EFFECT OF SITAGLIPTIN ON THE WORKING MEMORY
AND REFERENCE MEMORY IN TYPE 2 DIABETIC SPRAGUE-DAWLEY RATS:
POSSIBLE ROLE OF ADIPONECTIN RECEPTORS 1
and Medical Physiology Department, College of Medicine, King Khalid University, Saudi Arabia
INTRODUCTION
Dementia represents a variety of symptoms and signs, including a reduction in memory, thinking and communication skills with a gradual loss in the ability to fulfill activities of daily living. The commonest forms are Alzheimer’s disease (AD) and vascular dementia (VaD). VaD is considered one of the most common types of dementia in older adults, however because it is difficult to discern definitively, several studies examining its popularity may be incorrect. The incidence of dementia increases exponentially for patients with cerebrovascular risk factors such as hypertension, cardiac disease, diabetes, smoking, alcoholism and hyperlipidemia (1).
Type 2 diabetes mellitus is the most prevalent metabolic disease worldwide. It usually occurs as a consequence of insulin resistance and obesity. Diabetes mellitus caused different end-organ damage including the brain, kidney as well as retina a result of both microangiopathy and macroangiopathy. Type 2 (non-insulin-dependent) diabetes may be associated with impaired cognitive function such as problem solving, planning, organization, insight, reasoning as well as intentions (2).
The recognized relationship between type 2 diabetes mellitus and macro- and microvascular disease is pertinent to the pathogenesis of dementia. The former could cause cognitive impairment because of the increased incidence of embolic stroke. Moreover, cerebral microvascular disease is more common in people with diabetes, which is implied by the lacunar infarcts and white matter changes that are unmistakable in neuroimaging studies (3, 4). Dipeptidase (DPP) is an enzyme secreted from endothelial cells that rapidly degrades both glucose dependent insulinotropic peptide (GIP) and glucagon like peptide -1 (GLP-1). Inhibiting DPP-4 activity can effectively protect GIP from degradation, resulting in enhanced insulinotropic activity of infused GIP (5). Furthermore, in the presence of the DPP-4 inhibitor (valine-pyrrolidide), the proportion of intact GLP-1 released from the perfused porcine ilium is increased under both basal and stimulated conditions (6).
The first experimental study that evaluated the role of the cholinergic transmission in the storage and retrieval of new data were performed by Deutsch (7). Moreover, the effects of cholinergic antagonists and lesions of cholinergic nuclei are often related to cognitive deficits similar to those observed in aging and dementia (8). Recently Croxson et al. (9) concluded that in the absence of acetylcholine innervations to inferotemporal cortex, the retrieval of episodic memory is impaired and the amnesia caused by the structural injury is more dangerous.
Adiponectin, a hormone secreted by adipose tissue, plays a key role in the control of energy homeostasis through the adjustment of glucose and fatty acids metabolism in peripheral tissues such as muscle and liver (10). Adiponectin is well-defined as an anti-diabetic and anti-atherogenic adipokine (11). It acts through two receptors: Adipor1 and Adipor2 due to its metabolic effects (12, 13). However, Adipor1 is more abundantly expressed in muscles while Adipor2 predominates in the liver (12). Recently, they have also been described in rodent and human hypothalamus (14-16). Adiponectin is protective in human neuroblastoma cells with amyloid B neurotoxicity. Adiponectin has been reported to be neuroprotective against 1-methyl-4-phenyl-piridinum ion (MPP) induced cytotoxicity (17), against acetaldehyde-induced apoptosis, in a mouse model of epilepsy against kainic acid-induced excitotoxicity (18) and the rat model of stroke against cerebral ischemia-reperfusion injury (19). A recent study reported that intense expression of AdipoR1 was noted in the hypothalamus and the nucleus basalis of Meynert in the basal forebrain which is frequently affected in AD (20).
In the present study, the holeboard food search task has been used in rats to analyze their learning ability and different types of memory, IC working and reference memory. Vawter and Van Ree (21) confirmed that the performance in the holeboard memory test is sensitive to the degree of food deficiency. A higher level of food deprivation resulted in a superior performance of the animals, but the processes implicated in learning and memory were less affected. The data obtained by them indicated that both external and internal characteristics can influence the results of the hole board food search task, and thus the calculated scores for learning and memory (21).
However, there is no single report in literature investigating the possible protective effect of sitagliptin on the cognitive function of the diabetic rats. The aim of the present work is to investigate the effect of type 2 diabetes mellitus and incretins treatment on the working memory and reference memory through the holeboard memory test as well as the acetylcholine content of the hypothalamus and plasma adiponectin level. Also, The adiponectin receptors (Adipo 1) expression was assayed in the hypothalamus.
MATERAIL AND METHODS
Experimental animals
Thirty six male Sprague-Dawley rats weighing 170–200 g, were purchased from the Vaccine and Immunization Authority (Helwan, Cairo, Egypt) and housed (Animal House, Medical Physiology Department, Faculty of Medicine, Mansoura University, Egypt) under controlled conditions (temperature of 23±1°C, and a 12 h light: 12 h dark cycle). The animals were allowed free access to food and tap water. Experiments were performed according to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85–23, revised 1996). All experimental procedures in this study were approved by the Medical Research Ethics Committee of Mansoura University, Egypt.
Experimental design
The animals were then divided randomly into two groups. Normal rats (n=12) were fed standard rat chow diet and remained untreated for the duration of the study. Experimental rats (n=24) were fed a diet enriched in fat (25% fat, 15% protein, 51% starch and 5% fiber). After 1 month on their respective diets, experimental rats were injected intraperitoneally with streptozotocin (25 mg /kg body weight) and normal rats were injected with vehicle (0.05 mol/ L citric acid, pH 4.5). Both the low dose of streptozotocin and the high-fat diet are essential elements of the model designed to induce type 2 diabetes with insulin resistance. After 1 month, rats were fasted overnight and given 20% glucose (3 g/kg body weight). Blood samples were taken from the tail vein to measure glucose and insulin levels at 2 and 12 hours after glucose administration (Fig. 2). The diabetic rats were subdivided into two groups: type 2 diabetes mellitus received orange juice and type 2 diabetes mellitus treated with sitagliptin in a dose of 10 mg/kg daily for a month (22). Sitagliptin was dissolved in orange juice and administered to rats in a dose of 10 mg/kg by orogastric gavage, with an appropriate feeding needle as a volume of 5 ml/kg at 9 p.m.
With the end of the treatment duration, the rats were exposed to the holeboard memory test to measure speed of learning and memory capacity in rats. The experimental procedures were undertaken according to EU directives and local ethical regulations. The rats were housed in standard cages with liquid and food available ad libitum, under an artificial reversed 12-h light-dark cycle with light off at 7 a.m. The psychometric tests and pharmacological treatments were carried out in the dark phase of the cycle.
Psychometric assessment (holeboard memory test)
To measure the speed of learning and memory capacity in rats, the holeboard memory test was used (22). The testing area of the holeboard apparatus contains 16 holes in a 4’4 array. The holes are 20 cm apart and are filled with plastic cups (2.5 cm deep, 3 cm in diameter) for sweetened cereal food pellets. Some were spilled on the floor to eliminate the impact of smell (olfactory stimuli) on the search for food. The rats had to collect pellets in specified periods of time.
The test consists of 2 phases: habituation and training. During the habituation and training phases, food was restricted for the rats to enhance their motivation to search for it (water was available ad libitum).
The habituation phase was for 4 consecutive days. Each hole in the board was baited with 50 mg of cereals. The trial started when the rat entered the testing area, and it was stopped after 10 min or earlier, i.e. the time necessary to collect all food pellets. The monitoring included the time and the number of visits and revisits to the baited holes. Rats which found at least 14 of 16 pellets on the 4th day qualified for the training phase (Fig. 1A).
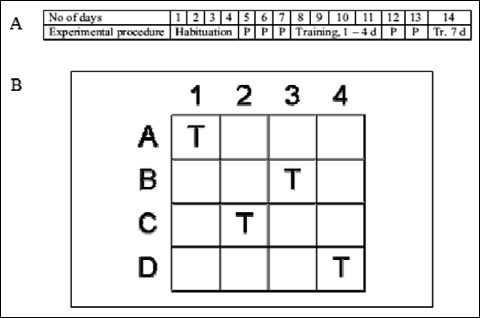 |
Fig. 1. (A) The experimental protocol for psychometric test. P – pause, Tr – training, d – day; see text for details. (B) Testing area of holeboard apparatus - position of baited holes during training phase (T) Modified after Lannert and Hoyer, 1998. |
Training phase started 3 days after habituation and continued for 7 consecutive days with a pause on the 5th and 6th day. The rats were trained to collect pellets form 4 holes: A1, B3, C2 and D4 (Fig. 1B). During one training session, four trials were carried out for each animal. The maximum time given to collect 4 food pellets was 5 min. There were intervals of about 1 min between the trials for cleaning and baiting A1, B3, C2 and D4 holes with pellets. Apart from the time of performance, the registration included latency time (time between the beginning of the trial and the first hole visit) and the number of visits and revisits to the baited and empty holes. Based on these parameters, two distinct memory functions - working and reference memory - were evaluated.
Working memory ratio (WM) was presented as a percentage of all visits to the baited set of holes that had been supplied with food (calculated as the number of food rewarded visits divided by the number of visits and revisits to the baited set of holes). Reference memory ratio (RM) was expressed by the number of visits to the baited set of holes as a percentage of the total number of visits to all holes (calculated as the number of visits and revisits to the baited set of holes divided by the number of visits and revisits to all holes). The parameters were evaluated based on results of the 7th day of training. Training on the holeboard apparatus gradually reduced latency, and the total time needed to complete the trial and markedly improved working (WM) and reference (RM) memories.
Blood and brain samples collection
The rats were euthanized after the the 7th training day. The blood samples were drawn by non-heparinized capillary tubes from the retrorbital plexus of the eye from anesthetized rats. The rat was anaesthetized with diethyl ether, and the tube was pointed at the inner corner of the eye, beside the eyeball. The tube was then slid a few millimetres forward, gently but firmly, along the side of the orbit to the ophthalmic venous plexus (orbital sinus), allowing blood to enter the capillarity tube. The eye was compressed for
30 s until bleeding stopped. These blood samples were collected without anticoagulant, left for 10 min, then centrifuged for 10 min at 4000 r/min to obtain serum, which was stored at –20°C until further biochemical analysis. The brain was quickly removed from the skull; under the dissecting microscope, first the cerebellum was cut off. Then, the olfactory pulp was dissected followed by removal of the frontal cortex by cutting at the anterior end of the corpus callosum. Another cut was performed at the tip of the anterior fornix and anterior commission fibers. This was followed by removal of the brain stem and midbrain to get good exposure of the hypothalamus. Tissue samples were immediately frozen in liquid nitrogen and were kept at –70°C until assayed.
Chemicals
Streptozotocin is (Trade name Zanosar) was purchased from Sigma chemical company (St. Louis, Mo, USA). The drug was dissolved in 0.1 M sodium citrate (pH 4.5). Sitagliptin was a gift from Merck (Rahway, NJ, USA).
Determination of hypothalamic acetylcholine
The hypothalamic homogenates were centrifuged , and the supernatant was stored at –20°C. The hypothalamic content of acetylcholine was measured by ELISA kits catalog number E0912r in duplicate.
Determination of serum glucose
Serum glucose was determined colorimetrically using a Randox reagent kit (Sigma-Aldrich), according to the method of Trinder (24).
Determination of serum adiponectin
Quantitative determination of serum adiponectin was performed using the mouse/rat adiponectin ELISA kit (B-Bridge international, Inc.; Cat# K1002-1), according to the manufacturer’s instructions.
Determination of serum insulin
Determination of serum insulin was performed using an ELISA kit (Diagnostic Systems Labs., Inc.)
Calculation of HOMA index
Homeostasis model of assessment - insulin resistance index (HOMA-IR): HOMA of IR was calculated by using the following equation: HOMA = fasting glucose (mmol L-1) ´ fasting insulin (mU L-1)/22.5. Typically, a HOMA value >2 is used to identify significant IR. Typically, a HOMA value >2 is used to identify significant IR (25).
Assessment of plasma lipids
Serum TG concentrations were measured by the peroxidase method using a commercial kit (SPINREACT, Spain). Serum TC measurement was performed using the cholesterol oxidase method where the pink color of quioneimine was measured at 500 nm. The used kit was supplied by SPINREACT, Spain. Serum HDL cholesterol was determined enzymatically after precipitation of apoB-containing lipoproteins by the phosphotungstic acid-Mg method with the kit supplied by SPINREACT, Spain. Serum LDL-Cholesterol was estimated with a commercial kit (SPINREACT, Spain), with a two-step technique (26). First, chylomicrons, VLDL and HDL were eliminated. Second, LDL was specifically measured through the action of peroxidase with the formation of pinkish color quinine. The intensity of the color formed is proportional to the LDL concentration in the sample.
Calculation of the atherogenic index (AI)
Atherogenic index (AI) = Log(TG/HDL_C) and “the zone of atherogenic risk”. Triglycerides and HDL-cholesterol in AI reflect the balance between the atherogenic and protective lipoproteins.
RNA isolation and cDNA synthesis
Hypothalamus tissues were collected from rats. Total RNA was extracted from hypothalamus homogenate using GStractTM RNA Isolation kit II (SA-40005, Maxim BioTech, Inc. San Francisco, USA) guanidium thiocynate method. The purity and concentration of RNA was quantified by spectrophotometry. Reverse transcription reaction was performed using oligo (dT) primers (USA). The 25 µl cDNA synthesis reaction consisted of 2.5 µl (5´) buffer with MgCl2, 2.5 µl (2.5 mM) dNTPs (Pharmacia Biotech), 1 µl (10 pmol) Oligo d(T) primer (Pharmacia Biotech), 2.5 µl RNA (2 mg/ml) and 0.5 unit reverse transcriptase enzyme (Qiagene, US). The mixture was incubated at 37°C for 1 hour. PCR amplification was performed in a thermal cycler (Applied Biosystems (ABI), USA) programmed at 42°C for 1 hour, 72°C for 10 min (enzyme inactivation) and the product was stored at 4°C until used.
Real time PCR and quantitative estimation of adiponectin R1 mRNA For qRT-PCR, a set of primers: sense:5’- AGGAGTTCGTATATAAGGTCTG -3’; antisense: 5’- ACATATTTGGTCTGAGCATGGT -3’) were designed from the published cDNA sequences of the rat AdipoR1 gene which amplified a 243 bp product. The reaction was carried out using Rotor-Gene 6000 system (Qiagen, USA) and consisted of 12.5 µl of 2X QuantitechSYBR® Green RT Mix (Fermentas, Germany), 1.0 µl of 25 pm/µl AdipoR1 primers, 2 µl cDNA (100 ng) and 9.25 µl of RNase free water. Samples were spun well before loading in the Rotor’s wells. The real time PCR program was performed as follows: initial denaturation at 95°C for 10 min.; 40 cycles of 95°C for 15 s; annealing at 60°C for 30 s and extension at 72°C for 30 s. Data acquisition performed during the extension step. This reaction was performed using Rotor-Gene 6000 system (Qiagen, USA). Negative controls were included in each set of PCR assays where cDNA was substituted with water as a control for contamination from exogenous sources. In addition, RT was omitted in some samples as a negative control for amplification of genomic DNA. Housekeeping gene, GDPH (glyceraldehyde 3-phosphate dehydrogenase) was amplified under similar conditions: sense: 5’-ATTGATCACTATCTGGGCAA-3’; antisense reverse primer 5’-GAGATACACTTCAATACTTTGACCT- 3’ for comparative quantification analysis using Rotor-Gene 6000 Series Software (Qiagen, USA). PCR products were electrophoresed on 1% agarose gel and visualized by ethidium bromide staining under UV light. The fold increase in the mRNA expression in all groups was calculated. Comparative quantification analysis was done using Rotor-Gene 6000 Series Software based on the following equation:
Fold change in reference gene expression (expt/control)
Statistical analysis
The results were expressed as means ± S.D. Statistical analysis was performed by using GraphPad Prism 6.0 (GraphPad Software Inc., San Diego, CA). The efficacy of training in the holeboard apparatus was assessed by the Two-way ANOVA, followed by the Tukey multiple comparison test. The biochemical data were analyzed by One-way ANOVA, followed by the Tukey multiple comparison test. P value of 0.05 or less was considered significant.
RESULTS
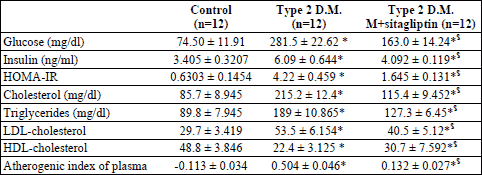
From Table 1, the serum glucose, insulin and HOMA-IR significantly increased in the diabetic rats as compared with the control group (p<0.05) and in response to sitagliptin treatment of the diabetic rats there were a significant reduction of the glucose, insulin and HOMA-IR (p<0.05) when compared with the diabetic rats. The lipid profile of the diabetic rats showed a significant increase in the cholesterol, triglycerides and LDL-cholesterol showed a significant increase, with a significant reduction in the HDL-cholesterol (p<0.05) as compared with the control group. The diabetic rats showed an increased risk of atherosclerosis as detected by the significant increase in the atherogenic index (p<0.05). In contrary, treatment of the diabetic rats with sitagliptin significantly lowered the circulating cholesterol, triglycerides and LDL-cholesterol (p<0.05) with a significant increase in the HDL-cholesterol (p<0.05). Also, the atherogenic index significantly decreased in response to sitagliptin treatment of the diabetic rats.
After feeding rats with high fat diet for one month and streptozotocin (25 mg /kg body weight) injection for induction of type 2 DM the serum glucose and insulin levels after 2 and 12 hours from i.p. injection of glucose showed that the plasma glucose after 2 h was 267.6±29.4 mg/dl and decreased to 151.8±17.2 mg/dl after 12 hours. Additionally, serum insulin after 2 hours was 4.89±0.41 ng/ml and decreased after 12 hours to 3.56±0.57 ng/ml (Fig. 2).
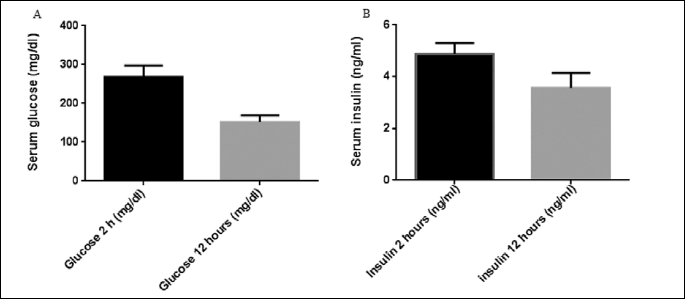
Fig. 3A showed that the serum adiponectin level was significantly reduced in the diabetic rats as compared with the control group (p<0.05). On the other hand, treatment of the diabetic rats with sitagliptin significantly increased the circulating adiponectin level (p<0.05). Also, Fig. 3B showed that the hypothalamic acetylcholine decreased significantly in the diabetic rats (p<0.05) , and it increased significantly (p<0.05) in response to treatment. Fig. 3C showed that the significant (p<0.001) positive correlation between serum adiponectin level and hypothalamic acetylcholine level (r=0.9449) (n=36), Fig. 3D showed that the significant (p<0.001) positive correlation between central acetylcholine levels and adiponectin R1 receptors (r=0.9488) (n=36).
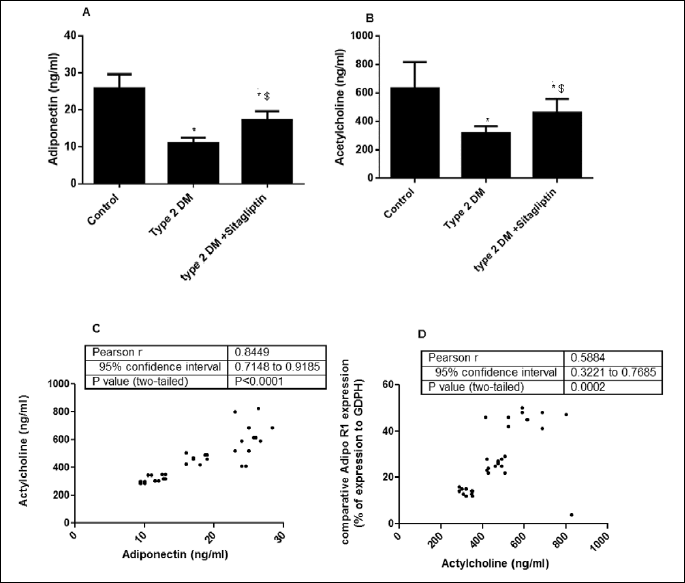
Fig. 4 showed that the AdipoR 1 mRNA expression was significantly decreased (p<0.05) in the diabetic rats by 63% as compared with the control group. While treatment of diabetic rats with sitagliptin significantly (p<0.05) increased the Adipo R1 expression by 55% versus the diabetic rats.
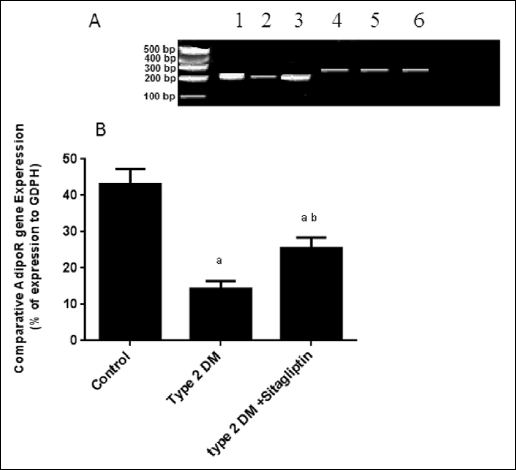 |
Fig. 4. (A) Gel electrophoresis of the quantitative reverse transcription polymerase chain reaction (QRT-PCR) amplification products for hypothalamic Adipo 1Rs mRNA using primers designed from the published cDNA sequences of the rat hypothalamic Adipo 1Rs gene that produced the expected 243 bp product. Samples 1–3 are products of Adipo 1 Rs gene expression. Samples 4–6 represents GDPH gene expression. 1 and 4: control rats; 2 and 5: type 2 DM rats, 3 and 6: type 2 DM rats treated with sitagliptin. (B) Hypothalamic Adipo 1Rs mRNA expression ratio in rats of the experimental groups. |
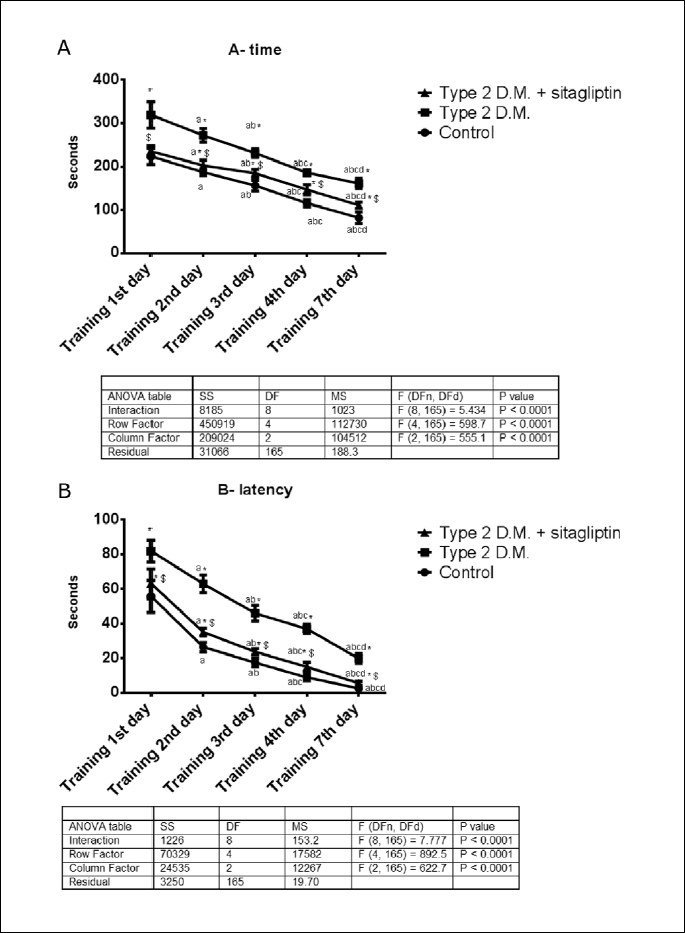
From Fig. 5A, the time needed to collect the pellets from the holes of the holeboard memory showed a significant decrease (p<0.05) on the 7th day as compared with the training sessions of the 1st, 2nd, 3rd and 4th days in the three experimental groups. In comparison with the control rats, the diabetic rats needed a significantly longer time (p<0.05) by 96%. Interestingly treatment of the diabetic rats with sitagliptin significantly (p<0.05) shortened the time to collect the food pellets on the 7th day from 161.3±11.96 seconds to 110.9±7.103 s. The F value for the interaction was 7.037.
From Fig. 5B , the time to visit the first hole (latency) is significantly shortened (p<0.05) in the three groups with the best time on the 4th and 7th days. In the diabetic rats, the latency is significantly (p<0.05) prolonged than the control rats from 2.583±0.3601 seconds to 19.83±2.858 seconds. While the treatment of the diabetic rats, latency was significantly (p<0.05) shortened from 19.83±2.858 seconds to 5.550±1.111 seconds. The F value for the interaction was 7.77.
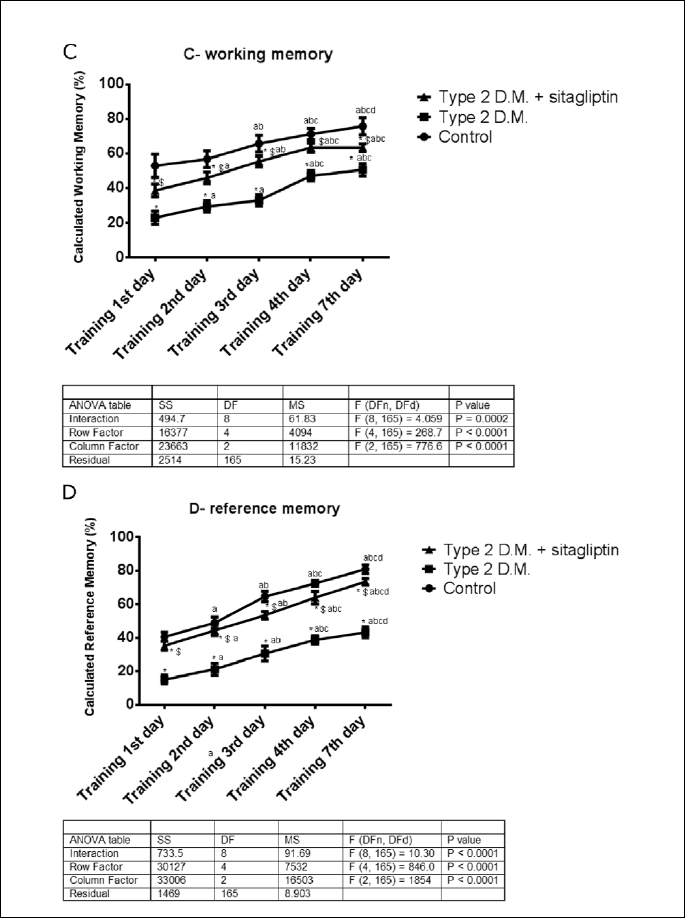
From Fig. 5C, working memory increased significantly (p<0.05) from the 1st day of training in the three groups in response to repeated training to reach the best on the 7th day. In the diabetic rats, working memory decreased significantly (p<0.05) by 23% as compared with the control rats. While treatment of the diabetic rats significantly (p<0.05) increased the working memory from 50.67±7.528 % to 63.5±2.168 %. The F value for the interaction was 10.59.
From Fig. 5D, reference memory increased significantly (p<0.05) in the experimental groups. In comparison between the diabetic rats versus the control, reference memory decreased significantly (p<0.05) to 43.33±3.266 % from 81.00±2.608 %. In response to sitagliptin treatment of the diabetic rats, reference memory increased significantly (p<0.05) by 69.76%. The F value for the interaction was 10.3.
DISCUSSION
Results of the present study revealed that diabetic rats showed a significant increase in fasting blood glucose, LDL, total serum cholesterol, and TGs levels, with a significant decrease in HDL. Furthermore, there was a significant increase in fasting insulin and HOMA-IR, with a significant decrease in adiponectin level when compared with control rats, indicating the development of insulin resistance, which is the main criterion in type 2 DM. These data are consistent with previous studies in animal models of high-fat feeding (27, 28) (Table 1).
In the current study, the diabetic rats needed longer time than the control rats to collect the food pellets in the holeboard memory test. Also, the time needed to start the collection of the food pellets was significantly prolonged than that of the control. Also, diabetic rats showed a significant reduction of the hypothalamic content of acetylcholine with decreased both working memory and reference memory. There are several mechanisms that explain cognitive impairment in diabetics such as cerebral microvascular disease which appeared also likely to be multifactorial. Chronic hyperglycemia produces structural and functional abnormalities in small blood vessels. Hypertension is also highly prevalent in people with type 2 diabetes and is associated with microangiopathy (29). Hyperinsulinemia may share in the pathogenesis of vascular dementia. Individuals with pre-diabetic states (e.g. impaired glucose tolerance or fasting hyperglycemia) or early type 2 diabetes typically have elevated circulating plasma insulin concentrations because of peripheral insulin resistance, which is in turn related to central obesity. Insulin receptors are found in high concentrations within the limbic system of the brain. In epidemiological studies of non-diabetic adults, hyperinsulinemia has been associated with poorer cognitive performance and an increased risk of AD (29).
The performance of the experimental rats might be affected by the degree of food deprivation. The deprivation for all the groups was the same. In agreement with our data, Moreira et al. (30) concluded that type 2 diabetic Goto-Kakizaki rats showed cognitive impairment induced by the progression of diabetes at 4 months age. They studied the operant behavior and positional discrimination in a lever-press task (LPT) for food in parallel with the evaluation of the exploratory activity and habituation to a novel environment in both GK and control Wistar rats. Holeboard memory test was previously used by Stasiak et al. (31), who it to assess the working and reference memory in a rat model of vascular dementia.
From the present data, the hypothalamic acetylcholine was significantly reduced in the diabetic rats when compared with the control rats (Fig. 3). These data are consistent with a previous study performed by Malouf and Birks (32) who concluded that the deficit in cholinergic neurotransmission, a characteristic of Alzheimer’s disease, has also been postulated to contribute to the cognitive disturbances of vascular disease of the brain in humans (32). Reduced acetylcholine production in diabetics has been reported by Irie et al. (33) who found the neuronal damage caused from increased oxidative stress and advanced glycation end products (amino-sugar compounds that are increased in hyper-glycemia and contribute to oxidative stress) leads to reduced acetylcholine production (33).
So far, several studies have documented that insulin resistance is linked to lack of adiponectin (34). They have proposed that obesity leads to hyperinsulinemia and reduction in adiponectin levels. This lack of adiponectin effect contributes to insulin resistance and type 2 DM. Also, Spiegelman’s group reported that adiponectin expression reduced in obese diabetic murine model db/db mice (35). Plasma levels of adiponectin have also been reported to be significantly reduced in obese/diabetic mice and humans (36, 37). Moreover, plasma adiponectin levels have been shown to be decreased in patients with cardiovascular diseases (38, 39), hypertension (40), or metabolic syndrome (41). Thus, reductions in plasma adiponectin levels are commonly observed in a variety of states often associated with insulin resistance.
In the present study, treating diabetic rats with sitagliptin produced a significant improvement of the metabolic dysfunctions (Table 1). The effects of sitagliptin on metabolic disturbances and insulin resistance in diabetic rats are consistent with Ahren et al. (42) who demonstrated that after 12 weeks of oral administration with DPP-4 inhibitor, vildagliptin in patients with type 2 diabetes, long-term glycaemic control, post- prandial plasma glucose, as well as fasting plasma glucose were significantly reduced (40). Previous clinical trials also indicated that a DPP-4 inhibitor such as vildagliptin improved critical B-cell function (43) and increased estimated insulin secretory rate after meal ingestion (44). Also, sitagliptin increased the adiponectin level suggests that adiponectin production may be one of the mechanisms of the insulin sensitizing action of sitagliptin in type 2 DM (Fig. 3). Our results were consistent with Lim et al. (45) who reported that treatment with sitagliptin induced increasing pattern of plasma concentration of adiponectin (45). This was also documented by Suzuki et al. who presented that sitagliptin treatment in Japanese type 2 diabetes patients caused increase of adiponectin (46).
The main aim of the present research is to examine the potential role of Adipo 1R in improving the working and reference memories. The data obtained from this study showed that type 2 DM rats exhibited a significant decrease in the expression of Adipo R1 receptors as compared with the control rats. In agreement with our data, a previous study reported that Adipo R1 expression were decreased in the diabetic rats as reviewed by Al-Hashim et al. (47) who documented that type 2 diabetes mellitus reduced the expression of Adipo R1 in the soleus muscle of Sprague-Dawley rats (47). While, treatment of the diabetic rats with sitagliptin increases the expression of the hypothalamic Adipo 1R mRNA significantly, as compared with the diabetic rats. Additionally, data showed that the acetylcholine content of the hypothalamus increased significantly in the treated group versus the diabetic rats. Post analysis showed a strong significant direct correlation between hypothalamic Adipo 1R mRNA expression and the acetylcholine level in the hypothalamus. Also, hypothalamic acetylcholine is directly correlated with serum adiponectin level. Furthermore, both working and reference memories disturbances are improved in the treated diabetic rats versus non-treated.
The physiological importance of the central adiponectin action is reinforced by the fact that low molecular forms of adiponectin can cross the brain blood barrier (BBB) and are also found in human cerebrospinal fluid (48, 49) and exerts several neuroprotective effects (17-20). As previously known, the hypothalamus is a brain region that plays a key role in memory formation and mood regulation. One unique feature of the hypothalamus is its capacity for neurogenesis. Hypothalamic disorders have been implicated in the pathophysiology of mood disorders and deficits in learning and memory (50). Thus, possibly the mechanism that explained the development of the cognitive function in the treated group could be attributed to the significant increase in the level of acetylcholine in the hypothalamus. Zhang et al. (50) reported that adiponectin via activation of Adipo 1R stimulates the proliferation of the adult hippocampal neurons through stimulation of the p38MAPK/GSK-3β/β-catenin signaling cascade. Furthermore, Zhang and his colleagues reported that apoptosis and differentiation of adult hNSCs into neuronal or glial lineage were not affected by adiponectin (50). With increased adiponectin level and through stimulation of Adipo R1, adiponectin has been shown to activate AMPK and p38MAPK signaling pathways in different cell lines and tissue (34) Adiponectin stimulates phosphorylation of GSK3β on Ser389, a key inhibitory site, and results in nuclear accumulation of the primary substrate of GSK3β, β-catenin.
Improvement of memory in the diabetic rats could be partially caused by GLP-1. GLP-1 exerts various proliferative, neogenic and anti-apoptotic effects on neuronal cells. GLP-1R agonist treatment of PC12 cells stimulates neurite outgrowth, enhances nerve growth factor induced differentiation and improves cell survival after nerve growth factor withdrawal (51). GLP-1R agonists prevent glutamate-induced apoptosis in cultured rat hippocampal neurons and restore cholinergic marker activity in the basal forebrain of ibotenic acid-treated rats, a rodent model of neurodegeneration (52). Furthermore, GLP-1R-activated pathways seem to be necessary for learning and memory. The GLP-1R agonist administration is associated with enhanced learning in rats, an effect that can be blocked by the co-administration of exendin (9, 42). In contrast, GLP-1RK/K mice exhibit deficits in learning that can be overcome by hippocampal Glp1r gene transfer (52). GLP-1RK/K mice are also more susceptible to kainate-induced seizures and hippocampal neuronal degeneration, whereas GLP-1R agonist treatment prevents kainate-induced apoptosis in wild type animals (53). These observations have led to the suggestion that GLP-1R agonists may potentially be useful for the treatment of neurological disorders, including the neuropathy that results as a secondary complication of diabetes (54). In contrast, endogenous GLP-1 has also been implicated in the pathogenesis of b-amyloid protein-induced neurotoxicity as a continuous coinfusion of a GLP-1R antagonist with b-amyloid protein prevented memory impairment and hippocampal apoptosis in rats. Additionally, the results produced by Abbas et al. (55) demonstrated that the murine GLP-1R plays an important role in the control of synaptic plasticity and some forms of memory formation in animal models of Alzheimer’s disease.
There are several factors that influence the cognitive function as well as working and reference memories such as human opiorphin which protects enkephalins from degradation by human neutral endopeptidase and aminopeptidase-N and inhibits pain perception in various behavioral rodent models of pain via endogenous enkephalin-related activation of opioidergic pathways (56). Also, the memory and learning could be disturbed in A.D., Va.D. (31) as well as liver cirrhosis (57).
This study showed that increased Adipo R1 mRNA expression in the hypothalamic neurons is response to sitagliptin treatment in type 2 diabetic rats. Possibly, Adipo 1R might be the cause of increased the acetylcholine level in the hypothalamus with the enhancement of working and reference memories. Further research is needed to explore the different effects of adiponectin on the hypothalamus.
Conclusion
Treatment of the diabetic rats with sitagliptin caused increase of adiponectin level and AdipoR1 mRNA expression, that possibly stimulates the formation of acetylcholine in the hypothalamic neurons. Increased acetylcholine level in the hypothalamus possibly improved working and reference memory.
Conflict of interest: None declared.
REFERENCES
- Zhao WQ, De Felice FG, Fernandez S, et al. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J 2008; 22: 246-260.
- Munshi M, Grande L, Hayes M, et al. Cognitive dysfunction is associated with poor diabetes control in older adults. Diabetes Care 2006; 29: 1794-1799.
- van Harten B, de Leeuw FE, Weinstein HC, Scheltens P, Biessels GJ. Brain imaging in patients with diabetes. A systematic review. Diabetes Care 2006; 29: 2539-2548.
- Tiehuis AM, van der Graaf Y, Visseren FL, et al. Diabetes increases atrophy and vascular lesions on brain MRI in patients with symptomatic arterial disease. Stroke 2008; 39: 1600-1603.
- Larsen J, Hylleberg B, Ng K, Damsbo P. Glucagon-like peptide-1 infusion must be maintained for 24 h/day to obtain acceptable glycemia in type 2 diabetic patients who are poorly controlled on sulphonylurea treatment. Diabetes Care 2001; 24: 1416-1421.
- Hansen L, Deacon CF, Orskov C, Holst JJ. Glucagon-like peptide-(7-36)amide is transformed to glucagon-like peptide-1-(9-36)amide by dipeptidyl peptidase IV in the capillaries supplying the L cells of the porcine intestine. Endocrinology 1999; 140: 5356-5363.
- Deutsch JA. The cholinergic synapse and the site of memory. Science 1971; 174: 788-794.
- Dawson GR, Heyes CM, Iversen SD. Pharmacological mechanisms and animal models of cognition. Behav Pharmacol 1992; 3: 286-297.
- Croxson PL, Browning PG, Gaffan D, Baxter MG. Acetylcholine facilitates recovery of episodic memory after brain damage. J Neurosci 2012; 32: 13787-13795.
- Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab 2002; 13: 84-89.
- Gil-Campos M, Canete R, Gil A. Adiponectin, the missing link in insulin resistance and obesity. Clin Nutr 2004; 23: 963-974.
- Yamauchi T, Kamon J, Ito Y, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003; 423: 762-779.
- Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and\metabolic actions. Nat Med 2007; 13: 332-339.
- Kos K, Harte AL, da Silva NF, et al. Adiponectin and resistin in human cerebrospinal fluid and expression of adiponectin receptors in the human hypothalamus. J Clin Endocrinol Metab 2007; 92: 1129-1136.
- Kubota N, Yano W, Kubota T, et al. Adiponectin stimulates AMP-activated protein kinase in the hypothalamus and increases food intake. Cell Metab 2007; 6: 55-68.
- Coope A, Milanski M, Araujo P, et al. AdipoR1 mediates the anorexigenic and insulin/leptin-like actions ofadiponectine in the hypothalamus. FEBS Lett 2008; 582: 1471-1476.
- Jung TW, Lee JY, Shim WS, Kang ES, Kim JS, et al. Adiponectin protects human neuroblastoma SH-SY5Y cells against MPP+-induced cytotoxicity. Biochem Biophys Res Commun 2006; 343: 564-570.
- Jeon BT, Shin HJ, Kim JB, et al. Adiponectin protects hippocampal neurons against kainic acid-induced excitotoxicity. Brain Res Rev 2009; 61: 81-88.
- Chen B, Liao WQ, Xu N, et al. Adiponectin protects against cerebral ischemia-reperfusion injury through anti-inflammatory action. Brain Res 2009; 1273: 129-137.
- Psilopanagioti A, Papadaki H, Kranioti EF, Alexandrides TK, Varakis JN. Expression of adiponectin and adiponectin receptor in human pituitary gland and brain. Neuroendocrinology 2009; 89: 38-47.
- Vawter MP, Van Ree JM. Hole board food search task in rats: effects of hole depths and food deprivation. Physiol Behav 1989; 45: 891-895.
- Mega C, de Lemos ET, Vala H, et al. Diabetic nephropathy amelioration by a low-dose sitagliptin in an animal model of type 2 diabetes (zucker diabetic fatty rat) Exp Diab Res 2011; 2011: 162092. doi: 10.1155/2011/162092.
- Lannert H, Hoyer S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci 1998; 112: 1199-1208.
- Trinder P. Determination of blood glucose using 4-amino phenazone as oxygen acceptor. J Clin Pathol 1969; 22: 246.
- Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and b-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28: 412-419.
- Friedwald, WT, Levy RI, Fredrickson DS. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972; 18: 499-502.
- Maebuchi M, Machidori M, Urade R, Ogawa T, Moriyama T. Low resistin levels in adipose tissues and serum in high-fat fed mice and genetically obese mice: development of an ELISA system for quantification of resistin. Arch Biochem Biophys 2003; 416: 164-170.
- Lopez IP, Milagro FI, Marti A, Moreno-Aliaga MJ, Martinez JA, De Miguel C. High-fat feeding period affects gene expression in rat white adipose tissue. Mol Cell Biochem 2005; 275: 109-115.
- Strachan MW, Reynolds RM, Frier BM, Mitchell RJ, Price JF. The relationship between type 2 diabetes and dementia. Br Med Bull 2008; 88: 131-146.
- Moreira T, Malec E, Ostenson CG, Efendic S, Liljequist S. Diabetic type II Goto-Kakizaki rats show progressively decreasing exploratory activity and learning impairments in fixed and progressive ratios of a lever-press task. Behav Brain Res 2007; 180: 28-41.
- Stasiak A, Mussur M, Unzeta M, Lazewska D, Kiec-Kononowicz K, Fogel WA. The central histamine level in rat model of vascular dementia. J Physiol Pharmacol 2011; 62: 549-558.
- Malouf R, Birks J. Donepezil for vascular cognitive impairment. Cochrane Database Syst Rev 1: CD004395. 2004.
- Irie F, Fitzpatrick A, Lopez O, et al. Enhanced risk for Alzheimer disease in persons with type 2 diabetes and apoE4. Arch Neurol 2008; 65: 89-93.
- Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev 1999; 26: 439-451.
- Hu E, Liang P, Spiegelman BM. AdipoQ is a novel adipose specific gene dysregulated in obesity. J Biol Chem 1996; 271: 10697-10703.
- Mori Y, Ide T, Murakami K, et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat Med 2001; 7: 941-946.
- Arita Y, Kihara S, Ouchi N, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 1999; 257: 79-83.
- Hotta K, Funahashi T, Arita Y, et al. Plasma concentrations of a novel, adipose-specific protein, adiponectin, in type 2 diabetic patients. Arterioscler Thromb Vasc Biol 2000; 20: 1595-1599.
- Kumada M, Kihara S, Sumitsuji S, et al. Osaka CAD Study Group. Association of hypoadiponectinemia with coronary artery disease in men. Arterioscler Thromb Vasc Biol 2003; 23: 85-89.
- Ouchi N, Ohishi M, Kihara S, et al. Association of hypoadiponectinemia with impaired vasoreactivity. Hypertension 2003; 42: 231-234.
- Trujillo ME, Scherer PE, Adiponectin: journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Intern Med 2005; 257: 167-175.
- Ahren B, Gomis R, Standl E, Mills D, Schweizer A. Twelve- and fifty two-week efficacy of the dipeptidyl peptidase IV inhibitor LAF237 in metformin-treated patients with type 2 diabetes. Diabetes Care 2004; 27: 2874-2880.
- Mari A, Sallas WM, He YL, et al. Vildagliptin, a dipeptidyl peptidase-IV inhibitor, improves model-assessed beta-cell function in patients with type 2 diabetes. J Clin Endocrinol Metab 2005; 90: 4888-4894.
- Ahren B, Pacini G, Foley JE, Schweizer A. Improved meal-related beta-cell function and insulin sensitivity by the dipeptidyl peptidase-IV inhibitor vildagliptin in metformin-treated patients with type 2 diabetes over 1 year. Diabetes Care 2005; 28: 1936-1940.
- Lim S, Choi SH, Shin H, et al. Effect of a dipeptidyl peptidase-IV inhibitor, des-fluoro-sitagliptin, on neointimal formation after balloon injury in rats. PLoS One 2012; 7: e35007.
- Suzuki K, Watanabe K, Suzuki T, et al. Sitagliptin improves vascular endothelial function in Japanese type 2 diabetes patients without cardiovascular disease JDM 2012; 2: 338-345.
- Al-Hashim F, Ibriahim I, Bastawy N, et al. Effect of insulin on adiponectin and adiponectin receptor-1 expression in rats with streptozocin induced type 2 diabetes. J Health Sci 2011; 57: 334-340.
- Kuminski CM, McTernan PG, SchrawT, et al. Adiponectin complexes in human cerebrospinal fluid: distinct complex distribution from serum. Diabetologia 2007; 50: 634-642.
- Norsted E, Gomuc B, Meister B. Protein components of the blood-brain barrier (BBB) in the mediobasal hypothalamus. J Chem Neuroanat 2008; 36: 107-121.
- Zhang D, Guo M, Zhang W, Lu XY. Adiponectin stimulates proliferation of adult hippocampal neural stem/progenitor cells through activation of the p38MAPK/GSK-3β/β-catenin signaling cascade J Biol Chem 2011; 286: 44913-4420.
- Perry T, Haughey NJ, Mattson MP, et al. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther 2002; 302: 881-888.
- Perry T, Lahiri DK, Chen D, et al. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther 2002; 300: 958-966.
- During MJ, Cao L, Zuzga DS, et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat Med 2003; 9: 1173-1179.
- Perry T, Greig NH. The glucagon-like peptides: a double-edged therapeutic sword? Trends Pharmacol Sci 2003; 24: 377-383.
- Abbas T, Faivre E, Holscher C. Impairment of synaptic plasticity and memory formation in GLP-1 receptor KO mice: interaction between type 2 diabetes and Alzheimer’s disease. Behav Brain Res 2009; 205: 265-271.
- Javelot H, Messaoudi M, Garnier S, Rougeot C. Human opiorphin is a naturally occurring antidepressant acting selectively on enkephalin-dependent delta-opioid pathways. J Physiol Pharmacol 2010; 61: 355-362.
- Ciecko-Michalska I, Wojcik J, Wyczesany M, et al. Cognitive evoked response potentials in patients with liver cirrhosis without diagnosis of minimal or overt hepatic encephalopathy. A pilot study. J Physiol Pharmacol 2012; 63: 271-276.
A c c e p t e d : September 10, 2013