DYNAMICS OF SODIUM RETENTION IN PREASCITIC CIRRHOTIC RATS ASSESSED THROUGH PARATHYROID HORMONE INJECTION
INTRODUCTION
Liver cirrhosis is complicated by renal sodium retention, which leads, eventually, to the ascitic stage of clinical decompensation. Before ascites development, sodium retention already occurs despite normal or even increased glomerular filtration rate (GFR) (1-6). The tubular segment where increased sodium reabsorption occurs in pre-ascitic cirrhosis is still debated.
Animals with preascitic cirrhosis and sodium retention have normal fluid delivery from the proximal convoluted tubule to the Henle’s loop (7-10). Lithium clearance, which is an established index of distal tubular delivery of fluid, is significantly reduced in standing preascitic cirrhotic patients, but is normal in supine ones (11-13). Yet patients with compensated cirrhosis while reclining show sodium retention in some tubular segment of the nephron (6, 12, 14).
Plasma levels of renin and aldosterone are either normal or slightly decreased in early cirrhosis (6, 15-17). This might mean that increased sodium reabsorption in aldosterone-sensitive nephron segments is unlikely, but mineralocorticoid-receptor antagonists delay ascites development (18), which led to the hypothesis that aldosterone sensitivity might be in some way increased in preascitic cirrhosis (19).
At least one alternative hypothesis to explain preascitic sodium retention may be suggested. In preascitic cirrhotic rats, Jonassen and colleagues described a selective hypertrophy of the thick ascending limb (TAL) of Henle’s loop in the outer medulla (8, 9). Those rats showed sodium retention at baseline, increased natriuretic responses to furosemide, and increased medullary interstitial sodium concentration (20, 21). Furthermore, rats with CCl4-induced liver cirrhosis have increased content of Na+-K+-2Cl– cotransporters in the TAL of Henle’s loop (22). Summarizing, so far increased preascitic sodium retention in the Henle’s loop has been repeatedly suspected but never demonstrated.
Parathyroid hormone (PTH) exerts physiological effects on the regulation of tubular electrolyte and water transport in the kidney. PTH infusion increases diuresis and natriuresis (23, 24), and primary hyperparathyroidism is associated with polyuria, polydipsia, and reduced urinary concentrating capacity (25, 26). PTH and hypercalcemia reduce the contents of Na+-K+-2Cl– cotransporters (BSC-1) in the ascending limb of Henle’s loop and of aquaporin-2 (AQP2) water channels in the collecting duct (24, 27).
Intravenous calcium loading leads to larger natriuresis and aquaresis in compensated cirrhotic patients than in healthy controls (14). This means that at least part of the sodium retention in pre-ascitic cirrhosis occurs in Ca++-sensitive segments of the nephron (i.e. in the Henle’s loop). The diuretic effect shown in calcium-loaded preascitic cirrhotic patients was attributed to stimulation of extracellular calcium/polyvalent cation-sensing receptors (CaRs) (14). This is reasonable because, in rats with experimental preascitic liver cirrhosis and sodium retention, stimulation of CaRs with calcimimetic agents returns sodium and free-water excretion to normality (28). In the kidney, CaRs are located mainly in the TAL of Henle’s loop and in the collecting duct (29). In the TAL of Henle’s loop, CaRs stimulation inhibits sodium-potassium-chloride co-transport and leads to increased natriuresis (24, 30-34).
PTH, calcium, and CaRs compose an actual Ca++-dependent diuretic system, the activity of which might be deranged in preascitic cirrhosis with sodium retention. We hypothesize that, in pre-ascitic cirrhosis, the Henle’s loop might be a key contributor to sodium retention. To dissect the actual contribution of Henle’s loop to sodium retention in rats with experimental preascitic cirrhosis, we have evaluated the renal biomolecular and functional effects that subcutaneous administration of PTH may cause through modulation of the content of CaRs and Na+-K+-2Cl– cotransporters (BSC-1) in renal tubules.
MATERIAL AND METHODS
Experiments in rats were performed in compliance with the procedures outlined in the Italian Ministry of Health guidelines (no. 86/609/EEC) and according to the Principles of Laboratory Animal Care (NIH no. 85-23, revised in 1985). The University of Torino ethics committee specifically approved this study.
Studies were performed on male adult Wistar rats with preascitic cirrhosis and related control animals (Harlan Italy, Udine, Italy). Both groups were fed ad libitum with standard chow and water. Preascitic cirrhosis was induced by carbon tetrachloride (CCl4) (Riedel de Haen, Sigma-Aldrich, Seelze, Germany) chronically administered by gavage twice weekly as described elsewhere (28, 35). Cirrhotic rats were studied 9 weeks after starting CCl4 administration, when cirrhosis was clearly developed (35) (Fig. 1). Control rats were studied following a similar period of standardized diet.
Groups of rats
Human PTH1-34 (Bachem Distribution Services GmbH, Weil am Rhein, Germany) was dissolved in 5% glucose solution (vehicle) and administered subcutaneously to ten cirrhotic rats (group G3) in five timely spaced doses (one dose every 12 hours for 2 days before the study day) of 0.5 ml vehicle containing 10 mcg/Kg PTH. Eight control rats (group G1) and ten cirrhotic rats (group G2) were injected subcutaneously with the same volumes of vehicle alone.
Study protocol
On the study day, the rats were anaesthetized as described elsewhere (36). Blood was sampled (time 0) by cardiac puncture (0.3 cc) and inulin (IN) 10% (w/v) (Laevosan-Gesellschaft, Linz/Donau, Austria) plus para-aminohippurate (PAH) 20% (w/v) (Nephrotest, BAG Gmbh, Munich, Germany) were administered into the caudal vein as a priming bolus (0.14 ml/kg and 0.03 ml/kg, respectively) followed by a continuous infusion of 0.09 ml/kg/h inulin and 0.025 ml/kg/h PAH for 150 min, in order to assess glomerular filtration rate (GFR) and renal plasma flow (RPF) at different times by means of their respective steady-state plasma clearances (CIN and CPAH) (28, 37, 38). The steady state technique implies these advantages over the urine collection clearance method: catheterization of the bladder and exact timing of serum and urine samples are unnecessary, less analytical work is needed, and more urine is available for further testing (38).
After 90 min of IN and PAH infusions (i.e. when the steady-state plasma levels of IN and PAH were reached) (28, 37), laparotomy was performed, the urinary bladder was emptied, and a clamp was positioned on the urethral orifice. Cardiac blood was then sampled (time 1) to assess basal values of CIN and CPAH, and then vehicle alone was injected as a single bolus (1 ml) into the right femoral vein of all rats (groups G1-G3). Cardiac blood was then sampled (0.3 cc) at precise intervals (20 min) for 1 hour (times 2, 3 and 4) to measure plasma osmolality and concentrations of inulin, PAH, sodium, potassium, and calcium. At each blood withdrawal, the volume of blood withdrawn was replaced with an equal volume of i.v. saline (28). Blood samples withdrawn at time 4 (i.e. 60 min after the injection of vehicle) were also used to measure plasma concentrations of PTH, vasopressin (AVP), plasma renin activity (PRA), aldosterone (A), norepinephrine (N), aspartate aminotransferase (AST), alanine aminotransferase (ALT), albumin and total bilirubin. At time 4, after collecting from the bladder the urine produced during the 60 min after vehicle administration, rats were killed by exsanguination through the aorta. This urine was then used to measure its osmolality and the excretion of sodium, potassium, and PGE2.
CaRs and BSC-1 protein concentrations in rat kidneys
Membrane fractions were prepared from kidneys removed from five rats in each experimental group (G1-G3) and the total protein content was assessed using Bradford Assay as described previously (28). Equal amounts of proteins (30 µg) were separated by SDS-PAGE and electrotransferred on nitrocellulose membrane (GE-Healthcare Europe, Milano, Italy). Membranes were probed with rabbit anti-CaRs (PA1-934, ABR Affinity BioReagents, Golden, CO, USA) and rabbit-anti BSC-1 (NKCC21-A, Alpha Diagnostic International, San Antonio, TX, USA) primary antibodies, followed by incubation with anti-rabbit IgG/HRP-conjugated secondary antibody (BioRad, Hercules, CA, USA).
Proteins were detected with Clarity Western ECL substrate (BioRad) and quantified by densitometry using analytic software (Quantity-One, Bio-Rad). Results were normalized with respect to β-actin densitometric value.
Kidney CaRs and BSC-1 indirect immunofluorescence
Indirect immunofluorescence was performed in a humidified chamber at room temperature, essentially as described elsewhere (28, 39), on kidney cryostat sections (6 µm thick). Mouse monoclonal anti-CD31, an endothelial marker (BD-Pharmingen, Erembodegem, Belgium), and rabbit polyclonal anti-CaRs and anti-BSC-1 primary antibodies were used (28, 40).
Plasma and urine analyses
Plasma and urinary concentrations of electrolytes, and IN and PAH concentrations in plasma were measured as described elsewhere (41, 42). AVP and PTH systemic concentrations were measured on EDTA plasma by RIA (Buhlmann Laboratories AG, Postfach, Switzerland). PRA was measured using RIA for angiotensin I (Renin Maia Kit, Biodata, Rome, Italy). Aldosterone was evaluated by RIA (Coat-A-Count Aldosterone kit, Diagnostic Products Corporation, Los Angeles, CA, USA), and norepinephrine was measured as described elsewhere (43, 44). Urine samples were assayed for PGE2 concentrations by ELISA (Neogen, Lexington, KY, USA). Plasma AST, ALT, albumin and total bilirubin levels were determined by means of automated Roche/Hitachi Cobas equipment.
Calculations
Sodium clearance (CNa) and potassium clearance (CK) were calculated through the formula:
Cx = (U-x x V)/P-x
where U-x is the urinary concentration of x, P-x is the plasma concentration of x, and V is the urinary output (ml/min). Inulin clearance (CIN) and para-aminohippurate clearance (CPAH) were calculated through the steady-state plasma clearance formula as:
Cx = Infusion rate (x)/ssP-x
where ssP-x is the steady-state plasma concentration of x. CIN and CPAH were taken as measures of GFR and RPF (37, 38). Filtered sodium load (FlNa) was derived, following Boer et al. (45), as:
FlNa = Sodium plasma concentration (P-Na) x CIN
Fractional sodium excretion (FENa) and fractional potassium excretion (FEK) were calculated, respectively, from the ratios of CNa and CK to CIN x 100.
Free-water clearance (F-WCl) was calculated, following Rose and Post (46), through the formula:
F-WCl = V – Cosm
where V is the urinary output (ml/min); Cosm is the osmolar clearance, which was computed via the usual formula:
Cosm = (Uosm x V)/Posm
where Uosm and Posm are urine and plasma osmolalities, respectively.
Finally, in order to evaluate aldosterone function on the distal tubular nephron, the trans-tubular concentration ratio of potassium in the cortical collecting duct (TTKR) was calculated according to the following formula (47, 48):
TTKR = [K+]urine /(U/P)osm
[K+]serum
This parameter provides a semiquantitative reflection of the ratio of [K+] in the tubule fluid to that in the plasma of the adjacent vessels in the cortical collecting duct, an aldosterone-dependent segment of the tubular nephron.
All renal function parameters were derived by computing the mean of three determinations of osmolality, inulin, PAH, sodium, potassium, and calcium in plasma during the 60 min urine collection period (blood sampling times 2, 3, and 4).
Morphological liver studies
Livers were removed from all rats receiving CCl4, and hepatic tissue samples for light microscopy were placed in buffered 4% formaldehyde solution (pH 7.4). The sections were stained with hematoxylin and eosin to assess fibrosis. Silver-impregnated liver sections were used to observe portal-central or central-central bridging fibrosis.
Statistical analysis
Results are expressed as means ± S.D. All comparisons among groups of rats were made by one-way ANOVA test, followed by the Bonferroni test. The analyses were processed by the SPSS® 13.0 software (Chicago, USA), using a significance level of 5%.
RESULTS
Morphological liver studies
Micronodular cirrhosis with hepatocellular necrosis and steatosis was found in all the livers removed from CCl4-treated rats (Fig. 1).

Content and immunostaining of renal CaRs and BSC-1
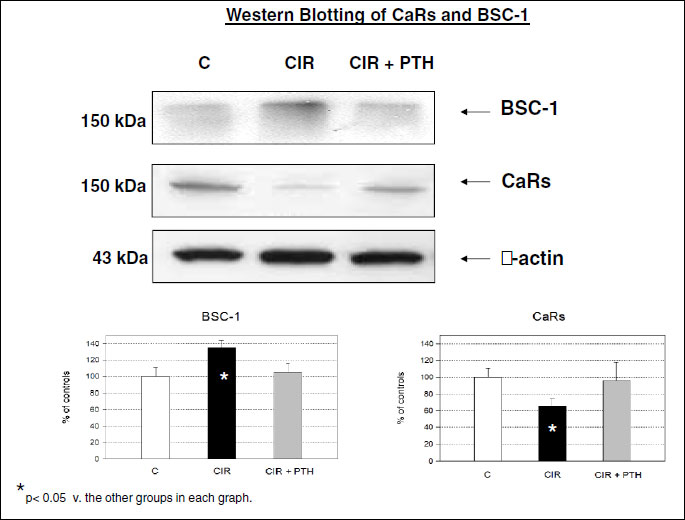
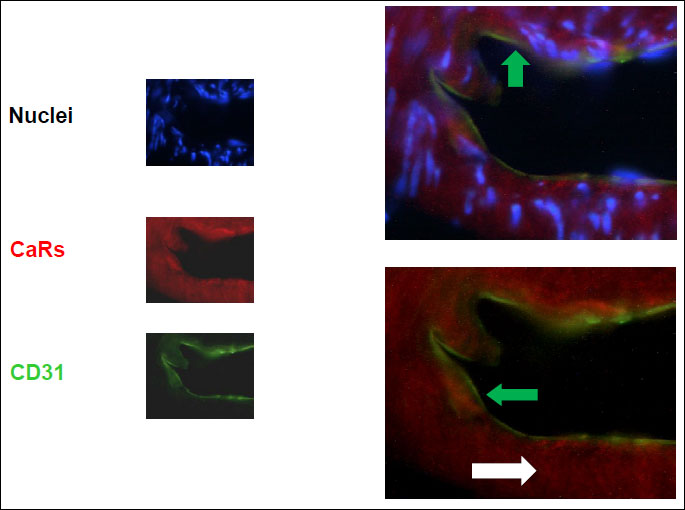
A comparative analysis of the membrane fraction of renal tissue homogenate showed that CaRs protein content was significantly reduced in cirrhotic vs. control animals, and protein content of BSC-1 was significantly increased in cirrhotic rats (Fig. 2). Protein content of CaRs and BSC-1 returned to control values in cirrhotic rats pre-treated with s.c. PTH (Fig. 2). CaRs- and BSC-1-positive tubular cells were identified in the TAL of Henle’s loop of normal and cirrhotic rat kidneys, as shown previously (28). CaRs were located in the sub-endothelial layers of kidney medullary arterioles of cirrhotic rats (Fig. 3). In control and cirrhotic rats, CaRs were also located in medullary collecting ducts, as previously shown (28).
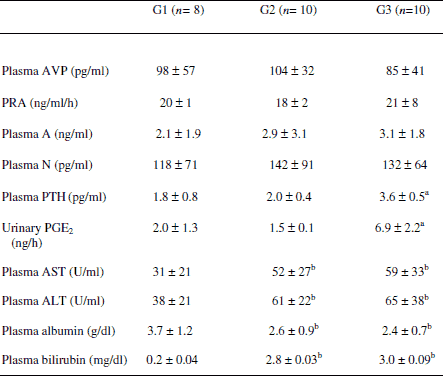
Hormonal status (Table 1)
Treatment with s.c. PTH had no effects on PRA, A, AVP, and N plasma levels in cirrhotic animals. PTH-treated cirrhotic rats (G3) had significantly higher plasma PTH concentrations and higher urinary excretion rate of PGE2 when compared to the untreated cirrhotic group (G2). Increased PGE2 urinary excretion is expected when hypercalcemia stimulates CaRs.
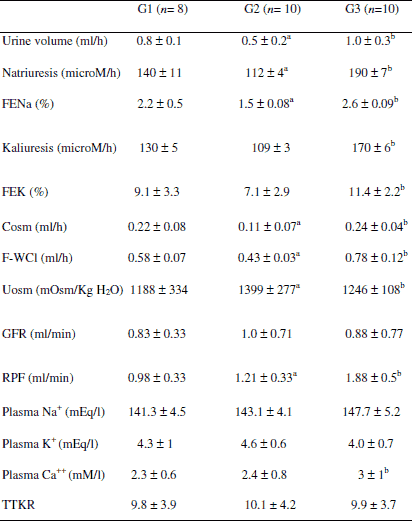
Renal function (Table 2)
Compared to control animals (G1), cirrhotic rats belonging to G2 showed reduced urine volume and absolute and fractional excretions of sodium. Free-water clearance was lower in cirrhotic animals than in control rats, and RPF was higher in cirrhotic rats than in controls, at baseline. In cirrhotic rats, s.c. PTH significantly improved urine volume, absolute and fractional excretion of sodium and potassium, which means that the TAL of Henle’s loop is affected, free-water clearance, which means the collecting duct is affected, and renal plasma flow. The values of TTKR were equal in control and cirrhotic rats. PTH did not affect GFR in cirrhotic rats, and, as expected, PTH significantly increased plasma Ca++ levels in cirrhotic rats.
DISCUSSION
In experimental preascitic cirrhosis, retention of sodium and fluid is associated with increased contents of Na+-K+-2Cl– cotransporters in the TAL of Henle’s loop. Sodium retention is also associated with decreased contents of CaRs (the targets of extracellular Ca++ and calcimimetic drugs) in Henle’s loop and collecting ducts (Fig. 2). PTH administration restores the physiological protein content of CaRs and BSC-1, and this increases urinary sodium and water excretion of cirrhotic rats up to the values measured in controls (Table 2). It is reasonable to say that perhaps little sodium retention would occur in this early stage of experimental preascitic cirrhosis if tubular content of CaRs and, mostly, BSC-1 were normal.
In this study, aldosterone function was assessed through the computation of the trans-tubular concentration ratio of potassium in the cortical collecting duct (TTKR) (47, 48). TTKR, which reflects the ratio between K+ concentrations in the tubule fluid and in the plasma of the adjacent vessels, was equal in control and cirrhotic rats. This indicates that mineralocorticoid-mediated hyperstimulation of tubular sodium re-absoprtion in exchange with potassium secretion has not occurred yet at this early stage of compensated cirrhosis. In other words, increased sodium re-absorption in the Henle’s loop precedes aldosterone-dependent sodium retention.
Subtle interactions between PTH systemic concentrations and renin-angiotensin-aldosterone system (RAAS) activity have been repeatedly described. For instance, infusion of angiotensin II or aldosterone causes significant increase in the rate of PTH secretion in human health and disease (49, 50); more importantly, in healthy human subjects short-term PTH infusion stimulates renin release and aldosterone secretion (51, 52), but also increases the rate of renal hydroxilation of vitamin D, which, in turn, may negatively regulate renin expression and RAAS activity (50, 53). On the whole, in this study PTH administration to cirrhotic rats caused a slight, statistically insignificant, increase of both PRA and plasma aldosterone, along with the natriuretic effects due to BSC-1 down-regulation. Once again, this underlines the modest, if any, role of aldosterone in the control of sodium handling in this stage of cirrhotic disease.
PTH exclusive capacity to down-regulate BSC-1 and to up-regulate CaRs was extremely useful to ascertain the dynamic connections between the expression of the above proteins and the patterns of tubular sodium handling. No other hormone can modulate expression of CaRs and sodium channels in the tubular nephron so promptly. Moreover, the choice of experimental animal models is the only available option to study the dynamics of expression of tubular sodium channels since it would be unethical and dangerous to submit human cirrhotic patients to repeated administrations of PTH.
This dynamic approach led to the conclusion that excessive sodium retention takes place in the Henle’s loop of preascitic cirrhotic rats. Previous literature had already suggested the existence of such a mechanism of sodium retention in cirrhotic rats (8, 21, 22, 28) based on the following findings: i) the TAL of Henle’s loop is hypertrophic; ii) the natriuretic response to furosemide and the medullary interstitial sodium concentration are increased. Moreover, in patients with compensated cirrhosis, the natriuretic responses to intravenous calcium loading are larger than in controls (14) and aldosterone-independent sodium retention takes place after the proximal convoluted tubule (6, 54).
There are analogies between the effects of PTH and intravenous calcium loading (14), and this indicates that the results presented in this study depend on the systemic hypercalcemia induced by PTH. This also means that the stimulation of CaRs in Henle’s loop and collecting duct, as recently shown with the use of calcimimetic agents (28), represents the actual trigger leading to the normalization of sodium and fluid excretion in experimental preascitic cirrhosis.
Following the studies of Wang and associates (24), we chose to inject a moderate dose of PTH to avoid early death of cirrhotic rats due to excessive hypercalcemia. Yet this PTH dose increased the urinary excretion of PGE2 (Table 1), the second messenger generated after specific stimulation of CaRs (32), and caused natriuresis and aquaresis (Table 2).
PTH injection in cirrhotic rats also caused a considerable renal vasodilatation (Table 2). This was due to the large amount of CaRs located in the sub-endothelial layers of renal arterioles of the outer medulla (Fig. 3). This renal vasodilatation might be useful in the advanced stages of cirrhosis, when renal vasoconstriction leads to reduced GFR (55, 56).
Our results may also suggest a method of investigation of two apparently unrelated processes: the hypercalciuria of mineralocorticoid escape (57) and the tubular pattern of sodium handling in chronic renal failure (CRF).
Mineralocorticoid escape indicates the occurrence of reduced reabsorption of sodium in aldosterone-independent tubules when the kidney faces chronically elevated levels of plasma aldosterone. The hypercalciuria that coincides with aldosterone escape might trigger decreased tubular sodium re-absorption in the Henle’s loop and compensatory natriuresis. Further on, increased expression of CaRs in peripheral blood monocytes of patients with diabetes mellitus and peripheral artery disease (58) might mean that this calcium-dependent homeostatic mechanism is already somehow operative in that clinical setting.
The already established association of CRF and systemic down-regulation of CaRs (59, 60), suggests the explanation of a common clinical observation: CRF is characterized by low GFR and increased fractional excretion of sodium, while functional renal failure (FRF) in liver cirrhosis is characterized by low GFR and avid tubular sodium retention with low fractional sodium excretion. The secondary hyperparathyroidism occurring in CRF (61), but not in cirrhotic FRF, might contribute to the completely different tubular handling of sodium that is found in these two diseases.
In conclusion, in sodium-retaining preascitic cirrhotic rats we confirmed increased content of Na+-K+-2Cl– cotransporters in the TAL of Henle’s loop and down-regulation of renal CaRs, the receptors that mediate Ca++-dependent diuresis. The injection of PTH restored the physiological content of these tubular proteins and returned renal sodium handling to normal. In this context, the action of parathyroid hormone-related peptide (PTHrP) (62) or exogenous compounds that affects both PTH and renal tubular function (e.g. genistein) (63) would be worth some experimental focus.
The findings described in this study represent experimental evidence to support the hypothesis that sodium retention in preascitic cirrhosis occurs first in the TAL of Henle’s loop and later in aldosterone-dependent tubular segments. A third mechanism of sodium retention, increased reabsorption in the proximal convoluted tubule, is operative in preascitic cirrhotic patients while standing (11, 12). This paves the way to potential new strategies of prevention of ascites: orally active calcimimetic agents (28, 61) or oral calcium supplement (14), due to their diuretic, aquaretic and renal vasodilating properties, might represent further means to delay ascites development when administered chronically in patients with compensated cirrhosis.
Abbreviations: A: aldosterone; AQP2: aquaporin 2; AST: aspartate aminotransferase; ALT: alanine aminotransferase; AVP: vasopressin; BSC-1: Na+-K+-2Cl– cotransporter; CCl4: carbon tetrachloride; CaRs: extracellular calcium/polyvalent cation-sensing receptors; CIN: inulin clearance; CK: potassium clearance; CNa: sodium clearance; CPAH: para-aminohippurate clearance; CRF: chronic renal failure; FEK: fractional excretion of potassium; FENa: fractional excretion of sodium; FlNa: filtered sodium load; FRF: functional renal failure; F-WCl: free-water clearance; GFR: glomerular filtration rate; IN: inulin; N: norepinephrine; PAH: para-aminohippurate; PRA: plasma renin activity; PTH: parathyroid hormone; PTHrP: parathyroid hormone-related peptide; RAAS: renin-angiotensin-aldosterone-system; RPF: renal plasma flow; SDS: sodium dodecyl sulfate; TAL: thick ascending limb of Henle’s loop; TTKR: trans-tubular concentration ratio of potassium in the cortical collecting duct.
Acknowledgements: Supported in part by grants from the Ministry of University and of Scientific Research (60%), 2008. No additional external funding was received for this study. The authors have no competing interest to disclose. This study was presented orally at the 2010 annual meeting of the European Association for the Study of the Liver (EASL) in Vienna, Austria.
Authors’ contributions: G.S. and M.P. contributed to study concept and design, study supervision as well as analysis and interpretation of data and drafting of the manuscript. M.A. and R.M. contributed to acquisition of data as well as to analysis and interpretation of data (including statistical analysis). All authors were involved in writing and in critical revision of the final manuscript.
Conflict of interests: None declared.
REFERENCES
- Wong F, Massie D, Hsu P, Dudley F. Renal response to a saline load in well-compensated alcoholic cirrhosis. Hepatology 1994; 20: 873-881.
- Wood LJ, Massie D, McLean AJ, Dudley FJ. Renal sodium retention in cirrhosis: tubular site and relation to hepatic disfunction. Hepatology 1988; 8: 831-836.
- Bernardi M, Di Marco C, Trevisani F, et al. Renal sodium retention during upright posture in preascitic cirrhosis. Gastroenterology 1993; 105: 188-193.
- Wong F, Liu P, Tobe S, Morali G, Blendis L. Central blood volume in cirrhosis: measurement with radionuclide angiography. Hepatology 1994; 19: 312-321.
- Lewis FW, Adair O, Rector WG. Arterial vasodilatation is not the cause of increased cardiac output in cirrhosis. Gastroenterology 1992; 102: 1024-1029.
- Sansoe G, Ferrari A, Baraldi E, Castellana CN, De Santis MC, Manenti F. Renal distal tubular handling of sodium in central fluid volume homoeostasis in preascitic cirrhosis. Gut 1999; 45: 750-755.
- Levy M. Sodium retention and ascites formation in dogs with experimental portal hypertension. Am J Physiol 1977; 233: F572-F585.
- Jonassen TE, Marcussen N, Haugan K, et al. Functional and structural changes in the thick ascending limb of Henle’s loop in rats with liver cirrhosis. Am J Physiol 1997; 273: R568-R577.
- Jonassen TE, Christensen S, Sorensen AM, et al. Effects of chronic octreotide treatment on renal changes during compensated liver cirrhosis in rats. Hepatology 1999; 29: 1387-1395.
- Jonassen TE, Christensen S, Kwon TH, Langhoff S, Salling N, Nielsen S. Renal water handling in rats with decompensated liver cirrhosis. Am J Physiol 2000; 279: F1101-F1109.
- Wong F, Liu P, Blendis L. The mechanism of improved sodium homeostasis of low-dose losartan in preascitic cirrhosis. Hepatology 2002; 35: 1449-1458.
- Sansoe G, Biava AM, Silvano S, et al. Renal tubular events following passage from the supine to the standing position in patients with well compensated liver cirrhosis: loss of tubuloglomerular feedback. Gut 2002; 51: 736-741.
- Anastasio P, Frangiosa A, Papalia T, et al. Renal tubular function by lithium clearance in liver cirrhosis. Semin Nephrol 2001; 21: 323-326.
- Sansoe G, Wong F. Natriuretic and aquaretic effects of intravenously infused calcium in preascitic human cirrhosis: physiopathological and clinical implications. Gut 2007; 56: 1117-1123.
- Wilkinson SP, Smith JK, Clarke M, et al. Intrarenal distribution of plasma flow in cirrhosis as measured by transit renography: relationship with plasma renin activity and sodium and water retention. Clin Sci Mol Med 1977; 52: 469-475.
- Wilkinson SP, Smith JK, Williams R. Changes in plasma renin activity in cirrhosis: a reappraisal based on studies in 67 patients and “low renin” cirrhosis. Hypertension 1979; 1: 125-129.
- Trevisani F, Bernardi M, Gasbarrini A, et al. Bed-rest-induced hypernatriuresis in cirrhotic patients without ascites: does it contribute to maintain “compensation?” J Hepatol 1992 16; 190-196.
- Jimenez W, Martinez-Pardo A, Arroyo V, et al. Temporal relationship between hyperaldosteronism, sodium retention and ascites formation in rats with experimental cirrhosis. Hepatology 1985; 5: 245-250.
- Jonassen TE, Petersen JS, Sorensen AM, Andreasen F, Christensen S. Aldosterone receptor blockade inhibits increased furosemide-sensitive sodium reabsorption in rats with liver cirrhosis. J Pharmacol Exp Ther 1998; 287: 931-936.
- Jonassen TE, Nielsen S, Christensen S, Petersen JS. Decreased vasopressin-mediated renal water reabsorption in rats with compensated liver cirrhosis. Am J Physiol 1998; 275: F216-F225.
- Jonassen TE, Sorensen AM, Petersen JS, Andreasen F, Christensen S. Increased natriuretic efficiency of furosemide in rats with carbon tetrachloride-induced cirrhosis. Hepatology 2000; 31: 1224-1230.
- Fernandez-Llama P, Ageloff S, Fernandez-Varo G, et al. Sodium retention in cirrhotic rats is associated with increased renal abundance of sodium transporter proteins. Kidney Int 2005; 67: 622-630.
- Kimmel PL, Rivera A, Khatri P. Effects of nonrenal hormones on the normal kidney. In: Principles and Practice of Endocrinology and Metabolism. Becker KL (ed.) Philadelphia, Lippincott Williams & Wilkins, 2001, pp. 1885-1895.
- Wang W, Li C, Kwon TH, et al. Reduced expression of renal Na+ transporters in rats with PTH-induced hypercalcemia. Am J Physiol 2004; 286: F534-F545.
- Spiegel AM. Pathophysiology of primary hyperparathyroidism. J Bone Miner Res 1991; 6 (Suppl. 2): S15-S17.
- Slatopolsky E, Hruska KA. Disorder of phosphorus, calcium, and magnesium metabolism. In: Diseases of the Kidney and Urinary Tract. Schrier RW (ed.) Philadelphia, Lippincott Williams & Wilkins, 2001, pp. 2607-2660.
- Wang W, Li C, Kwon TH, Knepper MA, Frokiaer J, Nielsen S. AQP3, p-AQP2, and AQP2 expression is reduced in polyuric rats with hypercalcemia: prevention by cAMP-PDE inhibitors. Am J Physiol 2002; 283: F1313-F1325.
- Sansoe G, Aragno M, Tomasinelli E, di Bonzo LV, Wong F, Parola M. Calcium-dependent diuretic system in preascitic liver cirrhosis. J Hepatol 2010; 53: 856-862.
- Riccardi D, Park J, Lee WS, Gamba G, Brown EM, Hebert SC. Cloning and functional expression of a rat kidney extracellular calcium/polyvalent cation-sensing receptor. Proc Natl Acad Sci USA 1995; 92: 131-135.
- Wang D, Pedraza PL, Abdullah HI, McGiff JC, Ferreri NR. Calcium-sensing receptor-mediated TNF production in medullary thick ascending limb cells. Am J Physiol 2002; 283: F963-F970.
- Ray K, Northup J. Evidence for distinct cation and calcimimetic compound (NPS 568) recognition domains in the transmembrane regions of the human Ca++ receptor. J Biol Chem 2002; 277: 18908-18913.
- Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev 2001; 81: 239-297.
- Procino G, Carmosino M, Tamma G, et al. Extracellular calcium antagonizes forskolin-induced aquaporin 2 trafficking in collecting duct cells. Kidney Int 2004; 66: 2245-2255.
- Wang WH, Lu M, Hebert SC. Cytochrome P-450 metabolites mediate extracellular Ca++-induced inhibition of apical Na+-K+-Cl- channels in the thick ascending limb of Henle. Am J Physiol 1996; 271: C103-C111.
- Proctor E, Chatamra K. High yield micronodular cirrhosis in the rat. Gastroenterology 1982; 83: 1183-1190.
- Sansoe G, Aragno M, Smedile A, Rizzetto M, Rosina F. Solute-free water retention in preascitic cirrhotic rats following intravenous water loading. J Physiol Pharmacol 2009; 60: 111-117.
- Cole BR, Giangiacomo J, Ingelfinger JR, Robson AM. Measurement of renal function without urine collection. A critical evaluation of the constant-infusion technique for determination of inulin and para-aminohippurate. N Engl J Med 1972; 287: 1109-1114.
- Schnurr E, Lahme W, Kuppers H. Measurement of renal clearance of inulin and PAH in the steady state without urine collection. Clin Nephrol 1980; 13: 26-29.
- Parola M, Leonarduzzi G, Robino G, Albano E, Poli G, Dianzani MU. On the role of lipid peroxidation in the pathogenesis of liver damage induced by long-standing cholestasis. Free Radic Biol Med 1996; 20: 351-359.
- Zamara E, Novo E, Marra F, et al. 4-Hydroxynonenal as a selective pro-fibrogenic stimulus for activated human hepatic stellate cells. J Hepatol 2004; 40: 60-68.
- Roe JH, Epstein JH, Goldstein NP. A photometric method for the determination of inulin in plasma and urine. J Biol Chem 1949; 178: 839-845.
- Smith HW, Finkelstein N, Alminosa L. The renal clearance of substituted hippuric acid derivatives and other aromatic acids in dog and men. J Clin Invest 1945; 24: 388-403.
- Eriksson BM, Persson BA. Determination of catecholamines in rat heart tissue and plasma samples by liquid chromatography with electrochemical detection. J Chromatogr 1982; 228: 143-152.
- Weicker H, Feraudi M, Hagele H, Pluto R. Electrochemical detection of catecholamines in urine and plasma after separation with HPLC. Clin Chim Acta 1984; 141: 17-25.
- Boer WH, Koomans HA, Dorhout-Mees EJ. Lithium clearance during paradoxical natriuresis of hypotonic expansion in man. Kidney Int 1987; 32: 376-381.
- Rose BD, Post TW. Clinical Physiology of Acid-Base and Electrolyte Disorders. New York, McGraw-Hill, 2001, pp. 285-298.
- Ethier JH, Kamel KS, Magner PO, Lemann J Jr, Halperin ML. The transtubular potassium concentration in patients with hypokalemia and hyperkalemia. Am J Kidney Dis 1990; 15: 309-315.
- Kamel KS, Quaggin S, Scheich A, Halperin ML. Disorders of potassium homeostasis: an approach based on pathophysiology. Am J Kidney Dis 1994; 24: 597-613.
- Brown JM, Williams JS, Luther JM, et al. Human interventions to characterize novel relationships between the renin-angiotensin-aldosterone system and parathyroid hormone. Hypertension 2014; 63: 273-280.
- Brown JM, Vaidya A. Interactions between adrenal-regulatory and calcium-regulatory hormones in human health. Curr Opin Endocrinol Diabetes Obes 2014; 21: 193-201.
- Jespersen B, Randlov A, Abrahamsen J, Fogh-Andersen N, Kanstrup IL. Effects of PTH (1-34) on blood pressure, renal function, and hormones in essential hypertension: the altered pattern of reactivity may counteract raised blood pressure. Am J Hypertens 1997; 10: 1356-1367.
- Tomaschitz A, Ritz E, Pieske B, et al. Aldosterone and parathyroid hormone interactions as mediators of metabolic and cardiovascular disease. Metabolism 2014; 63: 20-31.
- Gutierrez OM, Smith KT, Barchi-Chung A, Patel NM, Isakova T, Wolf M. (1-34) Parathyroid hormone infusion acutely lowers fibroblast growth factor 23 concentrations in adult volunteers. Clin J Am Soc Nephrol 2012; 7: 139-145.
- Sansoe G, Silvano S, Rosina F, Smedile A, Rizzetto M. Evidence of a dynamic aldosterone-independent distal tubular control of renal sodium excretion in compensated liver cirrhosis. J Intern Med 2005; 257: 358-366.
- Schrier RW, Niederberger M, Weigert A, Gines P. Peripheral arterial vasodilatation: determinant of functional spectrum of cirrhosis. Semin Liver Dis 1994; 14: 14-22.
- Arroyo V, Guevara M, Gines P. Hepatorenal syndrome in cirrhosis: pathogenesis and treatment. Gastroenterology 2002; 122: 1658-1676.
- Gehr MK, Goldberg M. Hypercalciuria of mineralocorticoid escape: clearance and micropuncture studies in the rat. Am J Physiol 1986; 251: F879-F888.
- Malecki R, Fiodorenko-Dumas Z, Jakobsche-Policht U, Malodobra M, Adamiec R. Altered monocyte calcium-sensing receptor expression in patients with type 2 diabetes mellitus and atherosclerosis. J Physiol Pharmacol 2013; 64: 521-527.
- Kifor O, Moore FD, Wang P, et al. Reduced immunostaining for the extracellular Ca++-sensing receptor in primary and uremic secondary hyperparathyroidism. J Clin Endocrinol Metab 1996; 81: 1598-1606.
- Mathias RS, Nguyen HT, Zhang MYH, Portale AA. Reduced expression of the renal calcium-sensing receptor in rats with experimental chronic renal insufficiency. J Am Soc Nephrol 1998; 9: 2067-2074.
- Colloton M, Shatzen E, Miller G, et al. Cinacalcet HCl attenuates parathyroid hyperplasia in a rat model of secondary hyperparathyroidism. Kidney Int 2005; 67: 467-476.
- Meyer R, Schreckenberg R, Kraus D, Kretschmer F, Schulz R, Schluter KD. Cardiac effects of osteostatin in mice. J Physiol Pharmacol 2012; 63: 17-22.
- Pantelic J, Ajdzanovic V, Medigovic I, et al. Genistein affects parathyroid gland and NaPi 2a cotransporter in an animal model of the andropause. J Physiol Pharmacol 2013; 64: 361-368.
A c c e p t e d : September 12, 2014