SJ et al. Effect of local application of growth factors on ulcer healing and mucosal expression of cyclooxygenase 1 and 2. Digestion 2001; 64: 15-29.
- Tsuji S, Sun WH, Tsuji M et al. Lanzoprazole induces mucosal protection through gastric receptor-dependent up regulation of cyclooxygenase-2 in rats. J Pharmacol Ther 2002; 303: 1301-1308.
- Tjandrawinata RR, Hughes-Fulford M. Upregulation of cyclooxygenase-2 by product-prostaglandin E2. Adv Exp Med Biol 1997; 407: 163-170.
- Tibble J, Sigthorsson G, Caldwell C, Palmer RH, Bjarnason I. Effects of NSAID on cryoprobe-induced gastric ulcer healing in rats. Aliment Pharmacol Ther 2001; 15: 2001-2008.
- Davies NM, Sharkey KA, Asfaha S, MacNaughton WK, Wallace JL. Aspirin causes rapid upregulation of cyclooxygenase-2 expression in the stomach of rats Aliment Pharmacol Ther. 1997; 11:1101-1108.
- Conn HO, Blitzer BI. Nonassociation of adrenocorticosteroid therapy and peptic ulcer. N Engl J Med 1976; 294: 473-479.
- Messer J, Reitman D, Sachs HS, Smith H Jr, Chalmers TC. Association of adrenocorticosteroid therapy and peptic ulcer disease. N Engl J Med 1983; 309: 21-24.
- Ellershaw JE, Kelly MJ. Corticosteroids and peptic ulceration. Palliat Med 1994; 8: 313-319.
- Conn HO, Poynard T. Corticosteroid and peptic ulcer: meta-analysis of adverse events during steroid therapy. J Intern Med 1994; 236: 619-632.
- Pecora PG, Kaplan B. Corticosteroids and ulcers is there an association. Ann Pharmacother 1996; 30: 870-872.
- Kuwayama H, Matsuo Y, Eastwood GL. Effects of prostaglandins on hydrocortisone-induced delayed healing of chronic gastric ulcers in the rat. J Clin Gastroenterol 1991; 13: 554-557.
- Luo JC, Shin VY, Liu ES et al. Non-ulcerogenic dose of dexamethasone delays gastric ulcer healing in rats. J Pharmacol Exp Ther 2003; 307: 692-698.
- Carpani de Kaski M, Rentsch R, Levi S, Hodgson HJ. Corticosteroids reduce regenerative repair of epithelium in experimental gastric ulcers. Gut 1995; 37: 613-616.
- Wallace JL. Glucocorticoid-induced gastric mucosal damage: inhibition of leukotriene, but not prostaglandin biosynthesis. Prostaglandins 1987; 34: 311-323.
- Takahashi M, Ota S, Hata Y. Hepatocyte growth factor as a key to modulate anti-ulcer action of prostaglandins in the stomach. J Clin Invest 1996; 98: 2604-2611.
- Ghosh AK, Hirasawa N, Niki H, Ohuchi K. Cyclooxygenase-2-mediated angiogenesis in carageeniu-induced granulation tissue in rats. J Pharmacol Exp Ther 2002; 295: 802-809.
- Konturek PC, Brzozowski T, Konturek SJ, Ernst H, Drozdowicz D, Hahn EG. Expression of epidermal growth factor and transforming growth factor alpha during ulcer healig. Time sequence study. Scand J Gastroenterol 1997; 32: 6-15.
- Konturek SJ. Role of growth factors in gastroduodenal protection and healing of peptic ulcers. Gastroenterology Clinics of North America 1990; 19: 41-65.
- Schmassmann A, Reubi JC. Cholecystokinin-B/gastrin receptors enhance wound healing in rat gastric mucosa. J Clin Invest 2000; 1066: 1021-1029.
- Li H, Helander HF. Hypergastrinemia increases proliferation of gastroduodenal epithelium during gastric ulcer healing in rats. Dig Dis Sci 1996; 41: 40-48.
- Brzozowski T, Konturek PC, Konturek SJ et al. Involvement of cyclooxygenase (COX)-2 products in acceleration of ulcer healing by gastrin and hepatocyte growth factor. J Physiol Pharmacol 2000; 51: 751-753.
- Brzozowska I, Targosz A, Sliwowski Z et al. Healing of chronic gastric ulcers in diabetic rats treated with native aspiryn, nitric oxide (NO)-derivative of aspirin and cyclooxygenase (COX)-2 inhibitor. J Physiol Pharmacol 2004; 55: 773-790.
- Brzozowski T, Konturek PC, Konturek SJ et al. Acceleration of ulcer healing by cholecystokinin (CCK): role of CCK-A receptors, somatostatin, nitric oxide, and sensory nerves. Reg Pept 1999; 82: 19-33.
- Konturek SJ, Brzozowski T, Dembinski A, Warzecha Z, Konturek PC, Yanaihara N. Interaction of growth hormone-releasing factor and somatostatin on ulcer healing and mucosal growth in rats: role of gastric and epidermal growth factor. Digestion 1988; 41: 121-128.
- Takeuchi K, Hirata T, Yamamoto H et al. Effects of S-0509, a novel CCKB/gastrin receptor antagonist on gastric acid secretion and experimental duodenal ulcers in rats. Aliment Pharmacol Ther 1999; 13: 87-96.
- Konturek PC, Brzozowski T, Burnat G et al. Role of brain-gut axis in healing of gastric ulcers. J Physiol Pharmacol 2004; 55: 179-192.
- Harsch IA, Brzozowski T, Bazela K et al. Impaired gastric ulcer healing in diabetic rats: role of heat shock protein, growth factors, prostaglandins and proinflammatory cytokines. Eur J Pharmacol 2003; 481: 249-260.
- Jaworek J, Brzozowski T, Konturek SJ. Melatonin as an organoprotector in the stomach and the pancreas. J Pin Res 2005; 38: 73-83.
- Brzozowska I, Konturek PC, Brzozowski T et al. Role of prostaglandins, nitric oxide, sensory nerves and gastrin in acceleration of ulcer healing by melatonin and its precursor, L-tryptophan. J Pineal Res 2002; 3, 149-162.
- Bandyopadhyay D, Biswas K, Bhattacharyya M, Reiter RJ, Banerjee RK. Gastric toxicity and mucosal ulceration induced by oxygen-derived reactive species: protection by melatonin. Curr Mol Med 2001; 1, 501-513.
- Cabeza J, Motilva V, Martin MJ, de la Lastra CA. Mechanisms involved in gastric protection and melatonin against oxidant stress by ischemia-reperfusion in rats. Life Sci 2001; 68: 1405-1415.
- Konturek PC, Brzozowski T, Burnat G, Kwiecien S, Pawlik WW, Hahn EG, Konturek SJ. Role of brain-gut axis in healing of gastric ulcers. J Physiol Pharmacol 2004; 55: 179-192.