LIPID PEROXIDATION, REACTIVE OXYGEN SPECIES AND ANTIOXIDATIVE FACTORS IN THE PATHOGENESIS OF GASTRIC MUCOSAL LESIONS AND MECHANISM OF PROTECTION AGAINST OXIDATIVE STRESS - INDUCED GASTRIC INJURY
2Gastroenterology and Hepatology Clinic, Jagiellonian University Medical College, Cracow, Poland
INTRODUCTION
The gastric mucosa plays an essential role in maintaining the physiological functions of the stomach. This mucosa acts as gastric barrier, which protects deeper tissue against the damaging actions of the gastric juice components and ingested mucosal irritants (1, 2). In the classic approach, a gastric mucosal barrier is composed of cells from the gastric epithelium with intracellular tight junctions as well as adjacent layer of mucus. Gastric blood flow plays a crucial role in the maintenance of gastric integrity (3, 4). The undisturbed gastric blood flow is regulated by many physiological factors and mechanisms, including nitric oxide (NO), afferent capsaicin-sensitive C fibers and products of cyclooxygenase (COX) activity (5, 6).
NO is produced and released from the vascular endothelium, epithelial cells and sensory nerve endings (5, 7) via the activity of NO synthase (NOS). A substrate for this enzyme is aminoacid L-arginine and NO-synthase (NOS) puts molecules of oxygen (O2) into molecule of L-arginine, capable of producing NO (7). NO diffuses from the endothelium to smooth muscles, located in the vascular wall, where NO reacts with guanylyl cyclase, leading to cellular enhancement of cyclic guanosine monophosphate (cGMP), acting as a second messenger. The increment of cGMP activity, in smooth endothelial muscle, causes relaxation of vascular wall, accompanied by an increase of blood flow through this vessel (8). This vasodilatatory effect is mimicked by exogenous administration of nitrates, namely NO donors, such as 3-morpholinosydnonimine (SIN-1), S-nitroso-N-acethyl-D,L-penicylamine (SNAP), gliceryl trinitrate (GTN) or NO-releasing aspirin (NO-ASA) (9). Other vasodilators, for example, pentoxifylline (PTX) may act on smooth myocytes, causing their relaxation and this effect seems to be NO independent (10).
The vasodilatatory effect of NO contributes to the maintenance of gastric mucosal barrier integrity and the inhibition of NO production by a nonspecific N-nitro-L-arginine (L-NNA) was shown to markedly impair important functions of a gastric mucosa including gastric secretion and gastric motility (11, 12). The inhibition of NOS by the administration of exogenous synthetic inhibitors such as L-NNA, L-NAME or by the endogenous NOS inhibitor, ADMA have been shown to exacerbate the acute gastric mucosal lesions and delayed the healing of chronic gastric ulcers (10, 12, 14, 15). The adverse effect of blockade of NOS on gastric integrity by L-NNA or the aggravatory effect of ADMA on gastric mucosal lesions can be reversed by administration of L-arginine, a substrate for this enzyme, administered in the presence of these inhibitors (10, 14, 15) (Fig. 1).
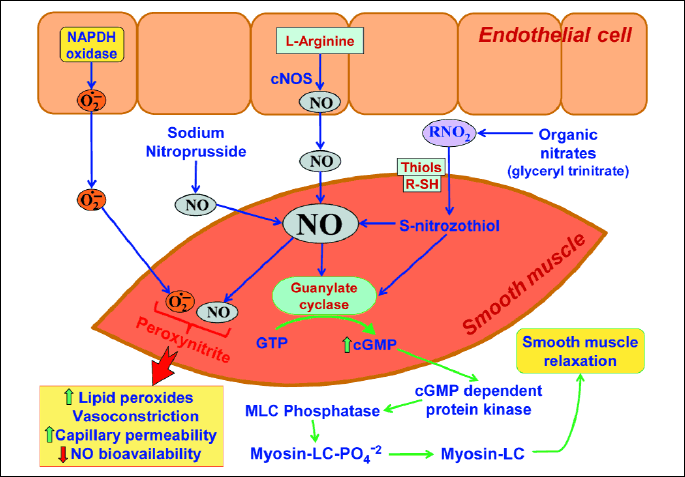
After stimulation of endothelial cells, constitutive NO synthase (cNOS) is activated, which transforms L-arginine (L-Arg) to nitric oxide (NO). NO diffuses to smooth muscle cells of gastric blood vessels. Inside smooth muscle, NO activates guanylate cyclase, transforming guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). This cGMP, acting via myosin light chains (MLC) phosphatase leads to relaxation of smooth muscle cell and subsequent increase of vessel diameter and larger blood flow. NO may be also deliver by exogenous NO donors, such as sodium nitrate, nitroprusside or other organic nitrates and then it acts the same way, as endogenous NO. Thiols (R-SH), for example, glutathione (GSH) cooperate with NO.
The maintenance of gastric mucosa integrity and the gastroprotection depend upon the activity of afferent capsaicin-sensitive C fibers (16-18). Sensory nerves are involved in the regulation of blood microcirculation in the gastric mucosa, which is densely innervated by capsaicin-sensitive afferent neurons, containing vasodilator peptides, such as calcitonin gene-related peptide (CGRP) (8, 16, 17). The C fibers are sensitive to capsaicin administration, because the low doses of capsaicin stimulate of sensory nerves accompanied by the release of CGRP, whereas high doses of capsaicin lead to functional ablation of these fibers (10). Therefore, the ablation of sensory nerves by high doses of capsaicin provides the opportunity to determine their role in the regulation of gastric integrity (13).
The next component, undoubtedly essential for gastric mucosal barrier physiology, is prostaglandin cyclooxygenase (COX), which converts arachidonic acid, a substrate for COX-1 and COX-2 to prostaglandins, especially prostaglandin E2 (PGE2) (6, 8, 9). Prostaglandins (PGs) prevent damage of deeper structures due to an increase of mucus secretion, intensification of bicarbonate anions production (HCO3-), which neutralise acidic gastric content and the stimulation of mucosal phospholipids (6, 19, 20). PGs were shown to evoke increment of gastric blood flow (4, 21, 22) (Fig. 2), thus enhancing oxygen and nutrients delivery to the gastric mucosa. Two isoforms of COX: constitutive isoform, called COX-1, as well as inducible isoform, called COX-2 were proposed (3, 6, 8). Classic approach to their functions revealed that PGs derived from COX-1 exert gastroprotective effects, while high levels of PGs, generated via COX-2 are associated with inflammation causing an increase of vessels permeability, pain and fever (8, 23-25). The administration of non-selective COX inhibitors, for instance aspirin, which exerts a potent anti-inflammatory effect, resulting from COX-2 inhibition, can also cause side effects such as bleeding and haemorrhagic lesions of the gastrointestinal mucosa, predominately due to COX-1 inhibition (10, 13).
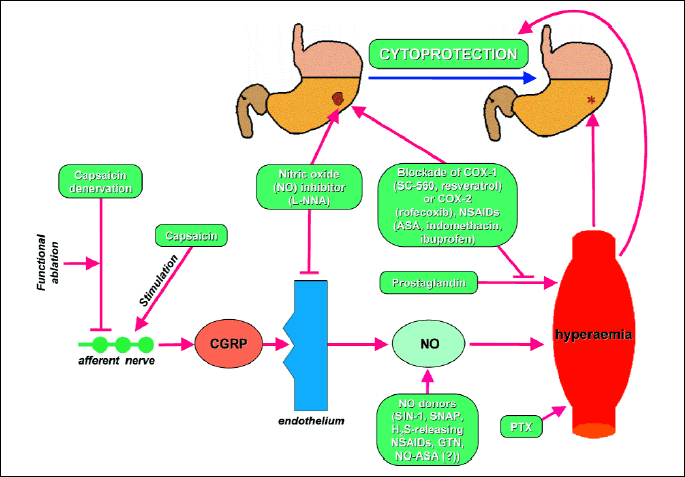
Calcitonin gene related peptide (CGRP) is released at the ending of an afferent nerve. Then, CGRP stimulates an endothelium to release nitric oxide (NO). NO influences a blood vessel, resulting in hyperemia. Hyperemia plays an important role in a cytoprotection. This pathway may be activated or blocked at different levels. Low dose of capsaicin stimulates an afferent nerve. High dose of capsaicin produces capsaicin denervation and blocks an afferent nerve. N-nitro-L-arginine (L-NNA) inhibits nitric oxide synthase (NOS) in an endothelium. NO donors, such as SIN-1 (3-morpholinosyndnoimine), SNAP (S-nitroso-N-acetyl-DL-penicillamine), nitroglycerin (GTN), NO-aspirin (NO-ASA) deliver NO exogenously. Pentoxifylline (PTX) and prostaglandins, similarly to NO, cause hyperaemia. COX (cyclooxygenase), producing prostaglandins, may be blocked by SC-560, rofecoxib, resveratrol or aspirin (ASA).
These adverse effects of aspirin's action and other NSAIDs ingested by patients with inflammatory disorders such as rheumatoid arthritis brought about the question, if new derivative of aspirin, chemically linked to NO moiety, and called NO-releasing NSAIDs (NO-aspirin, NO-naproxen) can counteract the mucosal damage and micro-bleeding associated with this novel NSAID therapy compared with classic NSAIDs therapy. Both novel NO-releasing NSAIDs were shown to possess COX-1-inhibitory and NO donating properties, thus diminishing side effects including both gastroduodenal bleedings and hemorrhagic lesions of gastrointestinal mucosa (24, 26, 27).
REACTIVE OXYGEN SPECIES
As mentioned above, the reactive oxygen species (ROS) contribute to the pathogenesis of gastric damage and many agents were shown before to afford protection of the gastric mucosa via inhibition of the oxygen metabolic pathways (13, 28-30). The ROS are atoms or molecules, which exhibit higher chemical activity than molecular oxygen in the basic state (31). The most important ROS's include free radicals, such as superoxide radical anion (O2●-), hydroperoxyl radical (HO2●-) and hydroxyl radical (OH●) (32, 33). They exhibit high reactivity due to unpaired electron in the outermost shell. Non-free radicals, such as hydrogen peroxide (H2O2), ozone (O3) and singlet oxygen (1O2) also belong to a class of ROS, because of their high oxidative reactivity. Interestingly, the ROS could be generated intracellulary and extracellulary (34-37). Intracellular mechanism of ROS production is predominantly based on local ischemic episodes within tissues followed by reperfusion (18, 21). Cellular ischemia results in diminished synthesis and release of adenosine triphosphate (ATP) in mitochondria. In this conditions ATP is breaking down to adenosine monophosphate (AMP) and then adenine and hypoxanthine. At the same time the mitochondria releases calcium ions (Ca2+) from their internal space into cytoplasm. The increment of the cytoplasmatic pool of Ca2+ activates intracellular protease, which converts xanthine dehydrogenase (XDH) into xanthine oxidase (XO) (33). The XDH uses nicotinamide adenine dinucleotide (NAD+) as the electron acceptor for the oxidation of hypoxanthine and xanthine into uric acid. This process is not accompanied by the generation of ROS, rather, XO uses molecular oxygen, which is delivered during reperfusion as an electron acceptor to produce superoxide radical anion. Another mode of ROS generation is related to the activation of the mitochondrial respiratory chain (38). Part of the total number of oxygen molecules, involved in the function of mitochondrial respiratory chain, is reduced in single - electron reaction, because electrons "leak" from the electron mitochondrial transport chain and this leads to the formation of superoxide radical anion (30) (Fig. 3).
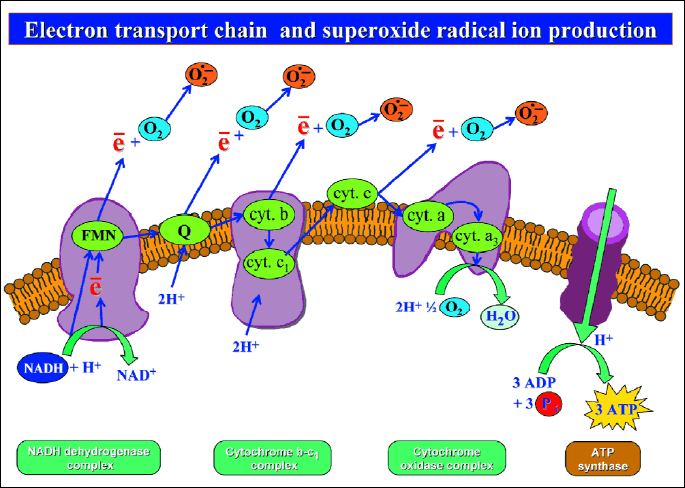
Reactive oxygen species generation is related to the activation of mitochondrial respiratory chain. Correct pathway of mitochondrial respiratory chain involves NADH (reduced nicotinamide adenine dinucleotide) dehydrogenase complex, cytochrome (cyt.) b - c1 complex and cytochrome oxidase complex. Electron transport along these complexes results in complete reduction of oxygen molecule (O2), in presence of hydrogen ions (H+), to water molecule (H2O). Part of total number of oxygen molecules, involved in the function of mitochondrial respiratory chain is reduced in single electron reaction, because electrons "leak" from the electron mitochondrial transport chain and this leads to formation of superoxide radical anion.
In the extracellular model of ROS generation, O2●- is released from the outer surface of the cellular membrane to the extracellular fluid. Classic example of this process is superoxide (O2●-) production by neutrophils. A neutrophil possesses in its cellular membrane a specific enzyme NADPH oxidase, which is composed by two subunits: flavoprotein and cytochrome b558 (30, 39). This enzyme catalyzes a double - electrons reduction of oxygen molecule (O2), finally leading to generation of O2●-. The source of electrons, in this process, is reduced nicotinamide adenine dinucleotide (NADPH) (36, 38). NADPH oxidase manifests higher affinity to NADPH, than NADH, so NADPH is only substrate for this enzyme in a cell. Neutrophil derived O2●- diffuses to adjacent tissues. Due to stability of O2●- in physiological pH, it can reach distant organelles from its place of generation (28, 36). Kasazaki et al. (40) have documented the association between both extracellular and intracellular sources of ROS. The radical O2●- produced by XO (intracellular mechanism) facilitates tissue infiltration by neutrophils and this effect leads to an augmentation of O2●- generation from extracellular sources.
Further transformations of O2●- take place in tissues, because the two O2●- radical react to each other leading to the formation of hydrogen peroxide (H2O2) (41, 42). This reaction can occur spontaneously or it could be catalyzed by an enzyme - superoxide dismutase (SOD). H2O2 reacts with O2●-, resulting in the generation of OH● according to the Haber-Weiss reaction. This process is accelerated by the presence of iron (Fe2+) ions (the Fenton reaction) (41, 46). The formation of ROS may serve as a prerequisite for damage to the surrounding tissues. Using electron paramagnetic resonance with spinal trapping, Kasazaki et al. (40) and Yasukawa et al. (42) have revealed that OH seems to play a major role in the formation of gastric mucosal injury.
LIPID PEROXIDATION
Irrespective of ROS type, the first stage of ROS-mediated cellular damage is peroxidation of cellular membrane components, especially membrane lipids in the process, called lipid peroxidation (47, 48). This process particularly involves the ROS-mediated oxidative degradation of components of cellular membrane phospholipids, such as polyunsaturated fatty acids (PUFA). In first step of lipid peroxidation, ROS detaches hydrogen the atom from a chain of PUFA, followed by the reduction of ROS to water and the transformation of fatty acid to free radical. This radical of fatty acid attaches to oxygen molecule loading to the generation of peroxyl radical. Free peroxyl radical of fatty acid has the ability to detach hydrogen atoms from other PUFAs to generate lipid peroxides. Compared with lipids, lipid peroxides are less stable and may break down to free radicals. This process is accelerated by the presence of iron and copper ions (Fe2+, Cu2+) (46, 49, 50). High reactivity of peroxyl radicals with lipids molecules, as well as chemical instability of lipid peroxides, contribute to positive feedback in lipid peroxidation, thus quickly involving of majority of lipids at cellular membrane (Fig. 4). Lipid peroxides are metabolized, via β-oxidation pathway to malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) (51-53). Other constituents of cellular membrane, as aminoacids or proteins, are also involved in the process of lipid peroxidation, however, in contrast to peroxidation of lipids, the speed of this reaction is slowed down (33, 47, 48).

The products of lipid peroxidation, MDA and 4-HNE, are advisable to be used as indicators of ROS-dependent tissue damage in various organs including stomach and intestine (33, 47, 54). Lipid peroxidation products modify properties of cellular membranes, by inserting polar groups into phospholipid molecules, located inside the lipid bilayer; this way the lipid internal part of the membrane becomes hydrophobic and more permeable (47). Lipid peroxidation also causes alterations in the membrane potential toward a depolarization. Moreover, the peroxidation inhibits activity of protein transporters, leading to a derangement of the active transport through the membrane (32, 51). The products of lipid peroxidation uncouples the respiratory chain phosphorylation within the mitochondria, resulting from an increase in permeability of internal mitochondrial membrane for protons. This mechanism creates the equilibrium of proton concentrations between both sides of the internal mitochondrial membrane (39, 52). Aldehyde products of lipid peroxidation, such as MDA, react with amine groups of membrane protein to yield Schiff bases and therefore cellular membrane becomes more stiff. Moreover, reaction between MDA and cellular membrane protein changes its antigenic characteristic. Thiols also undergo oxidation causing inactivation of the active enzymatic centers (33, 47, 48).
The next product of lipid peroxidation, namely 4-HNE, was also shown to participate in the disorder of cellular functioning during oxidative stress (46, 52, 53). This toxic product of lipid peroxidation is probably involved in the pathogenesis of many diseases including e.g. atherosclerosis, Alzheimer disease and peptic ulcer disease (52, 54). Moreover, 4-HNE modifies cellular growth and exhibits signaling properties (53). 4-HNE easily diffuses within the tissues making distant lesions and, similarly to MDA may react with thiol and amine groups of cellular proteins, resulting in cell metabolic disturbances. Other important properties of 4-HNE are stimulation of neutrophil chemotaxis, activation of phospholipase C (PLC) and the activation of adenylyl kinase. Stimulation of neutrophil chemotaxis facilitates ROS - dependent tissue destruction, resulting from intracellular generation of ROS (52, 53). In conclusion, lipid peroxidation products (MDA and 4-HNE) are responsible for a plethora of cellular pathophysiological events, therefore measurement of their concentrations, which reflects the damage induced by oxidative stress can be considered to be a useful tool in the experimental and clinical settings (39, 48, 53, 55).
SUPEROXIDE DISMUTASE
Superoxide radical anions (O2●-) are transformed to hydrogen peroxide (H2O2) during spontaneous or a catalyzed reaction, named a dismutation (43-45). SOD is an enzyme, which catalyzes the dismutation of O2●- into less noxious H2O2 (49). Three types of SOD can be distinguished: cytoplasmatic, mitochondrial or extracellular ones, all containing metal ions in their catalytic active site. Both the cytoplasmatic and extracellular SOD contains copper and zinc ions; while the mitochondrial SOD is equipped with manganous ion. Another isoform of SOD may contain iron ion in the catalytic active site, but this SOD is predominantly present in prokaryotes, for example, in bacteria (50, 51). The cytoplasmic form of SOD consists of two identical, chemical stable subunits. Chemical stability of this SOD is maintained by hydrogen bonds, disulfide linkages inside subunit, as well as the presence of zinc ion in the catalytic active site (45), while copper ion plays a crucial role in the chemical activity of this enzyme. The activity of this Cu/Zn-SOD enzyme involves the oxidation of cuprous ion (Cu+) and reduction of cupric ion (Cu2+) by O2●-, in the presence of hydrogen ions (H+), leading to H2O2 generation. To augment effectiveness of O2●- neutralization, SOD possesses a complex of amino acids with electric charges on the enzyme molecule surface, necessary for the creation of an electrical gradient. This gradient drags O2●- directly to the active center of the enzyme (49, 50, 57).
SOD plays a crucial role in scavenger cascade responsible for ROS neutralization (55). H2O2 formed due to SOD activity is further broken down by antioxidazing enzymes: a catalase or a glutathione peroxidase (13, 53, 58). The catalase accelerates H2O2 breakdown to water and oxygen. The second biochemical pathway of H2O2 metabolism depends on the activity of glutathione peroxidase, which cooperates with the action of glutathione reductase. Glutathione peroxidase - induced breakdown of H2O2 to water and is accompanied by transformation of reduced glutathione (GSH) to its oxidized form (GSSG) (Fig. 5). Glutathione peroxidase has a major affinity to H2O2, suggesting the potent antioxidizing activity of this enzyme in the maintenance of physiological conditions and cell homeostasis (56, 58-60).
REDUCED FORM OF GLUTATHIONE
Reduced form of GSH is believed to act as a main intracellular antioxidative buffer with multifaceted action against tissue oxidative stressors. Chemically, glutathione is a peptide composed of 3 amino acids, namely: glutaminic acid, cysteine and glycine (γ-glutamylcysteinylglycine), and all containing thiol (SH) groups originating from cysteine. The availability of the glutathione SH group to oxidative action of ROS leads to the formation of glutathione free radical (GS●) or, as mentioned above, glutathione disulphide (GSSG), also known as oxidized glutathione. Both forms undergo further biochemical transformations, leading to regeneration of a reduced form of GSH with a SH group, possessing the activity of digestive peptidases (33). Isopeptic bond in the GSH molecule, composed of γ-carboxylic residue of glutaminic acid with an amine group of cysteine protects GSH from intracellular degradation (58).
GSH is a substrate for glutathione peroxidase (GPx) - an enzyme which reduces H2O2. Both, GPx and GSH may also inhibit lipid peroxidation directly or indirectly by mediation of lipid peroxides peroxidase (10, 33). The important function of GSH is protection of cellular proteins against oxidative injury. ROS oxidizes proteins both through formation of protein free radicals and oxidation of protein thiols. This latter action can be dangerous to enzymatic proteins, because of their inactivation. GSH reduces protein free radicals yielding glutathione free radical (GS●). In the case of oxidation of protein thiols, activity of GSH is mediated by specific enzymes called thiol transferases, which acts as catalyzed, and are involved in the reduction of protein thiols by GSH, then transformed to an oxidized form GSSG (31, 34). GSH can act as a substrate for glutathione transferases (61, 62). These enzymes conjugate GSH to xenobiotics, which enables their removal from an organism. Glutathione transferases contribute to elimination of lipid peroxidation products, namely GSH and 4-HNE. GSH+4-HNE complex may be removed from a cell by active membrane transport (61, 62). GSH can also cooperate with SOD to neutralize ROS. Reactions between GSH and ROS yield glutathione free radical (GS●), as described above. GS● reacts with GSH to yield free radical of glutathione disulphide (GSSG●), which in turn donates an electron to the oxygen molecule, converting it to O2●-, and is then eliminated by SOD (31, 33). This clearly suggest that GSH and SOD cooperate in cell protective action against oxidizing stress that may lead to the formation of gastric mucosal lesions in the gastric mucosa caused by cellular damage via reactive oxygen metabolites such as O2●- and lipid peroxidation products (63, 64).
EXPERIMENTAL MODELS OF GASTRIC MUCOSA INJURY
The intragastric application of ethanol (3, 5, 63), the exposure of rodents to water immersion and restraint stress (8, 10, 13, 26) as well as the ischemia followed by reperfusion (I/R) (21, 43, 47, 64) are widely accepted model of experimental injury to gastric mucosa. In the majority of these methods, the development of inflammation, often hemorrhagic type of inflammation, serves as a prerequisite for mucosal erosions and even ulcers. The mechanism of gastroprotection against gastric mucosal lesions induced by ethanol, stress or I/R can involve the alterations in gastric blood flow, mucus production and the role of prostaglandins (21, 63-65), nitric oxide (9, 63, 65), growth factors (66, 67), appetite controlling peptides such as nesfatin-1 (15), leptin (19) or ghrelin (68-70). The pathogenesis of mucosal damage include the effect of damaging agents on gastric acid secretion and neural regulation via brain-gut axis (15, 19) and the participation of microorganism infecting human stomach, such as Helicobacter pylori (37, 50 71) and Candida albicans (72, 73). The ROS and oxidative metabolism can contribute to a disturbances of the gastric mucosal barrier and the formation of gastric lesions and their role in pathogenesis of gastric mucosal injury has been described in a numerous studies (10, 20, 26, 33, 47, 65). The neutrophil induced gastric tissue infiltration have also been documented, as well as the increase of tissue MDA, 4-HNE levels and diminution of antioxidative mechanisms. The alteration in antioxidative status of gastric mucosa is accompanied by the decrease of SOD activity, as well as the depletion of the GSH pool, both implicated in the pathogenesis of I/R gastric lesions (21, 43, 47). In rats exposed to 3.5 hours of water cold stress (WRS), numerous gastric mucosal bleeding erosions accompanied by the decrease of gastric blood flow (GBF) were observed (10, 13, 26, 47). Moreover, in a majority of these studies, an increase in MDA and 4-HNE considered as an indicator of lipid peroxidation with a decrease in gastric mucosal expression and activity of antioxidative enzymes SOD and GSH were also notified. The blockade of COX-1 and COX-2 activity by administration of SC-560 and rofecoxib, respectively, and the capsaicin denervation had magnified the number of gastric lesions and these effects were accompanied by a further reduction of GBF (Table 1), SOD activity, GSH concentration and enhancement in lipid peroxidation, as reflected by higher MDA and 4-HNE levels, when compared to animals exposed to WRS only (Table 2). On the other hand, NO donors, such as SIN-1, SNAP, glyceryl trinitrate and NO-releasing aspirin (26, 28) or antioxidazing compounds, such as resveratrol (65, 74) and pentoxifylline (75), afforded the protection of the gastric mucosa against WRS (Table 3), in part via activation of antioxidative parameters involving a decrease of MDA and 4-HNE, and an increase of SOD and GSH activities (Table 4). Although resveratrol inhibits COX-1 (65), it acts as a radical scavenger within mucosa injured by stress, thus affording some protection (Table 3 and Table 4). On the contrary, aspirin possesses scavenger's properties (Table 2), but this NSAID aggravates stress-induced gastric lesions (Table 1), suggesting that its damaging action depends rather on the more potent inhibition of prostaglandin production than the scavenging activity against the formation of ROS.
Some of these compounds can exert beneficial influence on the gastric mucosa (glyceryl trinitrite , pentoxifylline) (75) or they act as metabolites of medications (e.g. SIN-1 is a metabolite of molsydomine) (10, 13). The adverse effects in the stomach such as the acute microbleedings and gastric hemorrhagic lesions evoked by stress are potentiated in the presence of NSAID such as aspirin or indomethacin. Thus, the more detailed determination of oxidative stress and the associated pathology in the GI tract may contribute in design of new, noninvasive method of prevention of gastrointestinal injury caused by various ulcerogenes.
The lipid peroxidation products, specifically the activity of (SOD) and the levels of GSH, play an important role as an indicator of tissue damage by ROS, known to contribute to the pathogenesis of gastrointestinal damage (31, 34, 76). The oxidative stress, amplified during stress (ulcerogenesis) was accompanied by an elevation of MDA + 4-HNE and diminution of SOD and GSH content. These ROS-mediated effects in gastric mucosa exposed to stress were reversed by gastroprotective substances releasing of NO, such as SIN-1, SNAP, nitroglycerin, NO-releasingASA, and the antioxidizing compound resveratrol resulting in reduction of stress-induced gastric damage and attenuation of MDA + 4-HNE, content and an increase in activity of antioxidizing enzymes SOD and GSH.
Acknowledgements: The experimental work described in this review received funds from Ministry of Science and Higher Education (K/ZDS/003805 to Dr. Kwiecien) and was in part, supported by the grant from the National Center of Science in Poland (Grant NO. UMO-2013/09/B/NZ4/01566) to Dr T. Brzozowski. Authors are grateful to Dr. Nily Osman for her helpful linguistic comments and corrections to the text of this paper.
Conflict of interests: None declared.
REFERENCES
- Laine L, Takeuchi K, Tarnawski A. Gastric mucosal defense and cytoprotection: bench to bedside. Gastroenterology 2008; 135: 41-60.
- Tarnawski A, Ahluwalia A, Jones MK. Gastric cytoprotection beyond prostaglandins: cellular and molecular mechanisms of gastroprotective and ulcer healing actions of antacids. Curr Pharm Des 2013; 19: 126-132.
- deFoneska A, Kaunitz JD. Gastroduodenal mucosal defense. Curr Opin Gastroenterol 2010; 26: 604-610.
- Brzozowski T, Konturek PC, Konturek SJ, Brzozowska I, Pawlik T. Role of prostaglandins in gastroprotection and gastric adaptation. J Physiol Pharmacol 2005; 56 (Suppl. 5): 33-55.
- Whittle BJR, Lopez Belmonte J, Moncada S. Regulation of gastric mucosal integrity by endogenous nitric oxide, interactions with prostanoids and sensory neuropeptides in the rat. Br J Pharmacol 1990; 99: 607-611.
- Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn't the stomach digest itself? Physiol Rev 2008; 88: 1547-1565.
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev 1991; 43: 109-142.
- Brzozowski T, Konturek PC, Pajdo R, et al. Physiological mediators in nonsteroidal anti-inflammatory drugs (NSAIDs)-induced impairment of gastric mucosal defense and adaptation. Focus on nitric oxide and lipoxins. J Physiol Pharmacol 2008; 59 (Suppl. 2): 89-102.
- Kwiecien S, Pawlik MW, Brzozowski T, et al. Nitric oxide (NO)-releasing aspirin and (NO) donors in protection of gastric mucosa against stress. J Physiol Pharmacol 2008; 59 (Suppl. 2): 103-115.
- Kwiecien S, Brzozowski T, Konturek PC, et al. The role of reactive oxygen species and capsaicin-sensitive sensory nerves in the pathomechanisms of gastric ulcers induced by stress. J Physiol Pharmacol 2003; 54: 423-437.
- Ishii K, Chang B, Kerwin JF, Huang ZJ, Murad F. N-nitro-L-arginine a potent inhibitor of endothelium-derived relaxing factor formation. Eur J Pharmacol 1990; 176: 219-223.
- Kwiecien S, Ptak-Belowska A, Krzysiek-Maczka G, et al.: Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, interacts with gastric oxidative metabolism and enhances stress-induced gastric lesions. J Physiol Pharmacol 2012; 63: 515-524.
- Kwiecien S, Konturek PC, Sliwowski Z, et al. Interaction between selective cyclooxygenase inhibitors and capsaicin-sensitive afferent sensory nerves in pathogenesis of stress-induced gastric lesions. Role of oxidative stress. J Physiol Pharmacol 2012; 63: 143-151.
- Szlachcic A, Krzysiek Maczka G, Pajdo R, et al. The impact of asymmetric dimethylarginine (ADAMA), the endogenous nitric oxide (NO) synthase inhibitor, to the pathogenesis of gastric mucosal damage. Curr Pharm Des 2013; 19: 90-97.
- Szlachcic A, Sliwowski Z, Krzysiek-Maczka G, et al. New satiety hormone nesfatin-1 protects gastric mucosa against stress injury. Mechanistic roles of prostaglandins, nitric oxide, sensory nerves and vanilloid receptors. Peptides 2013; 49: 9-20.
- Holzer P. Acid sensing by visceral afferent neurones. Acta Physiol (Oxf) 2011; 201: 63-75.
- Ohno T, Hattori Y, Komine R, et al. Roles of calcitonin gene-related peptide in maintenance of gastric mucosal integrity and in enhancement of ulcer healing and angiogenesis. Gastroenterology 2008; 134: 215-225.
- Brzozowska I, Konturek PC, Brzozowski T, et al. Role of prostaglandins, nitric oxide, sensory nerves and gastrin in acceleration of ulcer healing by melatonin and its precursor, L-tryptophan. J Pineal Res 2002; 32: 149-162.
- Tache Y. Brainstem neuropeptides and vagal protection of the gastric mucosal against injury: role of prostaglandins, nitric oxide and calcitonin-gene related peptide in capsaicin afferents. Curr Med Chem 2012; 19: 35-42.
- Takeuchi K. Pathogenesis of NSAID-induced gastric damage: importance of cyclooxygenase inhibition and gastric hypermotility. World J Gastroenterol 2012; 18: 2147-2160.
- Brzozowski T, Konturek PC, Konturek SJ, et al. Ischemic preconditioning of remote organs attenuates gastric ischemia-reperfusion injury through involvement of prostaglandins and sensory nerves. Eur J Pharmacol 2004; 499: 201-213.
- Brzozowski T, Konturek PC, Moran AP, et al. Involvement of capsaicin-sensitive afferent nerves and cholecystokinin 2/gastrin receptors in gastroprotection and adaptation of gastric mucosa to Helicobacter pylori-lipopolysaccharide. J Pharmacol Exp Ther 2004; 310: 116-125.
- Brzozowski T. Nonsteroidal anti-inflammatory drug-induced experimental gastropathy: is gastric acid the major trigger? Clin Exp Pharm Physiol 2010; 37: 651-653.
- Lai LH, Chan FK. Nonsteroid anti-inflammatory drug-induced gastroduodenal injury. Curr Opin Gastroenterol 2009; 25: 544-548.
- Park SH, Hong H, Han YM, et al. Non-steroidal anti-inflammatory drugs (NSAID) sparing effects of glucosamine hydrochloride through N-glycosylation inhibition; strategy to rescue stomach from NSAID damage. J Physiol Pharmacol 2013; 64: 157-165.
- Konturek PC, Brzozowski T, Ptak A, et al. Nitric oxide releasing aspirin protects the gastric mucosa against stress and promotes healing of stress-induced gastric mucosal damage: role of heat shock protein 70. Digestion 2002; 66: 160-172.
- Nemmani KV, Mali SV, Borhade N, et al. NO-NSAIDs: gastric-sparing nitric oxide-releasable prodrugs of non-steroidal anti-inflammatory drugs. Bioorg Med Chem Lett 2009; 19: 5297-5301.
- Kwiecien S, Pawlik MW, Brzozowska I, et al. Effect of vasodilators on lipid peroxidation and antioxidative mechanisms in gastric mucosa. Clin Exp Med Lett 2005; 46: 33-38.
- Yucel AF, Kanter M, Pergel A, Erboga M, Guzel A. The role of curcumin on intestinal oxidative stress, cell proliferation and apoptosis after ischemia/reperfusion injury in rats. J Mol Histol 2011; 42: 579-587.
- Bhattacharyya A, Chattopadhyay R, Mitra S, Crowe SE. Oxidative stress: an essential factor in pathogenesis of gastrointestinal mucosal diseases. Physiol Rev 2014; 94: 329-354.
- Bandyopadhyay D, Chattopadhyay A. Reactive oxygen species-induced gastric ulceration: protection by melatonin. Curr Med Chem 2006; 13: 1187-1202.
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. Am J Physiol 1998; 275: C1-C24.
- Kwiecien S, Pawlik MW, Brzozowski T, Pawlik WW, Konturek SJ. Reactive oxygen metabolite action in experimental, stress model of gastric mucosa damage. Gastroenterol Pol 2010; 17: 234-243.
- Naito Y, Yoshikawa T. Oxidative stress involvement and gene expression in indomethacin-induced gastropathy. Redox Rep 2006; 11: 243-253.
- Cacanyiova S, Dovinova F, Kristek F. The role of oxidative stress in acetylocholine-induced relaxation of endothelium-denuded arteries. J Physiol Pharmacol 2013; 64: 241-247.
- Vucevic D, Mladenovic D, Ninkovic M, et al. Influence of aging on ethanol-induced oxidative stress in digestive tract of rats. Hum Exp Toxicol 2013; 32: 698-705.
- Targosz A, Brzozowski T, Pierzchalski P, et al. Helicobacter pylori promotes apoptosis, activates cyclooxygenase (COX)-2 and inhibits heat shock protein HSP70 in gastric cancer epithelial cells. Inflamm Res 2012; 61: 955-966.
- Misra MK, Sarwat M, Bhakuni P, Tuteja R, Tuteja N. Oxidative stress and ischemic myocardial syndromes. Med Sci Monit 2009; 15: RA209-RA219.
- Nagano Y, Matsui H, Shimokawa O, et al. Biphosphonate-induced gastrointestinal mucosal injury is mediated by mitochondrial superoxide production and lipid peroxidation. J Clin Biochem Nutr 2012; 51: 196-203.
- Kasazaki K., Yasukawa K, Sano H, Utsumi H. Non-invasive analysis of reactive oxygen species generated in NH4OH-induced gastric lesions of rats using a 300 MHz in vivo ESR technique. Free Radic Res 2003; 37: 757-766.
- Zhang JY, Wu QF, Wan Y, et al. Protective role of hydrogen-rich water on aspirin-induced gastric mucosal damage in rats. World J Gastroenterol 2014; 20: 1614-1622.
- Yasukawa K, Kasazaki K, Hyodo F, Utsumi H. Non-invasive analysis of reactive oxygen species generated in rats with water immersion restraint-induced gastric lesions using in vivo electron spin resonance spectroscopy. Free Radic Res 2004; 38: 147-155.
- Konturek PC, Duda A, Brzozowski T, et al. Activation of genes for superoxide dismutase, interleukin-1, tumor necrosis factor-a and intercellular adhesion molecule-1 during healing of ischemia-reperfusion gastric injury. Scand J Gastroenterol 2000; 35, 452-463.
- Nagano Y, Matsui H, Shimokawa O, et al. Rebamipide attenuates nonsteroidal anti-inflammatory drugs (NSAID) induced lipid peroxidation by manganese superoxide dismutase (MnSOD) overexpression in gastrointestinal epithelial cells. J Physiol Pharmacol 2012; 63: 137-142.
- Mei X, Xu D, Xu S, Zheng Y, Xu S. Novel role of Zn(II)-curcumin in enhancing cell proliferation and adjusting proinflammatory cytokine-mediated oxidative damage of ethanol-induced acute gastric ulcers. Chem Biol Interact 2012; 197: 31-39.
- Lorrain B, Dangles O, Loonis M, Armand M, Dufour C. Dietary iron - initiated lipid oxidation and its inhibition by polyphenols in gastric conditions. J Agric Food Chem 2012; 60: 9074-9081.
- Kwiecien S, Brzozowski T, Konturek SJ. Importance of aldehyde products of lipid peroxidation in the formation of gastric lesions induced by aspirin, ischemia-reperfusion and stress. Gastroenterol Pol 2002; 9: 273-280.
- Matsuda T, Tao H, Goto M, et al. Lipid peroxidation-induced DNA adducts in human gastric mucosa. Carcinogenesis 2013; 34: 121-127.
- Getzoff ED, Tainer JA, Weiner PK, Kollman PA, Richardson JS, Richardson DC. Electrostatic recognition between superoxide and copper, zinc superoxide dismutase. Nature 1983; 306: 287-290.
- Farkas R, Selmeci L, Tulassay Z, Pronai L. Superoxide dismutase activity of the gastric mucosa in patients with Helicobacter pylori infection. Anticancer Res 2003; 23: 4309-4312.
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991; 11: 81-128.
- Dianzani MU. 4-hydroxynonenal from pathology to physiology. Mol Aspects Med 2003; 24: 263-272.
- Awasthi YC, Sharma R, Cheng JZ, et al. Role of 4-hydroxynonenal in stress-mediated apoptosis signaling. Mol Aspects Med 2003; 24: 219-230.
- Adamczyk-Sowa M, Sowa P, Pierzchala K, Polaniak R, Labuz-Roszak B. Antioxidative enzymes activity and malondialdehyde concentration during mitoxantrone therapy in multiple sclerosis patients. J Physiol Pharmacol 2012; 63: 683-690.
- Tamura M, Matsui YN, Nagano T, et al. Salt is an oxidative stressor for gastric epithelial cells. J Physiol Pharmacol 2013; 64: 89-94.
- Min SY, Kim HS, Jung EJ, Jung EJ, Jee CD, Kim WH. Prognostic significance of glutathione peroxidase 1 (GPX1) down-regulation and correlation with aberrant promoter methylation in human gastric cancer. Anticancer Res 2012; 32: 3169-3175.
- Korbut E, Ptak-Belowska A, Brzozowski T. Mechanisms promoting physiological cells progression into tumorigenesis. J Physiol Pharmacol 2012; 63: 565-570.
- Amstad P, Moret R, Ceruti P. Glutathione peroxidase compensates for the hypersensivity of Cu,Zn-superoxide dismutase overproducers to oxidant stress. J Biol Chem 1994; 269: 1606-1609.
- Ertug PU, Aydinoglu F, Goruroglu Ozturk O, Singirik E, Ogulener N. Comparative study of the quercetin, ascorbic acid, glutathione and superoxide dismutase for nitric oxide protecting effects in mouse gastric fundus. Eur J Pharmacol 2013; 698: 379-387.
- Liu L, Cui J, Song CJ, et al. H2S-releasing aspirin protects against aspirin-induced gastric injury via reducing oxidative stress. PLoS One 2012; 7: e46301.
- Winterbourn CC. Superoxide as an intracellular radical sink. Free Radic Biol Med 1993; 14: 85-90.
- Koppenol WH. A thermodynamic appraisal of the radical sink hypothesis. Free Radic Biol Med 1993; 14: 91-94.
- Brzozowski T, Konturek PC, Drozdowicz D, et al. Grapefruit-seed extract attenuates ethanol- and stress-induced gastric lesions via activation of prostaglandin, nitric oxide and sensory nerve pathways. World J Gastroenterol 2005; 11: 6450-6458.
- Brzozowski T, Konturek PC, Sliwowski Z, et al. Prostaglandin/cyclooxygenase pathway in ghrelin-induced gastroprotection against ischemia-reperfusion injury. J Pharmacol Exp Ther 2006; 319: 477-487.
- Pajdo R, Brzozowski T, Konturek PC, et al. Ischemic preconditioning, the most effective gastroprotective intervention: involvement of prostaglandins, nitric oxide, adenosine and sensory nerves. Eur J Pharmacol 2001; 427: 263-276.
- Konturek PC, Brzozowski T, Pierzchalski P, et al. Activation of genes for spasmolytic peptide, transforming growth factor alpha and for cyclooxygenase (COX)-1 and COX-2 during gastric adaptation to aspirin damage in rats. Aliment Pharmacol Ther 1998; 12: 767-777.
- Brzozowski T, Konturek PC, Konturek SJ, et al. Effect of local application of growth factors on gastric ulcer healing and mucosal expression of cyclooxygenase-1 and -2. Digestion 2001; 64: 15-29.
- Brzozowski T, Konturek PC, Sliwowski Z, et al. Neural aspects of ghrelin-induced gastroprotection against mucosal injury induced by noxious agents. J Physiol Pharmacol 2006; 57 (Suppl. 6): 63-76.
- Warzecha Z, Ceranowicz P, Dembinski M, et al. Involvement of cyclooxygenase-1 and cyclooxygenase-2 activity in the therapeutic effect of ghrelin in the course of ethanol - induced gastric ulcers in rats. J Physiol Pharmacol 2014; 65: 95-106.
- Xu L, Li Z, Guo F. Curcumin improves expression of ghrelin through attenuating oxidative stress in gastric tissues of streptozocin - induced diabetic gastroparesis rats. Eur J Pharmacol 2013; 718: 219-225.
- Handa O, Naito Y, Yoshikawa T. Helicobacter pylori: a ROS-inducing bacterial species in the stomach. Inflamm Res 2010; 59: 997-1003.
- Brzozowski T, Zwolinska-Wcislo M, Konturek PC, et al. Influence of gastric colonization with Candida albicans on ulcer healing in rats: effect of ranitidine, aspirin and probiotic therapy. Scand J Gastroenterol 2005; 40: 286-296.
- Niv Y, Banic M. Gastric barrier function and toxic damage. Dig Dis 2014; 32: 235-242.
- Solmaz A, Sener G, Cetinel S, Yuksel M, Yegen C, Yegen BC. Protective and therapeutic effects of resveratrol on acetic acid-induced gastric ulcer. Free Radic Res 2009; 43: 594-603.
- Kwiecien S, Pawlik MW, Sliwowski Z, et al. Involvement of sensory afferent fibers and lipid peroxidation in the pathogenesis of stress-induced gastric mucosa damage. J Physiol Pharmacol 2007; 58 (Suppl. 3): 149-162.
- Naito Y, Takagi T, Handa O, Yoshikawa T. Lipid hydroperoxide-derived modification of proteins in gastrointestinal tract. Subcell Biochem 2014; 77: 137-148.
A c c e p t e d : October 1, 2014