G-protein inward rectifier K+ (GIRK) channels are widely distributed in the cerebral cortex (5). They conduct K+ ions towards the interior of cells more easily than towards the exterior. Although GIRK channels have low open probability at rest, they play a role in tuning the resting membrane potential in mPFC pyramidal neurons (6-10).
GIRK channels possess protein kinase C (PKC) and protein kinase A (PKA) phosphorylation sites (6, 11, 12) thus making them susceptible to transduction systems activated by metabotropic receptors. Two types of GIRK channels, cardiac and neuronal, have been identified (6, 13). PKC activation leads to the inhibition of both types of GIRK channel-mediated currents (10, 14-17). In contrast, activation of the cAMP/PKA-dependent transduction system increases the activity of cardiac-type GIRK channels (12, 18, 19). The effects of cAMP/PKA on neuronal GIRK channel currents have not been thoroughly tested. In this study, we investigated the effects of cAMP on neuronal-type GIRK channels in mPFC pyramidal neurons.
The experimental procedures used in this study were performed in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC). Three-week-old male Wistar rats (WAG Cmd) were decapitated and their brains were placed in a cold (4°C) oxygenated solution of (mM): sucrose (234), KCl (2.5), NaH2PO4 (1), glucose (11), MgSO4 (4), HEPES-Cl [N-(2-hydroxyethyl)piperazine-N-(2-ethanesulfonic acid)] (15), and CaCl2 (0.1). In all solutions applied in this study, the pH was adjusted to 7.4 and the osmolality to 310 mOsm/kg H2O. Coronal slices were prepared from cerebral prefrontal tissue using a vibrotome (Vibrotome 1000, Pelco International, CA).
Voltage-clamp recordings from dissociated neurons
The slices were stored at room temperature (2122°C) in a solution containing (mM): NaCl (118), KCl (5.3), CaCl2 (1.8), MgSO4 (0.4), NaHCO3 (26), and NaH2PO4 (0.9) bubbled with 95% O2 and 5% CO2. Regions of the slices that corresponded to the mPFC in rats were dissected (2.23.5 mm anterior to Bregma, 35 mm below the upper cortical surface, and 0.60.9 mm from the midline (20)), then transferred to a solution bubbled with O2 that contained (mM): NaCl (135), HEPES-Cl (10), KCl (5), MgSO4 (1) and glucose (10), and incubated at 32°C with protease type XIV (1 mg/ml, Sigma Aldrich) for 18 min. Next, parts of the slices were mechanically dispersed. The suspension of neurons was transferred to a recording chamber placed on the stage of a Nikon inverted microscope (Nikon Instech Co., Ltd., Kawasaki, Kanagawa, Japan). Cells were identified under Hoffman optics (magnification 400x) according to criteria described previously (see (10) and Fig. 1B in (21)) and by other authors (22-25)). The pyramidal neurons that were selected for channel current recordings had a smooth, three-dimensional appearance, triangular shape, residual apical and basal dendrites, and a short axon at the base (10, 21).
During channel current recordings the recording chamber was perfused with a solution containing (mM): KCl (145), CaCl2 (2), MgCl2 (2), glucose (15), HEPES-Cl (10), TTX (Tetrodotoxin citrate, 0.001), and LaCl3 (0.003). PH and osmolality of the solution were 7.4 and 320 mOsm/kg, respectively. The membrane potential of the tested cells was clamped at approximately 0 mV in this external solution. The pipette solution contained (mM): potassium acetate (120), HEPES-Cl (10), MgCl2 (2), CaCl2 (2) and blockers of voltage-gated Na+ channels TTX (0.001), K+ channels (tetraethylammonium chloride, 10; 4-aminopyridine, 5) and Ih channels (ZD7288, 50 µM), pH 7.4 and osmolality 280 mOsm/kg. The reversal potential for K+ ions was close to 0 mV. Between the channel current recordings the extracellular solution contained (mM): NaCl (145), CaCl2 (2), MgCl2 (2), glucose (10), and HEPES-Cl (10). Ph and osmolality of the solution were 7.4 and 320 mOsm/kg, respectively.
Channel currents were recorded in the cell-attached configuration using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA) at room temperature (2123°C) using the pClamp 10 software package. The data were digitised at 20 kHz through a 4-pole low-pass Bessel filter (2 kHz) and stored on a PC computer. The data were collected for 60 s under control conditions as well as during application and washout of the tested compounds. The channel current traces were idealised with pClamp 10.0. The channel conductance, channel open probability and mean open time were measured. Single channel conductance was calculated as the slope of the best-fit line to the I-V plot in the linear regimen: Q=I/V, where Q is the channel conductance, I is the maximum channel current amplitude, and V is the membrane potential. The channel open probability (Po) was calculated as follows: Po=topen/ttotal, where topen is the total channel open time and ttotal is the analysis time. All potentials were expressed in terms of the cytoplasmic side of the patch relative to the patch pipette. The data shown in the figures were further digitised at 1 kHz.
The recording chamber was washed out (2 ml/min) with extracellular solution free of test compounds. At the same time, the tested cell was continuously washed out using tubing placed in close proximity (inside diameter 250 µm, EVH-9, Bio-Logic Science Instruments, France). The tubing delivered either the extracellular control solution or the extracellular control solution plus the test compound to the neuron.
The pipettes for voltage clamp recordings were fabricated from borosilicate glass capillaries (O.D., 1.5 mm, I.D. 0.86 mm; Harvard Apparatus, Edenbridge, UK) using a P-87 puller (Sutter Instruments, Inc., CA), and then fire-polished. The pipette resistance in the bath was 78 M
The compounds ZD7288 (4-(N-ethyl-N-phenylamino)-1,2 dimethyl-6-(methylamino) pyrimidinium chloride), H-89 (N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide) and 8CPT-2Me-cAMP 8-(4-chlorophenylthio)-2-O-methyladenosine-3,5-cyclic monophosphate sodium salt were dissolved in distilled water and stored as 10 µl stock solutions until use. 8-Br-cAMP (8-bromoadenosine-3,5-cyclic monophosphate) was dissolved in distilled water and stored as 100 µl stock solution. Forskolin and KT 5720 ((9S,10S,12R)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3,2,1-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid hexyl ester) were dissolved in DMSO and stored as 10 µl stock solutions. The compounds were stored at 20°C for up to 1 month.
The majority of the chemical compounds were purchased from Sigma Aldrich. H-89 and KT 5720 were purchased from Tocris Bioscience (UK), forskolin was purchased from Ascent Scientific (UK).
Statistical analysis
All results presented throughout the paper and in the figures are shown as means ± standard errors of the mean (S.E.M.). Either the Friedman test (non-parametric repeated measures ANOVA) followed by the post-hoc Dunn test or the analysis of variance (ANOVA) followed by the Tukey-Kramer post-hoc test were used to analyse repeated measurements. GraphPad InStat software v3.06 (GraphPad Software, Inc., CA) was used for statistical analyses.
This study included only inwardly rectifying channel currents (n=86) with sparse resting activity (10) (Fig. 1A). The currents had a mean conductance of 31.9±1.5 pS, mean current amplitude of 2.21±0.12 pA, mean open time of 0.55±0.02 ms and mean Po of 2.0±0.17x103 at 75 mV (n=86). These features were identical to the properties of the GIRK channel currents recorded in the soma of mPFC pyramidal neurons (10), in the soma and dendrites of hippocampal pyramidal neurons (26) and in cardiac myocytes (27). Additionally, it was previously demonstrated that these channel currents were blocked by tertiapine and activated by baclofen (10). Therefore, it was assumed that the inward rectifier channel current we observed was likely mediated by the GIRK-like channels. During recordings also other types of potassium conductances were observed, including inward rectifier channel currents with high conductance and open probability that were probably mediated by Kir 2.x channels and various types of leak K+ channel currents. These conductances were described previously (10).
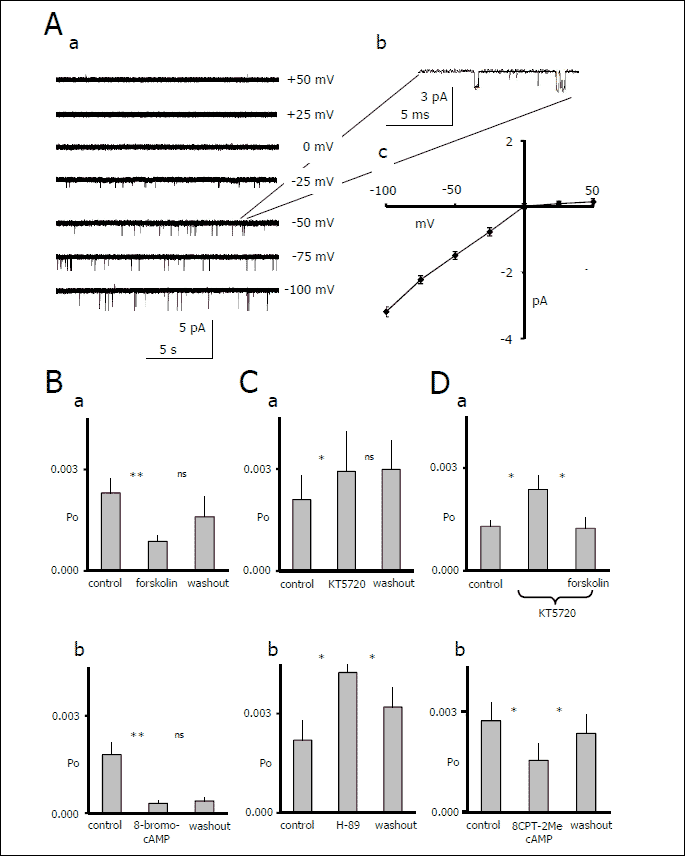 |
| Fig.
1. Effects of cAMP on GIRK-like channel currents in mPFC pyramidal
neurons (* - p<0.05, ** - p<0.01, ns - not significant). [A] (a) Channel currents recorded at different membrane potentials. (b) The segment of the trace recorded at –50 mV is extended and shown with an overlapping idealised trace generated by pClamp 10.0. c. The I/V relationship of the channel currents. [A] Mean open probability (Po) of the channel current before and during forskolin (10 µM, a) and 8-Br-cAMP (100 µM, b) application, and after washout of the compounds. [B] Mean open probability of channel current before and during KT5720 (0.5 µM) application and after washout (a), and before and during H-89 (10 µM) application and after washout (b). [C] Mean open probability of channel current before and during KT5720 (0.5 µM) alone and together with the application of forskolin (10 µM) (a). Mean open probability of channel current before, during and after washout of 8CPT-2Me-cAMP (10 µM) (b). |
In order to test whether cAMP modulates this GIRK-like channel current, forskolin (10 µM), a membrane-permeable activator of adenylyl cyclase that augments cytosolic cAMP levels, was applied. Forskolin administered for 9 min significantly decreased GIRK-like channel activity. Po decreased by 61% from the control, from 2.3±0.4 x 103 to 0.9±0.2 x 103 (n=13, p<0.0042, Friedman test Fr =10.9 followed by the Dunn test, p<0.01). After 12 min of forskolin washout, the Po of the GIRK-like channel current was 1.6±0.9 x 103 (Dunn test, p>0.05, Fig. 1Ba). To validate this effect, a membrane-permeable cAMP analogue, 8-Br-cAMP (100 µM), was applied for 9 min. 8-Br-cAMP decreased the Po of the GIRK-like channels by 83%, from 1.8±0.4 x 103 to 0.3±0.1 x 103(n=7, p=0.0004, ANOVA F(2, 20) =16.58, Tukey-Kramer post-hoc test p<0.001). Activity did not recover after the 9 min washout (Po =0.4±0.1 x 103, Tukey-Kramer post-hoc test p>0.05, Fig. 1Bb).
The obtained results indicate that cAMP leads to inhibition of the GIRK-like channel current.
We then attempted to assess the molecular mechanism linking cAMP to the GIRK channels. PKA belongs to the cellular effectors of cAMP. The application of the membrane-permeable and irreversible PKA inhibitor KT5720 (0.5 µM) significantly increased the Po of the tested channels. Po increased from 2.1±0.7 x 103 (control) to 2.9±1.2 x 103 after 9 min of KT5720 application (n=8, p=0.02, ANOVA(2, 23) =4.98, Tukey-Kramer test p<0.05). The effect was not abolished after the 9 min washout (n=8, Po=3.0±0.8 x 103, Tukey-Kramer test, p>0.05, Fig. 1Ca). To confirm this finding, another membrane-permeable PKA inhibitor, H-89, was applied at concentrations of 5 and 10 µM. H-89 at 5 µM did not affect the Po of the GIRK-like channels. In this case, Po was 2.2±0.6 x 103 in control conditions, 2.3±0.7 x 103 after 6 min of H-89 application and 2.4±0.6 x 103 after 6 min of washout (n=10, p>0.05, Friedman test Fr =1.6). However, the application of H-89 at a higher concentration (10 µM) significantly increased GIRK-like channel activity. The Po was 2.4±0.55 x 103 under control conditions and 4.2±1.25 x 103 after 9 min of H-89 application (Friedman test, n=6, p=0.029 Fr=7, Dunn test p<0.05). Activity partially recovered after a 15 min washout (3.3±0.98 x 103, n=6, Dunn test P>0.05, Fig. 1Cb). These data indicate that PKA can modulate the activity of the GIRK channels.
Therefore, we tried to establish whether GIRK-like channel activity inhibition by cAMP is abolished by a prior PKA blockade. First, the irreversible kinase A inhibitor KT5720 (0.5 µM) was applied for 6 min. As expected, GIRK-like channel activity increased after PKA inhibition. Po rose from 1.3±0.18 x 103 (control) to 2.3±0.4 x 103 after 9 min of KT5720 application (n=5, ANOVA(2, 14), p=0.0176, Tukey-Kramer post test, p<0.05). After application of the PKA inhibitor alone, we delivered KT5720 together with forskolin (adenylyl cyclase activator) for 9 min. The application of forskolin, despite PKA inhibition, still decreased Po by 52% to 1.2±0.3 x 103 (n=5, Tukey-Kramer post test, p<0.05, Fig. 1Da). This finding suggested that GIRK-like channel current activity might be inhibited by cAMP also in a PKA-independent manner.
Another pathway by which cAMP may regulate the activity of the ionic channels involves the activation of a guanine nucleotide exchange factor, Epac (28). While there are no known inhibitors of Epac, activators are available. The effect of a specific activator of Epac, 8CPT-2Me-cAMP at 10 µM, on GIRK-like channel activity was tested. After 6 min of Epac activator delivery, Po decreased from 2.83±1.2 x 103 to 1.61±0.47 x 103 and subsequently recovered after 15 min of washout (n=5, 2.4±0.52, Friedman test, p=0.0085, Fr =8.4, Fig. 1Db). Therefore, Epac may be a potential candidate for mediating the effects of cAMP on GIRK-like channels in mPFC pyramidal neurons.
The purpose of this study was to clarify the effect of the intracellular second messenger, cAMP, on GIRK-like channel currents in mPFC pyramidal neurons.
We found that GIRK-like channel activity was inhibited during application of adenylyl cyclase activator, forskolin, or membrane permeable cAMP analogue, 8-bromo-cAMP. Both procedures increased the concentration of cAMP in the cytoplasm and could potentially activate protein kinase A. The application of the PKA inhibitors KT5720 and H-89 led to an increase in GIRK-like channel current activity. These results strongly suggest that the cAMP/PKA system in neurons modulates GIRK-like channel current activity.
Despite application of the PKA inhibitor (KT5720, 0.5 µM), the activation of adenylyl cyclase by forskolin still led to an inhibition of GIRK-like channel current activity. This result may be interpreted as insufficient inhibition of PKA by KT5720, as other authors applied KT5720 in concentrations ranging from 0.1 µM to a few micromoles in order to fully suppress PKA activity (29-32). It may also indicate that another transduction pathway, independent of PKA, is activated by cAMP. It was demonstrated that cAMP activates another cAMP-dependent protein, i.e. an exchange protein directly activated by cAMP (Epac). Epac transduces diverse cellular actions of the cAMP independently of the PKA (28). The results of our study indicate that the application of a specific activator of Epac, 8CPT-2Me-cAMP, also inhibits GIRK-like channel current activity.
The obtained results demonstrate that cAMP inhibits GIRK-like channel currents and that the inhibitory effect is presumably mediated by both PKA and Epac.
Previous studies have shown that activation of the cAMP/PKA transduction system increased the open probability of cardiac-type GIRK channels (12, 18, 19). Cardiac and neuronal GIRK channels have different structures. The neuronal channels are heteromultimers composed of GIRK1 and GIRK2 subunits, while the cardiac types are composed of GIRK1 and GIRK4 subunits (6, 13). Therefore, differences in channel structure might be responsible for the difference in their susceptibility to cAMP.
The working memory is a system for temporarily storing and managing information required to carry out learning, reasoning and comprehension (33). The proper functioning of the working memory is compromised during metabolic encephalopathies (34), mental (35) and neurodegenerative diseases (36). Prolonged depolarisation and the resulting persistent activity of the mPFC pyramidal neurons are both considered to be an electrophysiological substrate of the working memory (37). At present it is unclear how the cAMP-linked transduction systems affect the working memory. Aujla and Beninger (38) found that the cAMP/PKA transduction system supports the working memory; in contrast, Taylor et al. (39) suggested that this transduction system impairs the working memory.
Our results suggest that an increase in the concentration of cAMP leads to a decrease in GIRK-like channel activity. In turn, inhibition of the GIRK-like channels can induce mPFC pyramidal neuron depolarisation (10) similarly to the inhibition of other types of potassium channels (40). There are several G-protein-coupled receptors which control cellular function by adenylyl cyclase (41-46). Activation of these receptors may thus reinforce prolonged depolarisation and sustained activity in mPFC pyramidal neurons. This mechanism may promote the working memory.
Acknowledgements: This study was supported by the Ministry of Science and Higher Education (Warsaw, Poland) grant no. NN401 076537 and the Ministry of Science and Higher Education (Warsaw, Poland) grant no. NN401 030037.
Conflict of interests: None declared.
- Korf S, Stein DJ, Harvey BH. Cortico-striatal cyclic AMP-phosphodiesterase-4 signalling and stereotypy in the deer mouse: attenuation after chronic fluoxetine treatment. Pharmacol Biochem Behav 2009; 92: 514-520.
- Pain TA, Neve RL, Carlezon Jr WA. Attention deficit disorder and hyperactivity following inhibition of cAMP dependent protein kinase within the medial prefrontal cortex of rats. Neuropsychopharmacology 2009; 34: 2143-2155.
- Wan L, Su L, Xie X, Liu Y, Wang Y, Wang Z. Protein receptor for activated C kinase 1 is involved in morphine reward in mice. Neuroscience 2009; 161: 734-742.
- Raiteri M. Functional pharmacology in human brain. Pharmacol Rev 2006; 58: 162-193.
- Karschin C, Dissmann E, Stuhmer W, Karschin A. IRK(1-3) and GIRK(1-4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J Neurosci 1996; 16: 3559-3570.
- Dascal N. Signalling via the G protein activated K+ channels. Cell Signal 1997; 9: 551-573.
- Kobayashi T, Washiyama K, Ikeda K. Inhibition of G protein-activated inwardly rectifying K+ channels by ifenprodil. Neuropsychopharmacology 2006; 31: 516-524.
- Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 1997; 19: 687-695.
- Rossi P, Mapelli L, Roggeri L, et al. Inhibition of constitutive inward rectifier currents in cerebellar granule cells by pharmacological and synaptic activation of GABA receptors. Eur J Neurosci 2006; 24: 419-432.
- Witkowski G, Szulczyk B, Rola R, Szulczyk P. D1 dopaminergic control of GIRK-like channel current in pyramidal neurons of the medial prefrontal cortex. Neuroscience 2008; 155: 53-63.
- Medina I, Krapivinsky G, Arnold S, Kovoor P, Krapivinsky L, Clapham DE. A switch mechanism for Gbetagamma activation of Ik. Arch J Biol Chem 2000; 275: 29709-29716.
- Muellner C, Vorobiov D, Bera A, et al. Heterologous facilitation of G protein-activated K+ channels by b-adrenergic stimulation via cAMP-dependent protein kinase. J Gen Physiol 2000; 115: 547-558.
- Yamada M, Inanobe A, Kurachi Y. G protein regulation of potassium ion channels. Pharmacol Rev 1998; 50: 723-757.
- Brown SG, Thomas A, Dekker L, Tinker A, Leaney JL. PKC-d sensitizes Kir3.1/3.2 channels to changes in membrane phospholipid levels after M3 receptor activation in HEK-293 cells. Am J Physiol Cell Physiol 2005; 289: C543-C556.
- Leaney JL, Dekker LV, Tinker A. Regulation of G protein gated inward rectifying K+ channel by Ca2+ independent protein kinase. J Physiol 2001; 534: 367-379.
- Mao J, Wang X, Chen F, et al. Molecular basis for the inhibition of G-protein coupled inward rectifier K+ channels by protein kinase C. Proc Natl Acad Sci USA 2004; 101: 1087-1092.
- Nikolov EN, Ivanova-Nikolova TT. Coordination of membrane excitability through a GIRK1 signaling complex in the atria. J Biol Chem 2004; 279: 23630-23636.
- Bender K, Parastoo N, Timpert M, Bing L, Pott L, Kienitz M. A role for RGS10 in ß-adrenergic modulation of G-protein-activated K+ (GIRK) channel current in rat atrial myocytes. J Physiol 2008; 586: 2049-2060.
- Muellner C, Yakubovich D, Dessauer CW, Platzer D, Schreibmayer W. Single channel analysis of the regulation of GIRK1/GIRK4 channels by protein phosphorylation. Biophys J 2003; 84: 1399-1409.
- Ongur D, Price JL. The organization of networks within the orbital and medial prefrontal cortex of rats, monkeys and humans. Cereb Cortex 2000; 10: 206-219.
- Witkowski G, Szulczyk P. Opioid mu receptor activation inhibits sodium currents in prefrontal cortical neurons via a protein kinase A- and C-dependent mechanism. Brain Res 2006; 1094: 92-106.
- Dong Y, Cooper D, Nasif F, Hu X, White FJ. Dopamine modulates inwardly rectifying potassium currents in medial prefrontal cortex pyramidal neurons. J Neurosci 2004; 24: 3077 - 3085.
- Dong Y, Nasif FJ, Tsui JJ, et al. Cocaine-induced plasticity of intrinsic membrane properties in prefrontal cortex pyramidal neurons: adaptations in potassium currents. J Neurosci 2005; 25: 936-940.
- Maurice N, Tkatch T, Meisler M, Sprunger LK, Surmeier DJ. D1/D5 dopamine receptor activation differentially modulates rapidly inactivating and persistent sodium currents in prefrontal cortex pyramidal neurons. J Neurosci 2001; 21: 2268-2277.
- Peterson JD, Wolf ME, White FJ. Repeated amphetamine administration decreases D1 dopamine receptor-mediated inhibition of voltage-gated sodium currents in the prefrontal cortex. J Neurosci 2006; 26: 3164-3168.
- Chen X, Johnston D. Constitutively active G-protein-gated inwardly rectifying K+ channels in dendrites of hippocampal CA1 pyramidal neurons. J Neurosci 2005; 25: 3787-3792.
- Voigt N, Maguy A, Yeh Y, et al. Changes in IK,ACh single-channel activity with atrial tachycardia remodelling in canine atrial cardiomyocytes. Cardiovasc Res 2008; 77: 35-43.
- Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol 2006; 577: 5-15.
- Li Z, Ji G, Neugebauer V. Mitochondrial reactive oxygen species are activated by mGluR5 through IP3 and activate ERK and PKA to increase excitability of amygdala neurons and pain behavior. J Neurosci 2011; 31: 1114-1127.
- Nakamura K, Hirano J, Kubokawa M. An ATP-regulated and pH-sensitive inwardly rectifying K(+) channel in cultured human proximal tubule cells. Jpn J Physiol 2001; 51: 523-530.
- Fu Y, Neugebauer V. Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci 2008; 28: 3861-3876.
- Yao L, Sakaba T. Activity-dependent modulation of endocytosis by calmodulin at a large central synapse. Proc Natl Acad Sci USA 2012; 109: 291-296.
- Baddeley AD, Hitch GJ. Working memory. In: The Psychology of Learning and Motivation, GA Bower (ed.), Academic Press, 1974, pp 47-89.
- Ciecko-Michalska I, Wojcik J, Wyczesany M, et al. Cognitive evoked response potentials in patients with liver cirrhosis without diagnosis of minimal or overt hepatic encephalopathy. A pilot study. J Physiol Pharmacol 2012; 63: 271-276.
- Hains AB, Arnsten AFT. Molecular mechanisms of stress-induced prefrontal cortical impairment: Implications for mental illness. Learn Mem 2008; 15: 551-564.
- Panegyres PK. The contribution of the study of neurodegenerative disorders to the understanding of human memory. QJM 2004; 97: 555-567.
- ODonnell P. Dopamine gating of forebrain neural ensembles. Eur J Neurosci 2003; 17: 429-435.
- Aujla H, Beninger RJ. Hippocampal-prefrontocortical circuits: PKA inhibition of the prefrontal cortex impairs delayed non matching in the radial maze in rats. Behav Neurosci 2001; 115: 1204-1211.
- Taylor JR, Birnbaum S, Ubriani R, Arnsten A. Activation of cAMP-dependent protein kinase A in prefrontal cortex impairs working memory performance. J Neurosci 1999; 19: 23.
- Huang CW, Tsai JJ, Huang CC, Wu SN. Experimental and simulation studies on the mechanisms of levetiracetam-mediated inhibition of delayed-rectifier potassium current (KV3.1): contribution to the firing of action potentials. J Physiol Pharmacol 2009; 60: 37-47.
- Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev 2011; 63: 182-217.
- Williams JT, Christie MJ, Manzoni O. Cellular and synaptic adaptations mediating opioid dependence. Physiol Rev 2001; 81: 299-343.
- Cato AC, Nestl A, Mink S. Rapid actions of steroid receptors in cellular signaling pathways. Sci STKE 2002; 2002(138): re9.
- Pineyro G, Blier P. Autoregulation of serotonin neurons: role in antidepressant drug action. Pharmacol Rev 1999; 51: 533-591.
- Ruat M, Traiffort E, Leurs R, et al. Molecular cloning, characterization, and localization of a high-affinity serotonin receptor (5-HT7) activating cAMP formation. Proc Natl Acad Sci USA 1993; 90: 8547-8551.
- Tokarski K, Kusek M, Hess G. 5-HT7 receptors modulate GABAergic transmission in rat hippocampal CA1 area. J Physiol Pharmacol 2011; 62: 535-540.