THE ROLE OF LOCAL AND SYSTEMIC CYTOKINES IN PATIENTS
INFECTED WITH CLOSTRIDIUM DIFFICILE
INTRODUCTION
Clostridium difficile is a gram positive, anaerobic rod first isolated by Hall and O'Toole from the meconium of a healthy newborn in 1935 (1). The constellation of symptoms resulting from the presence of this bacteria and its toxins in the colon is called Clostridium difficile infection (CDI). In the last two decades CDI has become the most common nosocomial infection of the gastrointestinal tract and the most common cause of nosocomial diarrhea (10–30% of cases). The clinical picture of CDI varies from the asymptomatic carrier state to different severities of diarrhea, to the most severe, systemic life-threatening infection (2, 3). It is widely accepted that the pathogenesis of CDI is multifactorial, dependent on pathogen virulence factors produced by the organism as well as disorders of the intestinal flora and the immune response of the host (4). This bacterium virulence factors are toxins and enzymes produced by the microorganism including collagenase, hyaluronidase and chondroitin-sulfatase, which impair the intestinal epithelial cytoskeleton. The most important virulence factors are bacterial toxin A (TcdA) and toxin B (TcdB), which show 63% structural similarity. The two toxins have the ability to act both as enterotoxins and cytotoxins, although toxin A is traditionally referred to as an enterotoxin while toxin B is referred to as a cytotoxin. Both strains producing and those strains not producing toxins can colonize the gastrointestinal tract of humans and other mammals, but only toxinogenic strains cause disease. There is a third toxin called a binary toxin, produced by some strains, but its role in the pathogenesis of CDI is still unclear (2, 4).
The aim of our study was to assess, in vivo, the dependence of plasma levels of cytokines on inflammatory and biochemical markers which are considered predictive factors of poor prognosis in CDI and the clinical course of this infection.
MATERIAL AND METHODS
The study was conducted in accordance with the Declaration of Helsinki (1975) and approved by the local ethics committee.
Patients and procedures
We prospectively studied 80 patients. Our study group included 40 patients aged 30–87 years (mean age 66.9 years) with CDI hospitalized at Infectious Diseases Department and Gastroenterology and Hepatology Department, University Hospital in Cracow, and 40 healthy volunteers aged 24–62 years (mean age 51.1 years). Blood samples were obtained no later than 48 hours after initiating antibiotic therapy, when therapeutic effects were not observed and patients still manifested clinical symptoms. The diagnosis of CDI was based on history, epidemiological data, physical examination, and laboratory tests. It was confirmed by the detection of the Clostridium difficile antigen and toxins in feces using the TOX A/B Quick Check Complete test (Wampol, TechLab, USA). Exclusion criteria included the presence of other acute or chronic inflammatory diseases, any hematologic disorder, or the use of immunosuppressive therapy. Blood samples were collected in the morning (from 7.00 am to 8.00 am) after an overnight fast, centrifuged (3500 g/15min) and stored at –80°C until assayed. Blood tests included white blood cells with differential leukocyte count, platelets (PLT) count, blood creatinine, alanine transaminase (ALT), and C-reactive protein (CRP). All tests were performed according to generally accepted standard methods. Serum concentrations of cytokines interleukin 1β (IL-1β), interleukin 6 (IL-6), interleukin 8 (IL-8), interleukin 10 (IL-10), tumor necrosis factor-α (TNF-α), myeloperoxidase (MPO), and prostaglandin E2 (PGE2) were measured using ELISA assays (Quantikine, R & D Systems, Minneapolis, MN., USA).
Statistical analysis
Results were statistically analyzed. Normal distribution of variables was checked using the Shapiro-Wilk test. We used the student's t-test for analysis of independent variables and the U Mann-Whitney test for analysis of dependent variables. Correlation between selected variables was evaluated using a Pearson correlation coefficient; in the absence of normal distribution, Spearman's rank correlation was used. Statistical analysis was performed using Statistica 10 software (StatSoft ® Inc., USA). The threshold of statistical significance included values of P<0.05.
RESULTS
Patients characteristics
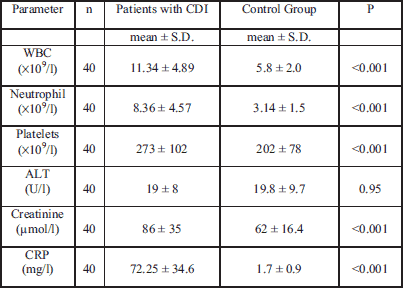
In the CDI study group, women held a slight majority (57.5%), similar to the distribution in the control group. In 37.5% of patients the recurrence of diarrhea was noticed and this recurrence was slightly more than average, due to the fact that we recruited a large percentage of hospitalized patients in our study. Clinical analysis of the study group is shown in Table 1.

Biochemical tests show that patients with CDI suffer from very intensive systemic inflammatory reactions, which was manifested by the increase in inflammatory markers. Patients with CDI were characterized by significantly higher values of WBC, platelets, creatinine and CRP compared to the control group. There were no statistically significant changes in ALT values (Table 2).
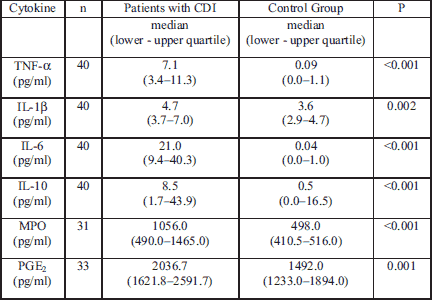
Plasma cytokines and myeloperoxidase levels in CDI patients
The largest increase of pro-inflammatory cytokines in blood included TNF-α and IL-6, however, we also observed an increase in concentrations of the anti-inflammatory cytokine IL-10 in the blood as well. Moreover statistically significantly higher levels of plasma IL-1β, MPO, and PGE2 were noticed in patients with CDI (Table 3). IL-8 was only detected in 6 patients with CDI and no patients in the control group (data not shown).
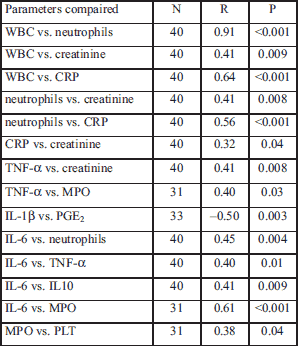
Selected correlation analyses
We performed correlation analysis on measured parameters and the results presenting the statistically significant correlations are shown in Table 4, Figs. 1-3. Other correlations performed on measured parameters were not statistically significant (data not shown).
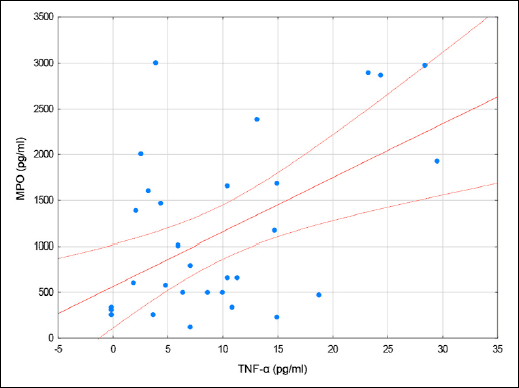 |
Fig. 1. A scatterplot of MPO vs. TNF-α in patients with CDI. |
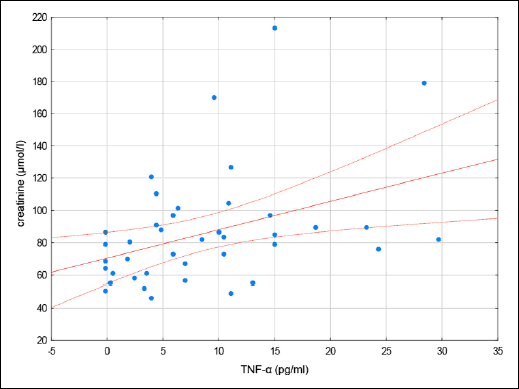 |
Fig. 2. A scatterplot of creatinine vs. TNF-α in patients with CDI. |
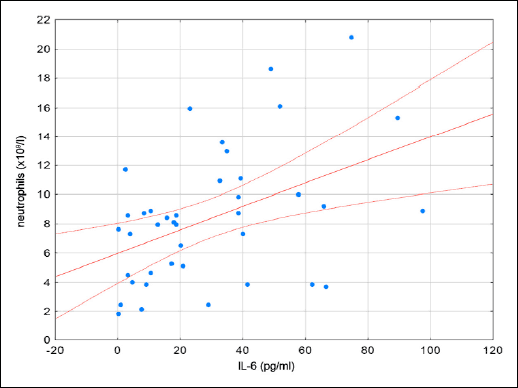 |
Fig. 3. A scatterplot of neutrophils vs. IL-6 in patients with CDI. |
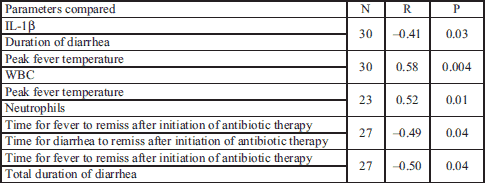
We also assessed the correlation between clinical characteristics and the parameters we measured in the blood (Table 5). We noted a significant inversely proportional correlation between plasma levels of IL-1β and duration of diarrheal symptoms (r= –0.41, P=0.03). Peak fever temperature correlated proportionally with the number of WBCs (r=0.58, P=0.004) and neutrophils (r=0.52, P=0.01) in the blood. We also noted an inverse relationship between the length of time it took fever to disappear after initiating antibiotic therapy and the length of time diarrhea persisted after initiating antibiotic therapy (r= –0.49, P=0.04). The shorter the amount of time that a fever persisted after antibiotic therapy, the longer diarrhea persisted. Also, there is an inverse relationship between the time of fever disappearance after initiating antibiotic therapy and the of diarrheal symptoms during the course of the illness (r= –0.5, P=0.04). Other correlations showed no statistically significant relationship.
DISCUSSION
Our study demonstrated the involvement of two cytokines IL-6 and TNF-α in vivo in the development of the systemic inflammation observed in the course of CDI. This finding might be of significance since the pathogenesis of CDI remains unknown but the incidence of CDI among hospitalized adults in the United States nearly doubled from 2001–2010 (2, 3). There is little evidence of improvement in patient mortality or hospital length of stay in individuals compromised with this infection. Our present results seem to indicate that IL-6 and TNF-α play a role in the inflammatory signaling network that drives neutrophil recruitment in response to CDI and may contribute to the perpetuation of intestinal mucosa infected with this bug (13, 14).
Bacterium Clostridium difficile is not considered part of the classic physiological intestinal flora, although it is estimated that this organism occurs in approximately 1–3% of the population. Clostridium difficile spores are widespread in the human environment, especially in health care environments. Contact with contaminated surfaces is the primary method of bacteria inoculation, and if favorable conditions are present, these bacterial spores transform into vegetative forms, producing toxins if they are toxicogenic strains. Antibiotic therapy causes a physiological imbalance of the natural flora of the gastrointestinal tract, promoting colonization and survival of Clostridium difficile (2, 5). All naturally occurring toxin-producing strains produce TcdB, while only some produce TcdA. This suggests that TcdB is dominant in the pathogenesis of CDI, as confirmed in both epidemiological studies (6) and animal models showing resistance to TcdB (7). Toxins bind to the membrane of host enterocytes, and are transported to the cytoplasm. Recent studies have shown that the toxins associated with the cell membrane are involved in the pathogenesis of disease (8, 9). Toxin penetration into the cytoplasm of the enterocytes inactivates GTP-ase, leading to disorders of the cell-signaling pathway, compromised cytoskeletal integrity, and increased permeability of the epithelial barrier (10). The toxin's effects are fast and cause an increase in epithelial permeability and fluid accumulation in the intestine. Disorders of enterocyte cytoskeletal structures lead to exfoliation, local inflammation, mast cell degranulation, epithelial cell death, and neutrophil recruitment (4). Bacterial toxins are the main virulence factors, but recent studies revealed that other factors such as surface layer proteins (SLP) or flagellin, may also be of important immunoregulatory importance (11).
The immune response in the course of CDI remains largely unexplored. An increase in epithelial cell cytokine production occurs within a few hours of inoculation. A major mediator of this process is IL-8, which promotes neutrophil chemotaxis towards the site of infection (4). Neutrophil infiltration is one of the basic drivers of the development of symptoms of CDI and it is believed that neutrophils play a major role in the pathogenesis of the disease (11). Local inflammation exposes deeper layers of the mucous membrane, further facilitating the effect of toxins on the cells of the immune system, exacerbating the inflammatory response, and increasing transport of fluids into the lumen (12). Both local and systemic neutrophil recruitment are observed in CDI (13, 14). The administration of CD18 monoclonal antibodies results in the inhibition of chemotaxis, adhesion, and tissue infiltration of neutrophils. Administration of CD18 monoclonal antibodies reduced the severity of disease in rabbits exposed to toxin A (14). Induction of neutropenia in rats is also associated with a reduction in symptoms (15). These studies suggest that neutrophils intensify the inflammatory response which leads to increased damage to the host (14, 15). Given that neutrophils are a key element of antimicrobial defense, their potential negative effect on the host requires an further study. Conversely, Jarchum et al. (16), demonstrated that activation and chemotaxis of neutrophils is an essential element in reducing bacterial translocation through the damaged mucosa and thus inhibits the development of CDI symptoms. In our study, we observed a statistically significant increase in the level of neutrophils in the blood of patients with CDI and directly proportional correlation to the concentration of IL-6, CRP, creatinine, and the degree of fever observed in these patients. Neutrophils are therefore a reflection of the severity of both general inflammation, as well as the antimicrobial response of the patient. In addition to IL-8 the important role of IL-1β, IL-6, TNF-α, IFN-γ, and leukotriene B4 (LTB4) has also been demonstrated in the pathogenesis of CDI (17, 18). So far, studies on the immune response in the course of CDI are mainly either in vitro studies or animal models. The only in vivo studies on the pathogenesis of CDI considered stool tests, which demonstrated an increase in IL-8 and lactoferrin (18). Feghaly et al. showed that patients with an increase in IL-8 in the feces had prolonged courses of diarrhea, which is also more common in patients in whom antibiotic therapy failed (19). In our study, we confirmed that the course of CDI is associated with a rapid systemic inflammatory response. We observed a statistically significant increase in the concentration of the following parameters: CRP, WBC, neutrophil, cytokines TNF-α, IL-1β, IL-6, IL-10, MPO, and PGE2. Interestingly, most patients with CDI showed no increase of IL-8, indicating that while IL-8 is essential in the development of local inflammatory responses, as shown by other authors (4, 18, 19), it does not play a direct role in more systemic inflammation. We observed that of the major importance in the cytokine systemic inflammatory response is the cytokine IL-6, which may interact with TNF-α, IL-10, MPO and contribute to the activation of neutrophils. The intensity of the immune response determines the severity of disease, duration of diarrhea and seems to correlate directly with poor prognosis (19-21). In our study, no relationships between the length of the duration of diarrhea, maximum fever, regression of diarrhea, fever after antibiotic therapy implementation, and plasma cytokines were observed. We only observed that when the longer diarrhea lasted before initiating treatment, the lower the plasma concentration of IL-1β was notified. In contrast, a clear correlation was observed between the values of fever and the number of WBC and neutrophils.
In recent years, many centers in the USA, Canada, and Europe, reported not only an increase in frequency, but also the severity of CDI. High leukocytosis and increased creatinine are confirmed factors of poor prognosis in patients with CDI (2, 3). Pepin et al. (22) showed that the factors of high significance were age, as well as significant WBC >20,000/µl and creatinine >99 µmol/L. WBC >50,000/L or lactate >5 mmol/l, were associated with a mortality rate of 75%. In contrast, both sex and fever failed to show such a relationship (22). In our study, we did not include patients with a high risk of mortality, and none of study participants died, so we cannot comment on any correlation of the parameters studied with mortality conditions.
In assessing the results of correlation analysis of clinical parameters with blood parameters studied, we noted a strong correlation between peak fever temperature, and the number of WBC and neutrophils counts. This suggests that this parameter is another factor that can predict the severity of CDI. An interesting relationship has been demonstrated between the time of disappearance of fever after the introduction of antibiotics and the length of duration of diarrhea. The longer the duration of diarrhea before treatment correlated with the faster fever relief. This confirms our own observations, already mentioned above, patients presented with an acute onset of high fever or intense diarrhea tend to have a worse clinical course, while patients who report milder symptoms for weeks before being diagnosed with CDI have a relatively low manifestation of the inflammatory process, and they responded to treatment almost immediately. This is most likely due to the individual patient's immune status and variability of cytokine production in response to inflammatory stimuli. Stress and excessive production of cytokines tend to a more severe course of CDI and other gastrointestinal disorders such as an exacerbation of mucosal injury and the delay in healing of gastric ulcers (23) as well as the perpetuation of certain infectious diseases, such as malaria or sepsis (24, 25).
In our study we observed a relationship between TNF-α and creatinine as well as between IL-6 and serum creatinine and neutrophils. As a result, these two cytokines TNF-α and IL-6 may play a predominant role as markers in CDI patients with a poor prognosis.
Clostridium difficile contains the surface layer proteins (SLPs) known to activate the pro-inflammatory reaction via TLR4, and flagellin which interacts via TLR5. SLPs seem to play a dominant role in the stimulation of the immune system. SLP initiate TLR4 activation through myeloid differentiation primary response gene 88 (MyD88), leading to IRF3 and NF B stimulation, which results in an increase in production of cytokines TNF-α, IL-6, IL-23 and interferon (26). TLR4-/- and MyD88-/- mice were more susceptible to the development of CDI, confirming that TLR plays the protective role against this infection (26). Other studies in vitro in dendritic cells from murine bone marrow and human dendritic cells have shown that the purified SLP induce the production of both proinflammatory cytokines TNF-α , IL-12, IL-23, and IL-1β as well as anti-inflammatory cytokines such as IL-10 (26). In this study, we observed an increase in plasma concentrations of both pro-inflammatory and anti-inflammatory cytokines in patients with CDI. Activation of TLR5 by flagellin, which also activates NFκB, stimulates the production of IL-8, and chemokines (CC-motif) ligand 20, which mediate the chemotaxis of neutrophils and lymphocytes (27). Another important pathway in the pathogenesis of CDI is the activation of NOD1. Clostridium difficile culture supernatants contain a high concentration of the NOD1 ligand while NOD1 deficient mice are characterized by a reduced production of chemokines, weakness of neutrophil migration to the site of infection, increased bacterial translocation, and increased mortality in mice compared to control (28).
PGE2 is a potent stimulator of chloride secretion into the intestinal lumen and fluid during bacterial infections (29). The concentration of PGE2 in intestinal secretions is elevated, both in animal models of bacterial diarrhea and in humans with acute bacterial infection (30). Administration of PGE2 analogues to healthy volunteers causes diarrhea (31). The increased COX-2 and PGE2 expression were observed in Salmonella infection (32), inducing similar effects as the cholera toxin. This effect can be inhibited by selective COX-2 inhibitors (33, 34). TcdA fed directly into the ileum of rabbits also results in the secretion of PGE2 and massive secretion of fluid (35). The mechanism of this phenomenon is not fully understood. It is estimated that the main signaling pathway for the induction of COX-2 by the action of TcdA results from the ROS-mediated activation of p38 MAPK, MSK-1, CREB, and ATF-1. Kim et al. (29) showed that toxin A induces expression of COX-2 and PGE2 in a dose- and time-dependent manner in cultured human epithelial cells NCM460. This induction was blocked by SB203580, the p38 MAPK inhibitor, which also reduced the phosphorylation of MSK-1, CREB/ATF-1, and COX-2 promoter activity after stimulation by TcdA (29). Activation of p38 MAPK enterocytes by TcdA appears to be essential in the pathogenesis of CDI. Interestingly, Warny et al. (36) reported a p38 MAPK-dependent effect on the expression of IL-8 in human monocytes dependent on TcdA. It is likely that PGE2 stimulates the gene expression of IL-8 in human enterocytes by a post-transcriptional mechanism (37). Based on these observations it is suggested that TcdA activates p38 MAPK, which further affects the expression of COX-2 in intestinal epithelial cells resulting in the secretion of PGE2, which in turn affects the transcription of the gene for IL-8 (29). In our study, we observed higher plasma concentrations of PGE2 in CDI patients than in healthy subjects, but also we noted an inverse correlation with the concentration of IL-1β. The prostaglandins, in particular of E series are an important mediator of the inflammatory process due to their recognized proinflammatory effect (38-40). However, several studies have shown that in certain situations prostaglandins of E series may exert an anti-inflammatory effect. Aronoff et al. (41) demonstrated that PGE2 inhibits macrophage phagocytosis in vitro and Pillinger et al. (42) revealed that PGE1 inhibits neutrophil adhesion, which is an important step in the development of the acute organ inflammatory response. Mor et al. (43) showed that PGE1 and PGE2 inhibit the secretion of matrix metalloproteinase 1 (MMP-1) mediated by IL-1β and TNF-α . The inhibition of IL-1β induced expression of monocyte chemoattractant peptide-1 (MCP-1) was reported by Largo et al. (44). In Pillinger et al. study (45), the effects PGE2 had depended on the presence or absence of other cytokines whereas Tchetin et al. (46) observed differing effects dependent on PGE2 concentration. In summary, the mechanism in which the prostaglandins of E series exhibit a divergent effect on the inflammatory process is not fully understood. It is postulated that these variable effects may be due to interaction with other cells, different concentrations of prostaglandins, receptor diversity on target cells, or possibly the interaction between all the above mentioned factors in conjunction with as yet unknown factors.
The MPO is an enzyme belonging to the peroxidase family which is produced in the primary granules of neutrophils, working in phagolysosomes, but it also is secreted outside the cell, where both can be combined with biological membranes and form micro-particles (47). MPO catalyzes the production of oxidizing substances, via the passage of chloride ions and hydrogen peroxide to hypochlorite, oxidation of halogen acid halides, and as a result of the above reactions compounds of the reactive oxygen species (ROS) are formed (47). ROS are toxic to bacteria, fungi, parasites, and tumor cells in addition to being the oxygen component of the killing mechanism by phagocytic cells (48). MPO is an important component of antimicrobial host defense, its deficiency results in an increased risk of morbidity and mortality rates due to infection (48-50). The concentration of MPO during the course of CDI in relation with alterations in intestinal microflora was evaluated in several animals studies in vivo. For instance, Hing et al. (51) showed that the expression of MPO in the large intestine was greater in mice infected with Clostridium difficile and this was confirmed by Kaur et al. (52, 53). Thus, the intestinal MPO activity in Clostridium difficile infected mice reflects the local activation of neutrophils and can serve as a marker for the severity of CDI (52, 53). In another study, loxoprofen which caused hemorrhagic lesions in the small intestine, resulted in the upregulation of enterobacterial invasion, MPO activity and iNOS/TNF-α expression as well as the downregulation of Muc2 expression (54). In our study there was no correlation between MPO levels and neutrophils, while MPO significantly correlated with TNF-α , confirming the role of MPO in the systemic inflammatory response. We also observed significant correlations with severity of disease, MPO and plasma platelet counts, which very often occurs in the course of systemic inflammatory response to multiple infections.
Little is known about the immune response to Clostridium difficile toxins, which are also characterized by immunogenicity (55, 56). Administration of TcdA in rabbit intestine stimulates an inflammatory response, resulting in increased concentrations of TNF-α , IL-1β, and NOS2 (56, 57). It is worth noting that another study showed a protective role of NOS2 in an animal model (57). It is not excluded that the polyphenols such as naringenin, naringin, hesperetin, hesperidin, quercetin, rutin, and catechin which have been shown to exert in vitro inhibitory influence on the growth of human intestine colonized by Bacteroides galacturonicus, Lactobacillus sp., Enterococcus caccae, Bifidobacterium catenulatum, Ruminococcus gauvreauii, and Escherichia coli (59) could be also effective against intestinal infection by Clostridium species but this hypothesis requires a confirmation in future studies.
In conclusion the local inflammatory process in the course of CDI is followed by a systemic inflammatory response: the clinical manifestation of fever and laboratory manifestation of leukocytosis, increased levels of neutrophils increased concentrations of TNF-α , IL-1β, IL-6, IL-10, MPO, and PGE2. Despite the well known important role of IL-8 in the local inflammatory process, we postulate that IL-6 is the cytokine that serves a central role in the promotion of systemic inflammation because most patients with CDI failed to demonstrate an increase in IL-8 concentrations. The cytokines tested demonstrated a relationship between creatinine and TNF-α in our study. Therefore, this cytokine appears to act as another prognostic factor of poor outcome in patients with CDI.
Acknowledgements: William Perucki is a student of the IV-year program at the Jagiellonian University School for Foreigners and the member of the Student Scientific Circle at Department of Infectious Diseases, Cracow, Poland.
Abbreviations: ALT, alanine transaminase; CDI, Clostridium difficile infection; CRP, C-reactive protein; IL-1β, interleukin 1β; IL-6, interleukin 6; IL-8, interleukin 8; IL-10, interleukin 10; LTB4, leukotriene B4; MCP-1, monocyte chemoattractant peptide-1; MMP-1, matrix metalloproteinase 1; MPO, myeloperoxidase; MyD88, myeloid differentiation primary response gene 88; PGE2, prostaglandin E2; PLT, platelets; ROS, reactive oxygen species; SLP, surface layer proteins; TNF-α, tumor necrosis factor-α.
Conflict of interests: None declared.
REFERENCES
- Hall IC, O'Toole E. Intestinal flora in newborn infants with description of a new pathogenic anaerobe. Am J Dis Child 1935; 49: 390-402.
- Stanley JD, Bartlett JG, Dart BW, Ashcraft JH. Clostridium difficile infection. Curr Probl Surg 2013; 50: 302-337.
- Knight CL, Surawicz CM. Clostridium difficile infection. Med Clin North Am 2013; 97: 523-536.
- Penichea AG, Savidge TC, Dann SM. Recent insights into Clostridium difficile pathogenesis. Curr Opin Infect Dis 2013; 26: 447-453.
- Vedantam G, Clark A, Chu M, McQuade R, Mallozzi M, Viswanatha VK. Clostridium difficile infection toxins and non-toxin virulence factors, and their contributions to disease establishment and host response. Gut Microbes 2012; 3: 121-134.
- Loo VG, Bourgault AM, Poirier L, et al. Host and pathogen factors for Clostridium difficile infection and colonization. N Engl J Med 2011; 365: 1693-1703.
- Savidge TC, Pan WH, Newman P, O'Brien M, Anton PM, Pothoulakis C. Clostridium difficile toxin B is an inflammatory enterotoxin in human intestine. Gastroenterology 2003; 125: 413-420.
- Chumbler NM, Farrow MA, Lapierre LA, et al. Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog 2012; 8: e1003072.
- Li S, Shi L, Yang Z, Feng H. Cytotoxicity of Clostridium difficile toxin B does not require cysteine protease-mediated autocleavage and release of the glucosyltransferase domain into the host cell cytosol. Pathog Dis 2013; 67: 11-18.
- Reinert DJ, Jank T, Aktories K, Schulz GE. Structural basis for the function of Clostridium difficile toxin B. J Mol Biol 2005; 351: 973-981.
- Madan R, Petri WA Jr. Immune responses to Clostridium difficile infection. Trends Mol Med 2012; 18: 658-666.
- Sun X, Savidge T, Feng H. The enterotoxicity of Clostridium difficile toxins. Toxins (Basel) 2010; 2: 1848-1880.
- Fujitani S, George WL, Murthy AR. Comparison of clinical severity score indices for Clostridium difficile infection. Infect Control Hosp Epidemiol 2011; 32: 220-228.
- Kelly CP, Becker S, Linevsky JK, et al. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J Clin Invest 1994; 93: 1257-1265.
- Qiu B, Pothoulakis C, Castagliuolo I, Nikulasson S, LaMont JT. Participation of reactive oxygen metabolites in Clostridium difficile toxin A-induced enteritis in rats. Am J Physiol 1999; 276: G485-G490.
- Jarchum I, Liu M, Shi C, Equinda M, Pamer EG. Critical role for MyD88-mediated neutrophil recruitment during Clostridium difficile colitis. Infect Immun 2012; 80: 2989-2996.
- Rocha MF, Maia ME, Bezerra LR, et al. Clostridium difficile toxin A induces the release of neutrophil chemotactic factors from rat peritoneal macrophages: role of interleukin-1βeta, tumor necrosis factor alpha, and leukotrienes. Infect Immun 1997; 65: 2740-2746.
- Steiner TS, Flores CA, Pizarro TT, Guerrant RL. Fecal lactoferrin, interleukin-1 and interleukin-8 are elevated in patients with severe Clostridium difficile colitis. Clin Diagn Lab Immun 1997; 4: 719-722.
- El Feghaly RE, Stauber JL, Deych E, Gonzales C, Tarr PI, Haslam DB. Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in Clostridium difficile infection. Clin Infect Dis 2013; 56: 1713-1721.
- Kelly CP, Kyne L. The host immune response to Clostridium difficile. J Med Microbiol 2011; 60: 1070-1079.
- Walker AS, Eyre DW, Wyllie DH, et al. Relationship between bacterial strain type, host biomarkers, and mortality in Clostridium difficile infection. Clin Infect Dis 2013; 56: 1589-1600.
- Pepin J, Vo TT, Boutros M, et al. Risk factors for mortality following emergency colectomy for fulminant Clostridium difficile infection. Dis Colon Rectum 2009; 52: 400-405.
- Konturek PC, Brzozowski T, Konturek SJ. Stress and the gut: pathophysiology, clinical consequences, diagnostic approach and treatment options. J Physiol Pharmacol 2011; 62: 591-599.
- McGuire W, Hill AV, Allsopp CE, Greenwood BM, Kwiatkowski D. Variation in the TNF-αlpha promoter region associated with susceptibility to cerebral malaria. Nature 1994; 371: 508-510.
- Kothari N, Bogra J, Abbas H, et al. Tumor necrosis factor gene polymorphism results in high TNF level in sepsis and septic shock. Cytokine 2013; 61: 676-681.
- Ryan A, Lynch M, Smith SM, et al. A role for TLR4 in Clostridium difficile infection and the recognition of surface layer proteins. PLoS Pathog 2011; 7: e1002076.
- Yoshino Y, Kitazawa T, Ikeda M, et al. Clostridium difficile flagellin stimulates toll-like receptor 5, and toxin B promotes flagellin-induced chemokine production via TLR5. Life Sci 2013; 92: 211-217.
- Hasegawa M, Yamazaki T, Kamada N, et al. Nucleotide-binding oligomerization domain 1 mediates recognition of Clostridium difficile and induces neutrophil recruitment and protection against the pathogen. J Immunol 2011; 186: 4872-4880.
- Kim H, Rhee SH, Kokkotou E, et al. Clostridium difficile toxin A regulates inducible cyclooxygenase-2 and prostaglandin E2 synthesis in colonocytes via reactive oxygen species and activation of p38 MAPK. J Biol Chem 2005; 280: 21237-21245.
- Matuchansky C, Mary JY, Bernier JJ. Further studies on prostaglandin E1-induced jejunal secretion of water and electrolytes in man, with special references to the influence of the ethacrynic acid, furosemide, and aspirin. Gastroenterology 1976; 71: 274-281.
- Lanza FL, Kochman RL, Geis GS, Rack EM, Deysach LG. A double-blind, placebo-controlled, 6-day evaluation of two doses of misoprostol in gastroduodenal mucosal protection against damage from aspirin and effect on bowel habits. Am J Gastroenterol 1991; 86: 1743-1748.
- Eckmann L, Stenson WF, Savidge TC, et al. Role of intestinal epithelial cells in the host secretory response to infection by invasive bacteria. Bacterial entry induces epithelial prostaglandin h synthase-2 expression and prostaglandin E2 and F2 alpha production. J Clin Invest 1997; 100: 296-309.
- Beubler E, Schuligoi R, Chopra AK, Ribardo DA, Peskar BA. Cholera toxin induces prostaglandin synthesis via post-transcriptional activation of cyclooxygenase-2 in the rat jejunum. J Pharmacol Exp Ther 2001; 297: 940-945.
- Beubler E, Schuligoi R. Mechanisms of cholera toxin-induced diarrhea. Ann NY Acad Sci 2000; 915: 339-346.
- Triadafilopoulos G, Pothoulakis C, O'Brien MJ, LaMont JT. Differential effects of Clostridium difficile toxins A and B on rabbit ileum. Gastroenterology 1987; 93: 273-279.
- Warny M, Keates AC, Keates S, et al. p38 MAP kinase activation by Clostridium difficile toxin A mediates monocyte necrosis, IL-8 production, and enteritis. J Clin Invest 2000; 105: 1147-1156.
- Yu Y, Chadee K. Prostaglandin E2 stimulates IL-8 gene expression in human colonic epithelial cells by a posttranscriptional mechanism. J Immunol 1998; 161: 3746-3752.
- Scher JU, Pillinger MH. The anti-inflammatory effects of prostaglandins. J Investig Med 2009; 57: 703-708.
- Legler DF, Bruckner M, Uetz-von Allmen E, Krause P. Prostaglandin E2 at new glance: novel insights in functional diversity offer therapeutic chances. Int J Biochem Cell Biol 2010; 42: 198-201.
- Korbecki J, Baranowska-Bosiacka I, Gutowska I, Chlubek D. The effect of reactive oxygen species on the synthesis of prostanoids from arachidonic acid. J Physiol Pharmacol 2013; 64: 409-421.
- Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol 2004; 173: 559-565.
- Pillinger MH, Philips MR, Feoktistov A, Weissmann G. Crosstalk in signal transduction via EP receptors: prostaglandin E1 inhibits chemoattractant-induced mitogen-activated protein kinase activity in human neutrophils. Adv Prostaglandin Thromboxane Leukot Res 1995; 23: 311-316.
- Mor A, Abramson SB, Pillinger MH. The fibroblast-like synovial cell in rheumatoid arthritis: a key player in inflammation and joint destruction. Clin Immunol 2005; 115: 118-128.
- Largo R, Diez-Ortego I, Sanchez-Pernaute O, et al. EP2/EP4 signalling inhibits monocyte chemoattractant protein-1 production induced by interleukin 1β in synovial fibroblasts. Ann Rheum Dis 2004; 63: 1197-1204.
- Pillinger MH, Marjanovic N, Kim SY, et al. Matrix metalloproteinase secretion by gastric epithelial cells is regulated by E prostaglandins and MAPKs. J Biol Chem 2005; 280: 9973-9979.
- Tchetina EV, Di Battista JA, Zukor DJ, Antoniou J, Poole AR. Prostaglandin PGE2 at very low concentrations suppresses collagen cleavage in cultured human osteoarthritic articular cartilage: this involves a decrease in expression of proinflammatory genes, collagenases and COL10A1, a gene linked to chondrocyte hypertrophy. Arthritis Res Ther 2007; 9: R75.
- Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol 2005; 77: 598-625.
- Hirche TO, Gaut JP, Heinecke JW, Belaaouaj A. Mieloperoxidase plays critical roles in killing Klebsiella pneumoniae and inactivating neutrophil elastase: effects on host defense. J Immunol 2005; 174: 1557-1565.
- Aratani Y, Koyama H, Nyui S, Suzuki K, Kura F, Maeda N. Severe impairment in early host defense against Candida albicans in mice deficient in myeloperoxidase. Infect Immun 1999; 67: 1828-1836.
- Kutter D, Devaquet P, Vanderstocken G. Consequences of total and subtotal myeloperoxidase deficiency: risk of benefit? Acta Haematol 2000; 104: 10-15.
- Hing TC, Ho S, Shih DQ, et al. The antimicrobial peptide cathelicidin modulates Clostridium difficile-associated colitis and toxin A-mediated enteritis in mice. Gut 2013; 62: 1295-1305.
- Kaur S, Vaishnavi C, Prasad KK, Ray P, Kochhar R. Effect of Lactobacillus acidophilus and epidermal growth factor on experimentally induced Clostridium difficile infection. Indian J Med Res 2011; 133: 434-441.
- Kaur S, Vaishnavi C, Ray P, Kochhar R, Prasad KK. Effect of biotherapeutics on cyclosporin-induced Clostridium difficile infection in mice. J Gastroenterol Hepatol 2010; 25: 832-838.
- Hayashi S, Kurata N, Kitahirachi E, et al. Cinacalcet, a calcimimetic, prevents nonsteroidal anti-inflammatory drug-induced small intestinal damage in rats. J Physiol Pharmacol 2013; 64: 453-463.
- Johnson S, Gerding DN, Janoff EN. Systemic and mucosal antibody responses to toxin A in patients infected with Clostridium difficile. J Infect Dis 1992; 166: 1287-1294.
- Ng J, Hirota SA, Gross O, et al. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 2010; 139: 542-552.
- Savidge TC, Urvil P, Oezguen N, et al. Host S-nitrosylation inhibits Clostridial small molecule-activated glucosylating toxins. Nat Med 2011; 17: 1136-1141.
- Qiu B, Pothoulakis C, Castagliuolo I, Nikulasson Z, LaMont JT. Nitric oxide inhibits rat intestinal secretion by Clostridium difficile toxin A but not Vibrio cholerae enterotoxin. Gastroenterology 1996; 111: 409-418.
- Duda-Chodak A. The inhibitory effect of polyphenols on human gut microbiota. J Physiol Pharmacol 2012; 63: 497-503.
A c c e p t e d : September 30, 2014