PTEROSTILBENE INDUCES ACCUMULATION OF AUTOPHAGIC VACUOLES FOLLOWED BY CELL DEATH IN HL60 HUMAN LEUKEMIA CELLS
INTRODUCTION
Autophagy is an evolutionarily conserved intracellular process in which cytoplasmic components including entire organelles are targeted for lysosomal degradation (1, 2). It occurs at low basal level in most cells and is rapidly upregulated in response to stress such as hypoxia and nutrient or growth factor depletion. Elevated autophagy may also appear after drug treatment. Autophagy is activated in order to maintain homeostasis and promote cell survival. Furthermore, autophagy facilitates cellular or tissue remodelling, e.g. in mammalian embryogenesis as well as in erythropoiesis and adipogenesis (3-5). It also appears to be important in cellular defence against pathogens (6, 7).
Noteworthy, autophagy disfunction contributes to the pathogenesis of many diseases such as neurodegenerative disorders, cancer, Danon's disease or Crohn's disease (8, 9). Autophagy participates in protein quality control and removal of misfolded and aggregated proteins. Its disruption results in accumulation of abnormal proteins and degeneration. Altered proteins were shown to accumulate in Parkinson's, Huntington's and Alzheimer's disease. In Parkinson's disease, degradation of mutant a-synuclein by chaperone-mediated autophagy (CMA) is disrupted (10). Despite a high affinity of this protein to the lysosomal membrane receptor LAMP-2A, it is poorly translocated into the lysosomal lumen. Impaired macroautophagy was shown to be implicated in Huntington's disease (11). Defects in the recognition and sequestration of cytosolic cargo results in an inefficient removal of mutant huntingtin and other substrates of autophagic degradation. Surprisingly, in the case of Alzheimer's disease macroautophagy was found to generate the amyloid β-peptide (12). Autophagic vacuoles, in which this toxic peptide accumulates, serve as its intracellular reservoir. Their removal is inefficient due to their impaired maturation (12, 13). There is evidence supporting the potential link between autophagy failure and Danon's disease (14, 15). It is related to the LAMP-2 deficiency. Several studies indicated that polymorphism in ATG16L1 autophagy gene may be associated with an increased risk of developing of Crohn's disease (16-18). Defects in ATG16L1 protein function results in dysfunction of secretory activity of Paneth cells (18). Defective autophagy has also been linked to cancer development (19-22). The essential autophagy Beclin 1 gene is monoallelicaly deleted in a high percentage of human breast, ovarian and prostate cancers (19). Studies in animal model showed that heterogenous disruption of this gene promotes tumorigenesis (20). In addition to Beclin 1, mutations of several other autophagy-related genes were found in various types of cancer (21, 22). The recent advances in the understanding of the role of autophagy in diseases revealed an opportunity of pharmacological modulation of this process for therapeutic purposes. However, further studies are needed to expand our knowledge of possible targets for effective therapy and identify selective therapeutic agents.
Recently, some natural polyphenolic compounds have been demonstrated to induce both autophagy and apoptosis in human cancer cell lines (23-25). Trincheri et al. reported that resveratrol (trans-3,4',5-trihydroxystilbene) induces autophagy in DLD1 human colorectal cancer cells as a prosurvival stress response that switches to caspase-dependent apoptosis after prolonged drug exposure (23). Pterostilbene (trans-3,5-dimethoxy-4'-hydroxystilbene), a structural analog of resveratrol, was found to induce both autophagy and apoptosis in human bladder and breast cancer cell lines (24, 25). Moreover, it was also shown to cause accumulation of autophagic vacuoles as well as promote cell death via a mechanism involving lysosomal membrane permeabilization in human melanoma, colon, lung and breast cancer cell lines (26). Pterostilbene has greater bioavailability than resveratrol (27). Results of many studies suggest that this bioactive component of grapes and blueberries may be a promising chemotherapeutic agent (28, 29). A clinical trial performed in humans revealed that pterostilbene is generally safe for use up to 250 mg/day (30). In this study, we demonstrate for the first time that pterostilbene induces accumulation of autophagic vacuoles followed by cell death in HL60 human leukemia cells. Understanding of the mechanisms of its action may help to identify new targets for effective cancer therapy.
MATERIAL AND METHODS
Chemicals
Pterostilbene was purchased from Sigma-Aldrich (USA). Pterostilbene stock solutions were prepared in dimethyl sulfoxide (DMSO) and diluted to indicated concentrations before use. Propidium iodide (PI) and H2DCFDA (2',7'-dichlorodihydrofluorescein diacetate) were obtained from Molecular Probes (USA). JC-1 (5,5',6,6'-tetrachloro-1,1,3,3'-tetraethylbenzimidazolylcarbocyanine iodide) was from Calbiochem (USA). MTT (3[4,5-dimethylthiazol-2-y1]-2,5-diphenyltetrazolium bromide), neutral red, 3-methyladenine, ethidium bromide, acridine orange, RNase A and Hoechst 33342 were purchased from Sigma-Aldrich (USA). Caspase inhibitors Z-LEHD-FMK and Z-IETD-FMK were purchased from BD Pharmingen (USA). Rabbit anti-LC3 primary antibodies were purchased from Medical&Biological Laboratories Co. (Japan). Mouse anti-β-tubulin primary antibodies were from Santa Cruz Biotechnology (USA). Horseradish peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies were purchased from Sigma-Aldrich (USA). Cy3-conjugated anti-rabbit secondary antibodies were obtained from Jackson ImmunoResearch Laboratories, Inc. (USA). All other reagents, obtained from commercial suppliers, were of analytical grade.
Cell culture
HL60 cell line (human promyelocytic leukemia cell line) was kindly provided by Dr. Grzegorz Stasilojc (Laboratory of Cell Biology, Intercollegiate Faculty of Biotechnology, University of Gdansk and Medical University of Gdansk, Poland). HL60 cells were maintained at 37°C in a humidified atmosphere containing 5% CO2, in RPMI 1640 medium (Sigma-Aldrich, USA) supplemented with 10% heat-inactivated fetal bovine serum (Sigma-Aldrich, USA), 100 IU/ml penicillin (Sigma-Aldrich, USA) and 100 µg/ml streptomycin (Sigma-Aldrich, USA).
Neutral red uptake assay
HL60 cells (1×105 cells per plate) were treated with various concentrations of pterostilbene for 72 hours. Control cells were treated with DMSO (solvent) alone. The concentration of DMSO never exceeded 0.1% (v/v) and did not affect cell growth. After 72 hours of treatment, cells were collected, washed with phosphate buffered saline (PBS) and suspended in neutral red solution (final concentration: 33 µg/ml) and then incubated for 2.5 hours at 37°C. Next, cells were washed with PBS and suspended in acetic acid/ethanol solution (1% acetic acid in 50% ethanol, v/v). Absorbance was measured at 540 nm using a microplate reader (Jupiter; ASYS Hitech GmbH, Austria). The cell viability was expressed as the percentage of control. A dose-response curve was plotted and used to calculate the concentration of pterostilbene required to inhibit cell growth by 90% (IC90).
MTT assay
HL60 cells were seeded in 96-well plates (15×103 cells per well) and exposed to pterostilbene. Control cells were incubated in the presence of DMSO (solvent). The concentration of DMSO never exceeded 0.5% (v/v) and did not interfere with cell growth. At the end of treatment, MTT (final concentration: 0.5 mg/ml) was added and cells were incubated at 37°C for 4 hours. The plates were then centrifuged (300×g/15 min/room temperature), supernatants were removed and DMSO was added to dissolve MTT formazan crystals. Absorbance was measured at 570 nm using a microplate reader (ELx800; BioTek Instruments, Inc., USA).
Trypan blue exclusion assay and vacuolated cells enumeration
After pterostilbene-treatment HL60 cells were collected, washed with PBS and suspended in complete culture medium. viable, dead and vacuolated cells were counted in the hemocytometer. The number of vacuolated cells was assessed without staining, whereas viable and dead cells were counted after trypan blue staining. Trypan blue-negative cells were considered viable. Trypan blue-positive cells were categorized as dead.
Cell cycle analysis
After treatment, 2×106 cells were collected, washed with cold PBS and fixed in ice-cold 70% ethanol at –20°C overnight. The fixed cells were washed with cold PBS and suspended in staining solution (50 µg/ml PI and 25 µg/ml DNase-free RNase A in PBS). After incubation in the dark at 37°C for 30 min, flow cytometric analyses were performed (Becton Dickinson FACScan, USA).
Detection of intracellular reactive oxygen species production
The intracellular production of reactive oxygen species (ROS) was evaluated using H2DCFDA. Cells were incubated with pterostilbene or DMSO (control). 30 min before the end of incubation, H2DCFDA (final concentration: 10 µM) was added. Next, cells were washed and suspended in cold PBS. Samples were analysed for DCF fluorescence by flow cytometry.
Annexin V-FITC/PI assay
Phosphatidylserine externalization was examined using Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen, USA). After treatment, 5×105 cells were stained with PI and FITC-conjugated annexin V according to the manufacturer's protocol. Samples were analysed by flow cytometry.
DNA fragmentation assay
DNA fragmentation was analysed by agarose gel electrophoresis as described previously (31). DNA fragments were fractionated on 1.8% agarose gel, stained with ethidium bromide and examined using Gel Doc 2000 (Bio-Rad, Italy).
Analysis of mitochondrial membrane potential
Changes in mitochondrial membrane potential (ΔΨm) were assessed by flow cytometry using the cationic lipophilic JC-1 dye as described previously (31).
Caspase-3 activity measurement
Caspase-3 activity was measured using FITC-conjugated Monoclonal Active Caspase-3 Antibody Apoptosis Kit I (BD Pharmingen, USA). Briefly, cells were stained with FITC-conjugated anti-active caspase-3 antibody according to the manufacturer's protocol. Samples were analysed by flow cytometry.
Inhibition of caspase-8,-9 activation
The caspase-9 inhibitor Z-LEHD-FMK and the caspase-8 inhibitor Z-IETD-FMK were dissolved in DMSO according to the chemicals' characteristics. HL60 cells were pretreated with either DMSO or 30 µM inhibitor for 2 hours and exposed to 43 µM pterostilbene or DMSO alone for the next 24 hours. The percentage of dead cells was assessed by flow cytometry using Annexin V-FITC Apoptosis Detection Kit (BD Pharmingen, USA).
Acridine orange/ethidium bromide staining
Morphological changes of pterostilbene-treated cells were examined after AO/EB staining. Acridine orange (AO) is a vital dye and stains both viable and non-viable cells. Ethidium bromide (EB) stains only dead cells. It is taken up by cells when cell membrane integrity is lost. By AO/EB staining viable cells appear green, whereas early apoptotic cells stain green and show chromatin condensation or nuclear fragmentation presented as bright green dots in nuclei. Late apoptotic and necrotic cells stain bright red/orange. Late apoptotic cells, in contrast to necrotic cells, show condensed or fragmented nuclei. AO and EB dyes were dissolved in PBS. AO/EB solution was prepared before use by mixing 1 part of 100 µg/ml AO and 1 part of 100 µg/ml EB. After treatment, cells were washed and suspended in PBS. 25 µl of cell suspension was mixed gently with 1 µl of AO/EB solution, samples were immediately placed onto microscopic poly-L-lysine coated slides (Sigma-Aldrich, USA) and covered with glass coverslips. The slides were examined using PCM 2000 (Nikon, Japan) confocal microscope system as well as the Nikon Eclipse 600 fluorescence microscope (Japan) with Radiance 2100 confocal system (Bio-Rad, UK). The confocal images were obtained using 60 × oil immersion objective lenses.
Neutral red staining
Neutral red (NR) staining is based on the ability of viable cells to incorporate the supravital dye neutral red into lysosomes or acidic vacuoles. NR stains the acidic structures red. After pterostilbene-treatment, cells were washed and suspended in PBS. Next, cells were stained with NR (33 µg/ml) and examined by phase contrast microscopy (Olympus CKX41, Japan).
Transmission electron microscopy
After pterostilbene-treatment, cells were fixed in a fixative solution (2% formaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4) at 4°C overnight. Next, cells were washed with 0.1 M sodium cacodylate buffer (pH 7.4) and postfixed in 1% OsO4 as described previously (32). Ultrathin sections were stained with uranyl acetate and lead citrate, and examined by transmission electron microscopy (JEM 1200 EX II, JEOL Ltd., Japan).
Western blotting analysis
After pterostilbene-treatment, cells were lysed. Cell lysates were centrifuged at 14,000×g for 20 min (4°C), supernantants were transfered to fresh tubes and pellets were discarded. Protein samples were separated electrophoretically by SDS-PAGE (12%) and transferred onto PVDF (polyvinylidene difluoride) membrane. The membrane was incubated with 5% non-fat dry milk in TBST (Tris-buffered saline tween 20) at room temperature (RT) for 1 hour. After washing with TBST, the membrane was incubated with specific primary antibodies (rabbit anti-LC3 antibodies, 1:4000; mouse anti-β-tubulin antibodies, 1:300) at 4°C overnight, and then incubated with horseradish peroxidase-conjugated secondary antibodies (anti-rabbit 1:10,000; anti-mouse 1:3000) for 2 hours (RT). The bound antibodies were detected by the enhanced chemiluminescence method and densitometric analysis of immunoreactive protein bands was performed.
Immunofluorescence analysis
After treatment, cells were washed with PBS and cytocentrifuged onto microscopic poly-L-lysine coated slides (Sigma-Aldrich, USA). Cells were then fixed and permeabilized in cold methanol (5 min, –20°C). Next, cells were washed with PBS, incubated for 30 min (RT) with 10% FBS (fetal bovine serum) in PBS. After washing with PBS, cells were incubated with specific rabbit anti-LC3 primary antibodies (1:500) for 1 hour (RT), washed with PBS and then incubated with Cy3-conjugated anti-rabbit secondary antibodies (1:600) for 1 hour (RT) in the dark. After washing with PBS, cells were stained with 1 µM Hoechst 33342 for 15 min (RT). Next, samples were mounted in mounting medium and covered with glass coverslips. The slides were examined by PCM 2000 (Nikon, Japan) confocal microscope system and FV1200 (Olympus, Japan) confocal microscope system. The confocal images were obtained using 60 × or 100 × oil immersion objective lenses.
Statistical analysis
Statistical analysis was performed using Statistica 9 software (StatSoft, Poland). Data are expressed as mean ± S.D. Each experiment was repeated at least three times in duplicates. Statistical differences between samples were evaluated using the non-parametric Mann-Whitney U test. Differences were considered significant at p<0.05, p<0.01, p<0.001, respectively.
RESULTS
Cytoplasmic vacuolation of pterostilbene-treated cells
In preliminary experiments we found that pterostilbene concentration required to inhibit growth of HL60 cells by 90% (IC90) was equal to 43 µM as assessed by the neutral red uptake assay (Fig. 1). The IC90 value was determined after 72 hours of pterostilbene-treatment (after about three population doublings of HL60 cells). AO/EB staining revealed that treatment of HL60 cells with 43 µM pterostilbene led to the intensive cytoplasmic vacuolation (Fig. 2a). Vacuoles appeared within the first 6 hours of incubation with this compound. They enlarged gradually and after 24 hours occupied most of the cell's cytoplasm. Nuclei of most cells were then located peripherally and did not show fragmentation or chromatin condensation. Confocal microscopy demonstrated that after 6 and 24 hours of incubation with pterostilbene majority of the vacuolated cells remained viable, stained green after AO/EB staining (Fig. 2a). Almost no vacuolated cells were visible after removing pterostilbene by washing the cells and further incubation with pterostilbene-free culture medium (Fig. 2b), suggesting that the cytoplasmic vacuolation was reversible. Results of the quantitative analysis indicated that after 24 hours of incubation with 43 µM pterostilbene vacuolated cells constituted about 67% of the total cell population (Fig. 2c), whereas after removing of the compound and further incubation with pterostilbene-free medium, the fraction of vacuolated cells dramatically decreased (Fig. 2c). The trypan blue exclusion assay revealed that after 24 hours of pterostilbene-treatment viable cells constituted 83% of the total cell population (Fig. 2c). After removing of the compound followed by 24, 48 and 72 hours of incubation with fresh medium viable cells constituted 71%, 78% and 86%, respectively (Fig. 2c).
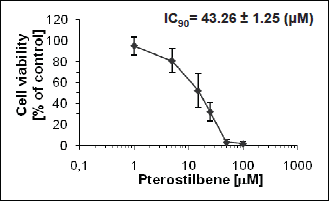 |
Fig. 1. Neutral red uptake assay. HL60 cells were treated with DMSO (control) or pterostilbene for 72 hours. Data are presented as mean ± S.D. of four independent experiments in duplicates. |
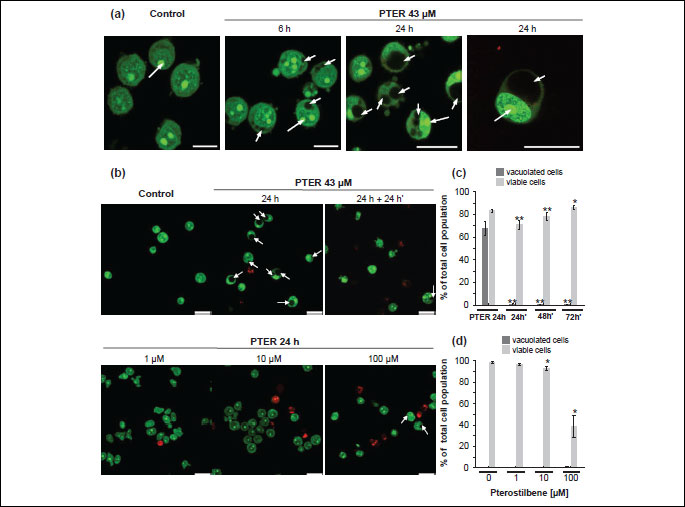
In contrast to the effects of 43 µM pterostilbene (Fig. 2b, 2c), after 24 hours-exposure of HL60 cells to 1 µM (Fig. 2b, 2d) and 10 µM pterostilbene (Fig. 2b, 2d) no vacuoles were visible. After treatment with 1 µM pterostilbene for 24 hours majority of cells remained viable, stained green after AO/EB staining and exhibited intact nuclear architecture (Fig. 2b). The fraction of viable cells was 97% as assessed by the trypan blue exclusion assay (Fig. 2d). Confocal microscopy indicated that following 24 hours of incubation with 10 µM pterostilbene (Fig. 2b), the fraction of red-stained dead cells slightly increased compared to the effects of 1 µM pterostilbene (Fig. 2b). Trypan blue-negative viable cells constituted 93% of the total cell population (Fig. 2d). After 24 hours of treatment with 100 µM pterostilbene, the fraction of vacuolated cells was about 0.6% (Fig. 2d). Exposure to 100 µM pterostilbene resulted in a significant increase in the number of red-stained dead cells, mostly late apoptotic with condensed or fragmented nuclei (Fig. 2b). The trypan blue exclusion assay revealed that viable cells constituted about 39% of the total cell population (Fig. 2d).
Effect of pterostilbene on cell growth and cell cycle progression
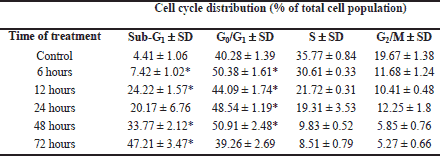
Studies on morphology of vacuolated HL60 cells revealed that the neutral red dye, which had previously been used to assess cytotoxicity of pterostilbene, accumulated in giant vacuoles (Fig. 3a). Therefore, in addition to the neutral red uptake assay the MTT cytotoxicity test was performed. The NR assay is based on the ability of viable cells to incorporate the NR dye into lysosomes, whereas the MTT assay is based on the ability of mitochondrial succinate dehydrogenase of viable cells to reduce the MTT tetrazolium salt. Treatment of HL60 cells with 43 µM pterostilbene for 72 hours resulted in a dramatic decrease in cell viability as assessed by the MTT assay (Fig. 3b). The cell cycle analysis revealed that pterostilbene-treated cells were arrested in the G0/G1-phase and stopped dividing (Fig. 3c, Table 1). Compared to the control (untreated cells), starting from 6 hours of treatment with 43 µM pterostilbene significantly more cells accumulated in the G0/G1-phase, which was accompanied by a corresponding decrease of the population of cells in S- and G2/M- phases (Fig. 3c, Table 1). Moreover, incubation with the compound for 12-72 hours led to a significant increase in the number of cells in the sub-G1 fraction (hypodiploid cells), indicative of apoptotic DNA degradation (Fig. 3c, Table 1).
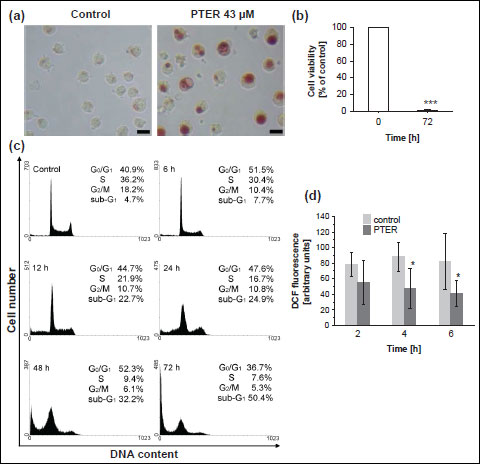 |
Fig. 3. Effect of pterostilbene on viability, cell cycle distribution and ROS production in HL60 cells. (a) Phase-contrast images of HL60 cells exposed to 43 µM pterostilbene for 24 hours (NR staining); Bar, 20 µm; PTER, pterostilbene. Data are representative of three independent experiments in duplicates. (b) MTT cytotoxicity assay. HL60 cells were treated with 43 µM pterostilbene for 72 hours. Data are presented as mean ± S.D. of three independent experiments in triplicates. ***p<0.001, vs. control (untreated cells). (c) Cell cycle analysis (PI staining, flow cytometry analysis). HL60 cells were treated with 43 µM pterostilbene for 0–72 hours. Data are representative of five independent experiments in duplicates. (d) Intracellular production of reactive oxygen species in pterostilbene-treated HL60 cells. Cells were exposed to 43 µM pterostilbene for 2–6 hours. 30 min before the end of incubation H2DCFDA was added and cells were analysed for DCF fluorescence by flow cytometry. PTER, pterostilbene. Data are presented as mean ± S.D. of three independent experiments in duplicates.*p<0.05, vs. control. |
Effect of pterostilbene on reactive oxygen species generation
To determine whether the mechanism of pterostilbene-induced vacuolation of HL60 cells could involve oxidative stress, we examined the effect of this polyphenolic compound on the intracellular production of reactive oxygen species. As shown in Fig. 3d, compared to the control, treatment with 43 µM pterostilbene greatly inhibited the formation of ROS. After 4 and 6 hours of pterostilbene-exposure, the intracellular ROS production decreased by about 2-fold, suggesting that pterostilbene did not induce oxidative stress.
Accumulation of autophagic vacuoles in pterostilbene-treated cells
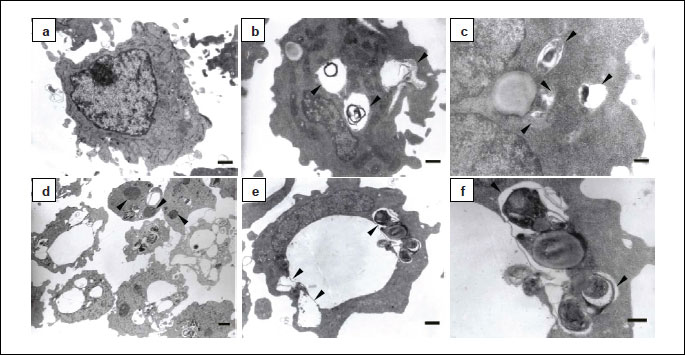
The mechanism of macroautophagy involves the sequestration and lysosomal degradation of cytosol and/or organelles (2). By definition autophagic vacuoles such as autophagosomes, amphisomes or autolysosomes formed during macroautophagy are membrane-bound vacuoles containing cytoplasmic material. Autophagosomes have a double or multiple limiting membrane, whereas autolysosomes are limited by a single membrane (2, 33). Amphisomes are considered to be intermediate autophagic compartments. In morphological studies using transmission electron microscopy autophagic vacuoles are often classified as initial or early autophagic vacuoles (AVi) and late or degradative autophagic vacuoles (AVd) (2). AVi can be autophagosomes and newly formed amphisomes containing morphologically intact cytoplasmic material, whereas AVd can be autolysosomes and amphisomes with partially degraded cytoplasmic content. Our results revealed accumulation of early and late autophagic vacuoles in pterostilbene-treated cells (Fig. 4). AVi were limited by a double or multiple membrane and contained recognizable cytoplasmic material (Fig. 4f). AVd with partially disintegrated content were mostly single membrane-bound (Fig. 4b-4d). In addition to relatively small AVi and AVd very large single membrane-bound vacuoles were observed which morphologically resembled enlarged or distended AVd (Fig. 4b, 4d, 4e). The unrecognizable, partially degraded, electron-dense cytoplasmic material they contained occupied only a very small part of their lumina, whereas the rest was electron-lucent. In comparison to 6 hours, after 24 hours of pterostilbene treatment the number of these vacuoles increased. Moreover, they seemed to enlarge in time. Transmission electron micrographs also showed that many vacuoles adjoined other vacuoles, indicating their fusion (Fig. 4e). After 6 hours of pterostilbene-exposure most cells showed intact nuclear architecture. After prolonged incubation time, some but very few cells showed fragmented nuclei (Fig. 4d).
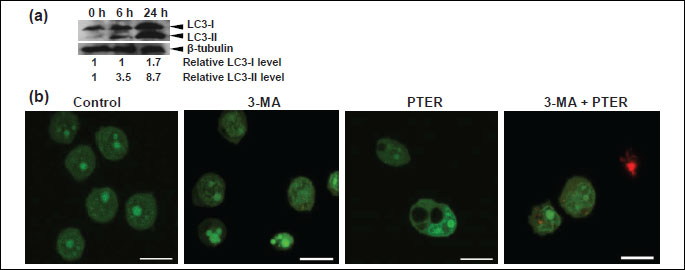
The microtubule associated protein 1 light chain 3 (LC3) plays a critical role in autophagy (2, 34). Induction of autophagy leads to conjugation of phosphatidylethanolamine to cytosolic LC3 (LC3-I) and formation of its non-soluble form (LC3-II) which associates with a phagophore/isolation membrane as well as both inner and outer membrane of autophagosomes (2, 34). To establish whether pterostilbene may influence autophagic pathways, the conversion of cytosolic LC3-I to membrane-bound LC3-II was examined. As shown in Fig. 5a, 6 and 24 hours of treatment of HL60 cells with 43 µM pterostilbene led to the processing of LC3-I and formation of LC3-II. The Western blotting analysis showed that 6 hours-exposure of HL60 cells to pterostilbene resulted in an increased level of LC3-II. After 24 hours, levels of both LC3-I and LC3-II were higher than after 6 hours of treatment (Fig. 5a).
To find out whether pterostilbene-induced vacuolation could be prevented by autophagy inhibitors, HL60 cells were pretreated with 3-methyladenine (3-MA) and the effects were studied using AO/EB staining (Fig. 5b). Confocal micrographs showed that after treatment with 3-MA for 1 hour and further 24 hours-incubation with pterostilbene no vacuoles were visible (Fig. 5b). Moreover, red-stained apoptotic cells were observed (Fig. 5b). The results suggest that pretreatment of HL60 cells with 3-MA prevented pterostilbene-induced vacuolation, but not cell death.
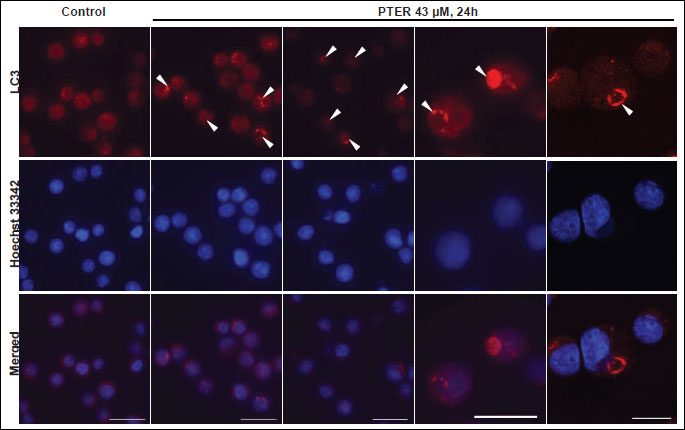
Interestingly, immunofluorescence staining with anti-LC3 antibodies revealed the presence of large LC3-positive ring-shaped structures in pterostilbene treated cells (Fig. 6). In some cells they occupied most of the cytoplasm. In control cells LC3-staining was diffuse, which is typical for cytosolic localization of LC3 (Fig. 6).
Induction of cell death
The cell cycle analysis revealed that treatment of HL60 cells with 43 µM pterostilbene resulted in a significant increase in the number of cells in the sub-G1 fraction, suggesting induction of apoptosis (Fig. 3c, Table 1). Therefore, to elucidate mechanisms of pterostilbene-induced cell death we examined markers of apoptosis such as phosphatidylserine externalization (Fig. 7a, 7b). The analysis of HL60 cells treated with 43 µM pterostilbene for 12 hours revealed that fractions of annexin-V+/PI- cells (corresponding to early apoptotic cells) and annexin-V+/PI+ cells (representing late apoptotic/necrotic cells) were about 4% and 3%, respectively (Fig. 7b). After 24 hours of treatment, annexin-V+/PI- and annexin-V+/PI+ cells constituted about 5% and 12%, respectively (Fig. 7b). Changes in the plasma membrane asymmetry and integrity were more prominent after 48 hours of pterostilbene-exposure. The fractions of annexin-V+/PI- and annexin-V+/PI+ cells were about 20% and 27%, respectively (Fig. 7b). Treatment of cells with pterostilbene for 72 hours resulted in a significant increase in the percentage of early apoptotic cells (58%) and late apoptotic/necrotic cells (29%), whereas the unchanged cells (annexin-V-/PI-) constituted only 9% of the total measured cell population (Fig. 7b).
As the characteristic cleavage of DNA into internucleosomal fragments is considered to be a hallmark of apoptosis, we investigated DNA fragmentation in pterostilbene-treated cells. Results shown in Fig. 7c demonstrate lack of DNA fragmentation in untreated cells. A weak ladder-like pattern, typical for internucleosomal DNA degradation appeared after 12–48 hours of incubation with 43 µM pterostilbene (Fig. 7c). After 72 hours, a smear of DNA fragments of various lengths was observed, indicative of nonspecific DNA cleavage (Fig. 7c).
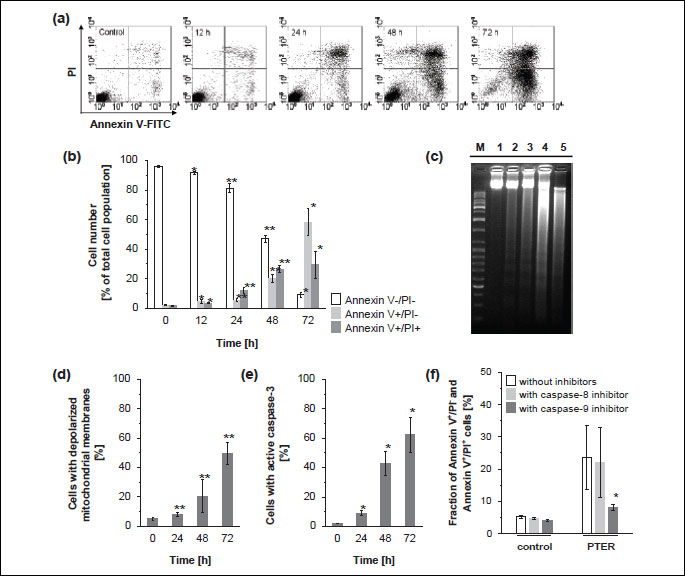
Since mitochondria are involved in the intrinsic apoptotic pathway, we investigated the effects of pterostilbene on changes of the mitochondrial membrane potential. As shown in Fig. 7d, a time-dependent increase in the number of cells with depolarized mitochondrial membranes was observed after treatment of HL60 cells with 43 µM pterostilbene. After 24 hours of incubation with this compound only about 8% of cells showed reduced/loss of mitochondrial membrane potential, whereas after 48 and 72 hours the fraction of cells with altered ΔΨm was about 20% and 49%, respectively (Fig. 7d).
After 24 hours of pterostilbene-exposure, the percentage of cells with active caspase-3, that plays an important role in the executive stage of apoptosis, was less than 10% of the total measured cell population (Fig. 7e). However, after 48 and 72 hours of pterostilbene-treatment 43% and 62% of cells, respectively, showed caspase-3 activity (Fig. 7e).
The activity of caspase-8 is required for the extrinsic, whereas caspase-9 is involved in the intrinsic apoptotic pathway. The caspase-8 inhibitor Z-IETD-FMK failed to block death of HL60 cells induced by pterostilbene-treatment for 24 hours (Fig. 7f) as assessed by annexin V-FITC/PI assay. However, the caspase-9 inhibitor Z-LEHD-FMK partially inhibited cell death (Fig. 7f). In its presence the number of dead cells decreased by 3-fold.
DISCUSSION
Autophagy plays a dual role in cancinogenesis. There is increasing data suggesting that it can act as a tumor suppressor (19, 20, 35-37). However, it is also well documented that in some cases it can promote tumor cell survival, metastasis and resistance to cancer therapy (38-42). Upregulation of autophagy in cancer cells promotes their survival under metabolic stress or hypoxia as well as protects them from anoikis (38-40). Recent studies have demonstrated that autophagy activated in cancer cells represents an adaptive mechanism to survive drug-induced cellular stress (41, 42). Downregulation or inhibition of autophagy was shown to prevent resistance to cancer therapy (41, 42). Therefore, the use of negative modulators of autophagy was proposed as a new approach in cancer treatment.
In the present study, we demonstrate that pterostilbene may act as a modulator of autophagy in HL60 human leukemia cells. The HL60 cell line is widely used as an experimental model in studies on cellular mechanisms (31, 43). We found that 43 µM pterostilbene (IC90) induced intensive cytoplasmic vacuolation in HL60 cells. The vacuolation was concentration-dependent and reversible upon removal of the drug. Pterostilbene did not induce oxidative stress. It decreased intracellular ROS production. Similar effects of pterostilbene we observed in MOLT4 human leukemia cells (44). Moreover, accumulating reports have shown that pterostilbene as well as other polyphenols exhibit antioxidant properties (29, 45, 46). The formation of vacuoles in pterostilbene-treated HL60 cells was blocked by 3-methyladenine, known to inhibit the sequestration step of macroautophagy, i.e. autophagosome formation (47). Although it prevented pterostilbene-induced vacuolation, it did not prevent cell death. Noteworthy, the Western blotting analysis revealed an increased level of LC3-II after 6 and 24 hours of pterostilbene-treatment. This indicates the conversion of LC3-I to LC3-II, typically observed after induction of autophagy (34, 48). It may reflect the increased number of autophagosomes due to an elevated activity of autophagic process (34). However, it may also indicate reduced turnover of autophagosomes resulting in their accumulation characteristic for impaired autophagy (34). Autophagic flux inhibition seems probable, because after 24 hours of treatment the LC3-II level did not decrease, but was even higher than after 6 hours-incubation time. Additional information could provide analysis of the level of p62/SQSTM1 protein (48). Mena S. et al. studied the levels of LC3-II and p62/SQSTM1 after 24 hours of pterostilbene-treatment in A375, A549, HT29 and MCF7 cancer cells (26). They observed an increased level of both proteins, suggestive of autophagic flux inhibition. The elevated amount of LC3-I in HL60 cells after 24 hours of treatment suggests that its processed LC3-II form is still required, e.g. due to the need to form new autophagosomes. Furthermore, the immuno-fluorescence analysis revealed that vacuoles accumulating in pterostilbene-treated cells were LC3-positive. We detected large ring-shaped structures, positively stained with anti-LC3 antibodies. LC3 protein serves as a marker of autophagic vacuoles (2, 34). It is present in both inner and outer membrane of autophagosomes. It may also be present in nascent amphisomes or nascent autolysosomes. In immunofluorescence staining with anti-LC3 antibodies autophagic vacuoles usually appear as fluorescent dots (puncta) or ring-shaped structures (48, 49). Transmission electron micrographs showed the presence of cytoplasmic material within large vacuoles accumulating in pterostilbene-treated HL60 cells, providing additional evidence for their autophagic origin. The vacuoles seemed to enlarge in time. After 24 hours of pterostilbene treatment they occupied most of the cell's cytoplasm. Results of electron microscopy analysis also suggested that larger vacuoles probably arose by fusion of smaller vacuoles. Swelling of the vacuoles should also be considered, because they contained very little electron-dense cytoplasmic material in relation to their electron-lucent area. Taken together, vacuolar structures accumulating in pterostilbene-treated HL60 cells presumably originated from autophagic vacuoles. Accumulation of autophagic vacuoles suggests delayed/arrested autophagy, probably due to defects in some of its step/s. Many chemical compounds, e.g. vinblastine, leupeptin or asparagine were reported to inhibit maturation of autophagic vacuoles and cause their accumulation (50-52). When autophagy is not disturbed, removal of autophagic vacuoles is efficient and their number is relatively low. Defective autophagy may result from impaired fusion of autophagosomes with lysosomes or disruption of lysosomal functions. Another possible explanation may be that the capacity of autophagy to degrade accumulating material was exceeded and subsequently removal of autophagic vacuoles became ineffective due to an elevated rate of their formation.
Noteworthy, the autophagic pathway was found to coalesce with the endocytic pathway generating amphisomes as soon as autophagosomes are formed (53). Some chemical compounds, e.g. leupeptin were shown to cause accumulation of amphisomes (50). Electron micrographs showed large AVd with partially degraded cytoplasmic material in pterostilbene-treated HL60 cells. Autolysosomes contain acid hydrolases (54). Amphisomes probably acquire some lysosomal enzymes through endocytic pathway and consequently may exhibit at least limited ability to degrade their content. Interestingly, the neutral red dye accumulated in pterostilbene-induced vacuoles, indicating an acidic pH within them. Neutral red primarily stains lysosomes, but it can also stain other acidic vesicles including acidic autophagic compartments (48, 55). It accumulates and becomes trapped in acidic intracellular vesicles due to protonation (55). The newly formed autophagosomes have the same pH as the cytoplasm, but it changes during their maturation (48, 54). Amphisomes as well as autolysosomes are acidic (48, 54). Thus, they can both be stained by neutral red. Taken together, is should be considered whether pterostilbene-induced vacuoles in HL60 cells originate from amphisomes or autolysosomes.
Treatment of HL60 cells with 43 µM pterostilbene resulted in the G0/G1 cell cycle arrest and cell death. Pterostilbene led to phosphatidylserine externalization as well as disruption of the mitochondrial membrane potential, activation of caspase-3 and internucleosomal DNA degradation. Pterostilbene-induced cell death was effectively inhibited by caspase-9 inhibitor, indicating the involvement of the mitochondrial apoptotic pathway. All the above findings provide evidence for induction of apoptosis. However, it is important to note that morphology of pterostilbene-treated HL60 cells was not strictly apoptotic. Intensive accumulation of autophagic vacuoles was also detected. It started before pronounced hallmarks of apoptosis appeared, suggesting that the autophagic pathway switched to the apoptotic pathway. Thus, morphology of pterostilbene-treated cells could be affected by both apoptosis and presumably defective autophagy. Because vacuolated cells expressed phosphatidylserine at the external cell surface, they would probably be recognized in vivo and removed by phagocytes without inducing an inflammatory response. There is growing evidence of a complex relationship between autophagy and apoptotic cell death (56-60). Several studies indicated that autophagy lies upstream of apoptosis (56-58). Interestingly, pterostilbene was reported to induce both autophagy and apoptosis in human bladder and breast cancer cell lines (24, 25). Moreover, another polyphenolic compound, resveratrol, induced autophagy followed by apoptosis in human colorectal DLD1 cancer cells (23).
In summary, our findings suggest that treatment of HL60 cells with 43 µM pterostilbene results in reduced rather than enhanced autophagic degradation, which in turn leads to cell death. Autophagy is initiated in pterostilbene-treated cells, but removal of autophagic vacuoles is probably inefficient. Further studies are required to fully explain this phenomenon. Our studies provide a new insight into the mechanisms of action of pterostilbene and its potential therapeutic use. Understanding of the molecular signaling pathways affected by pterostilbene in HL60 cells may help to identify new targets for effective cancer therapy or predict potential adverse drug effects.
Acknowledgements: This work was supported by grant no. W-110/2006-2008 from the Medical University of Gdansk (Poland) and grant no. N N204 132040 from the Polish Ministry of Science and Higher Education (Poland). We thank Professor J. M. Witkowski and Professor E. Bryl (Department of Pathophysiology, Medical University of Gdansk, Poland) for the access to the flow cytometry laboratory and scientific consultations during cytometric analyses. We are also grateful to Professor J. Kubasik-Juraniec (Department of Electron Microscopy, Medical University of Gdansk, Poland) for the access to the electron microscopy laboratory.
Conflict of interests: None declared.
REFERENCES
- Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol 2008; 9: 1004-1010.
- Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy 2005; 1: 1-10.
- Tsukamoto S, Kuma A, Murakami M, Kishi C, Yamamoto A, Mizushima N. Autophagy is essential for preimplantation development of mouse embryos. Science 2008; 321: 117-120.
- Mortensen M, Ferguson DJ, Edelmann M, et al. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc Natl Acad Sci USA 2010; 107: 832-837.
- Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA 2009; 106: 19860-19865.
- Nakagawa I, Amano A, Mizushima N, et al. Autophagy defends cells against invading group A Streptococcus. Science 2004; 306: 1037-1040.
- Orvedahl A, MacPherson S, Sumpter R Jr, Talloczy Z, Zou Z, Levine B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010; 7: 115-127.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132: 27-42.
- Kroemer G, White E. Autophagy for the avoidance of degenerative, inflammatory, infectious, and neoplastic disease. Curr Opin Cell Biol 2010; 22: 121-123.
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004; 305: 1292-1295.
- Martinez-Vicente M, Talloczy Z, Wong E, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 2010; 13: 567-576.
- Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy-a novel beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol 2005; 171: 87-98.
- Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 2005; 64: 113-122.
- Nishino I, Fu J, Tanji K, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000; 406: 906-910.
- Tanaka Y, Guhde G, Suter A, et al. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000; 406: 902-906.
- Prescott NJ, Fisher SA, Franke A, et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn's disease and is independent of CARD15 and IBD5. Gastroenterology 2007; 132: 1665-1671.
- Rioux JD, Xavier RJ, Taylor KD, et al. Genome-wide association study identifies new susceptibility loci for Crohn's disease and implicates autophagy in disease pathogenesis. Nat Genet 2007; 39: 596-604.
- Cadwell K, Liu JY, Brown SL, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456: 259-263.
- Aita VM, Liang XH, Murty VV, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 1999; 59: 59-65.
- Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 2003; 112: 1809-1820.
- Chen N, Debnath J. Autophagy and tumorigenesis. FEBS Lett 2010; 584: 1427-1435.
- Morselli E, Galluzzi L, Kepp O, et al. Oncosuppressive functions of autophagy. Antioxid Redox Signal 2011; 14: 2251-2269.
- Trincheri NF, Follo C, Nicotra G, Peracchio C, Castino R, Isidoro C. Resveratrol-induced apoptosis depends on the lipid kinase activity of Vps34 and on the formation of autophagolysosomes. Carcinogenesis 2008; 29: 381-389.
- Chen RJ, Ho CT, Wang YJ. Pterostilbene induces autophagy and apoptosis in sensitive and chemoresistant human bladder cancer cells. Mol Nutr Food Res 2010; 54: 1819-1832.
- Wang Y, Ding L, Wang X, et al. Pterostilbene simultaneously induces apoptosis, cell cycle arrest and cyto-protective autophagy in breast cancer cells. Am J Transl Res 2012; 4: 44-51.
- Mena S, Rodriguez ML, Ponsoda X, Estrela JM, Jaattela M, Ortega AL. Pterostilbene-induced tumor cytotoxicity: a lysosomal membrane permeabilization-dependent mechanism. PLoS One 2012; 7: e44524.
- Kapetanovic IM, Muzzio M, Huang Z, Thompson TN, McCormick DL. Pharmacokinetics, oral bioavailability, and metabolic profile of resveratrol and its dimethylether analog, pterostilbene, in rats. Cancer Chemother Pharmacol 2011; 68: 593-601.
- Adrian M, Jeandet P, Douillet-Breuil AC, Tesson L, Bessis R. Stilbene content of mature Vitis vinifera berries in response to UV-C elicitation. J Agric Food Chem 2000; 48: 6103-6105.
- Rimando AM, Kalt W, Magee JB, Dewey J, Ballington JR. Resveratrol, pterostilbene, and piceatannol in vaccinium berries. J Agric Food Chem 2004; 52: 4713-4719.
- Riche DM, McEwen CL, Riche KD, et al. Analysis of safety from a human clinical trial with pterostilbene. J Toxicol 2013; 2013: 463595.
- Augustin E, Mos-Rompa A, Skwarska A, Witkowski JM, Konopa J. Induction of G2/M phase arrest and apoptosis of human leukemia cells by potent antitumor triazoloacridinone C-1305. Biochem Pharmacol 2006; 72: 1668-1679.
- Karbowski M, Kurono C, Wozniak M, et al. Cycloheximide and 4-OH-TEMPO suppress chloramphenicol-induced apoptosis in RL-34 cells via the suppression of the formation of megamitochondria. Biochim Biophys Acta 1999; 1449: 25-40.
- Eskelinen EL. To be or not to be? Examples of incorrect identification of autophagic compartments in conventional transmission electron microscopy of mammalian cells. Autophagy 2008; 4: 257-260.
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19: 5720-5728.
- Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402: 672-676.
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100: 15077-15082.
- Marino G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, Lopez-Otin C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem 2007; 282: 18573-18583.
- Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006; 10: 51-64.
- Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25: 717-729.
- Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell 2008; 19: 797-806.
- Ma XH, Piao S, Wang D, et al. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin Cancer Res 2011; 17: 3478-3489.
- Marino ML, Fais S, Djavaheri-Mergny M, et al. Proton pump inhibition induces autophagy as a survival mechanism following oxidative stress in human melanoma cells. Cell Death Dis 2010; 1: e87.
- Kaczmarek M, Frydrychowicz M, Nowicka A, et al. Influence of pleural macrophages on proliferative activity and apoptosis regulating proteins of malignant cells. J Physiol Pharmacol 2008; 59(Suppl. 6): 321-330.
- Siedlecka-Kroplewska K, Jozwik A, Kaszubowska L, Kowalczyk A, Boguslawski W. Pterostilbene induces cell cycle arrest and apoptosis in MOLT4 human leukemia cells. Folia Histochem Cytobiol 2012; 50: 574-580.
- Spanier G, Xu H, Xia N, et al. Resveratrol reduces endothelial oxidative stress by modulating the gene expression of superoxide dismutase 1 (SOD1), glutathione peroxidase 1 (GPx1) and NADPH oxidase subunit (Nox4). J Physiol Pharmacol 2009; 60(Suppl. 4): 111-116.
- Hamlaoui S, Mokni M, Limam N, et al. Resveratrol protects against acute chemotherapy toxicity induced by doxorubucin in rat erythrocyte and plasma. J Physiol Pharmacol 2012; 63: 293-301.
- Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci USA 1982; 79: 1889-1892.
- Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445-544.
- Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007; 3: 323-328.
- Berg TO, Fengsrud M, Stromhaug PE, Berg T, Seglen PO. Isolation and characterization of rat liver amphisomes. Evidence for fusion of autophagosomes with both early and late endosomes. J Biol Chem 1998; 273: 21883-21892.
- Fengsrud M, Roos N, Berg T, Liou W, Slot JW, Seglen PO. Ultrastructural and immunocytochemical characterization of autophagic vacuoles in isolated hepatocytes: effects of vinblastine and asparagine on vacuole distributions. Exp Cell Res 1995; 221: 504-519.
- Hoyvik H, Gordon PB, Berg TO, Stromhaug PE, Seglen PO. Inhibition of autophagic-lysosomal delivery and autophagic lactolysis by asparagine. J Cell Biol 1991; 113: 1305-1312.
- Liou W, Geuze HJ, Geelen MJ, Slot JW. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J Cell Biol 1997; 136: 61-70.
- Dunn WA. Studies on the mechanisms of autophagy: maturation of the autophagic vacuole. J Cell Biol 1990; 110: 1935-1945.
- Dell'Antone P. Evidence for an ATP-driven "proton pump" in rat liver lysosomes by basic dyes uptake. Biochem Biophys Res Commun 1979; 86: 180-189.
- Espert L, Denizot M, Grimaldi M, et al. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J Clin Invest 2006; 116: 2161-2172.
- Crighton D, Wilkinson S, O'Prey J, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126: 121-134.
- Kunchithapautham K, Rohrer B. Apoptosis and autophagy in photoreceptors exposed to oxidative stress. Autophagy 2007; 3: 433-441.
- Zarzynska J, Motyl T. Apoptosis and autophagy in involuting bovine mammary gland. J Physiol Pharmacol 2008; 59(Suppl. 9): 275-288.
- Gonzalez-Polo RA, Boya P, Pauleau AL, et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci 2005; 118: 3091-3102.
A c c e p t e d : September 24, 2013