EFFECTS OF PHOSPHODIESTERASE 5 INHIBITOR SILDENAFIL ON THE RESPIRATORY PARAMETERS, INFLAMMATION AND APOPTOSIS IN A SALINE LAVAGE-INDUCED MODEL OF ACUTE LUNG INJURY
INTRODUCTION
A variety of insults can evoke an acute lung damage. While direct pulmonary impacts primarily damage the epithelial alveolar cells, indirect causes predominantly hit the pulmonary capillary endothelium. Regardless of etiology, the lung reacts to various types of insults in similar way, as a stereotypic response (1, 2). Diffuse damage to alveolar-capillary barrier leads to exudation of protein-rich fluid into the alveolar space, which manifests as non-cardiogenic pulmonary edema (3). Lung edema, dysfunction of pulmonary surfactant, atelectasis and ventilation-perfusion mismatch impair the gas exchange. Hypoxia-induced pulmonary vasoconstriction, thrombo-embolic occlusions in the pulmonary microvasculature, increased production of vasoconstrictive agents further augment pulmonary hypertension, significantly contributing to mortality in patients with acute respiratory distress syndrome (ARDS) (4). Damage to epithelial and endothelial cells triggers pulmonary neutrophil recruitment, which is mediated by interaction of primed and activated neutrophils with the lung microvascular endothelium (5). The development of acute lung injury (ALI) is associated with release of pro-inflammatory cytokines, proteases, reactive oxygen species (ROS) and reactive nitrogen species (RNS) (6, 7). When the local anti-inflammatory and antioxidant mechanisms have become exhausted, the uncontrolled inflammatory response in ALI contributes to further lung and systemic damage (3, 8).
3’5’-cyclic guanosine monophosphate (cGMP) as a second messenger participates in various signalling pathways and regulates many aspects of cell function. Besides the classical regulatory role of cGMP in the vascular tone regulation and smooth muscle relaxation, additional physiological roles including platelet and cardiac functions have been discovered (9). Because of ability to decrease pulmonary vascular resistance, improve pulmonary blood flow, increase right cardiac output and normalize lung development, sildenafil and other phosphodiesterase 5 (PDE5) inhibitors have been increasingly used in pulmonary arterial hypertension including persistent pulmonary hypertension in neonates and in bronchopulmonary dysplasia in preterm infants (9, 10). However, cGMP analogues can exert some anti-inflammatory activity as cGMP signalling is involved in downregulation of P-selectin expression and leukocyte recruitment (11) and inhibition of lipopolysaccharide (LPS)-induced tumor necrosis factor alpha (TNF-α) secretion (12, 13). Besides this, increased levels of intracellular cGMP have been found to inhibit the TNF-α-induced increase of nitric oxide (NO)-metabolite levels and inducible NO synthase (iNOS) expression in rat tracheal smooth muscle cells (14). The concentration of intracellular cGMP depends on a balance between synthesis by soluble guanylate cyclases (sGCs) and degradation by cyclic nucleotide phosphodiesterases (PDEs). The activity or expression of sGCs and PDEs could be modified in various situations. For instance, ROS, TNF-α and excessive NO were all indicated to reduce sGC expression (15, 16), while PDEs activities are largely increased in oxidative stress and inflammatory processes (17, 18). Therefore, sGC activators or PDE inhibitors may exhibit a promising therapeutic potential in the treatment of inflammatory diseases.
Among these pharmacological agents, PDE5 inhibitors appear to be particularly applicable in treating pulmonary diseases, since PDE5 is expressed in high levels in the lung tissue and is highly specific for hydrolysis of cGMP (19). Sildenafil has been beneficial in various forms of acute lung damage. For instance, sildenafil reduced both LPS-induced and ovalbumin-induced airway hyperresponsiveness, leukocyte influx and NO generation in two guinea pig models (20). In meconium-induced models of ALI, sildenafil treatment reversed an increase in pulmonary vascular resistance (21), attenuated lung injury score, reduced inflammation and oxidative stress and attenuated lung cell apoptosis (22). In addition, sildenafil decreased markers of oxidative stress and inflammation in bleomycin-induced model of lung fibrosis (23) and also in models of indirect lung injury (24, 25).
Promising results from these studies have inspired us to evaluate therapeutical potential of sildenafil in such form of ALI where surfactant depletion and lung edema are predominant, while inflammation, pulmonary vasoconstriction and lung remodelling are less important. The surfactant depletion model induced by repetitive saline lung lavage considerably imitates the situation in near-drowning or in preterm neonates with respiratory distress syndrome due to immature lung and insufficient production of surfactant. Despite the most obvious benefit of sildenafil is expected in forms of lung injury with serious pulmonary vasoconstriction,we have presumed that sildenafil thanks to its complex actions can positively influence migration of polymorphonuclears (PMNs), reduce inflammation, oxidative stress, epithelial cell apoptosis and formation of lung edema, and thereby enhance respiratory parameters also in the model of surfactant depletion.
MATERIALS AND METHODS
General design of experiments
Experimental protocols were performed in accordance with ethical guidelines and were authorized by the local Ethics Committee of Jessenius Faculty of Medicine and by the National Veterinary Board.
In the study, young New Zealand white rabbits of the mean body weight (b.w.) of 2.5 ± 0.3 kg were used. Animals were anesthetized with intramuscular tiletamine + zolazepam (15 mg/kg b.w.; Zoletil, Virbac, France) and xylazine (5 mg/kg b.w.; Xylariem, Riemser, Germany), followed by an infusion of tiletamine + zolazepam (10 mg/kg/h i.v.). A tracheotomy was performed and an endotracheal tube was inserted. Catheters into the femoral artery and right atrium for sampling the arterial and mixed venous blood and into the femoral vein to administer anesthetics were inserted. Animals were paralyzed with pipecuronium bromide (0.3 mg/kg b.w./30 min; Arduan, Gedeon Richter, Hungary) and subjected to ventilator Aura V (Chirana, Slovakia) and were ventilated conventionally with following settings: frequency (f.) of 40/min, fraction of inspired oxygen (FiO2) 1.0, time of inspiration (Ti) 50%, positive end-expiratory pressure (PEEP) 0.5 kPa, and peak inspiratory pressure (PIP) to achieve a tidal volume (VT) < 6 ml/kg b.w. After 15 min of stabilization, respiratory parameters were recorded. Samples of 0.5 ml of arterial and mixed venous blood were taken for measurement of blood gases by blood gas analyzer (RapidLab 348, Siemens, Germany). After the measurement, unused blood was returned back into circulation via femoral artery or jugular vein catheters, respectively. One group of animals served as healthy oxygen-ventilated and non-treated controls (Control group, n = 9). In other animals, the model of lung injury was induced according to modified protocol by other authors (26, 27). Briefly, in oxygenated animals repetitive saline lung lavage removes a significant portion of pulmonary surfactant what results into worsening of oxygenation and lung compliance. Thus, in our study saline (0.9% NaCl, 30 ml/kg b.w., 37°C) was instilled into the endotracheal cannula in the semi-upright right and left lateral positions of the animal and was immediately suctioned by a suction device. Lavage was performed 6 – 12 times, with 2 min. intervals between each lavage, during which animals were ventilated. When PaO2 decreased to < 26.7 kPa in 2 measurements at 5 and 15 min after the lavage and lung compliance decreased of < 30% of the initial value, the criteria of stable ALI model were full-filled. Respiratory parameters were recorded and blood samples taken again. Then, animals were treated with sildenafil citrate (SC-208388, Santa Cruz Biotechnology, USA; water solubility of 3.5 mg/ml declared by the producer (28), dissolved in normal saline at a dose of 1 mg/kg b.w. (ALI + Sil group, n = 8) which was given intravenously during 2 min., or were left without therapy (ALI group, n = 9). All animals were oxygen-ventilated (FiO2 1.0, f. 40/min, PEEP 0.5 kPa, VT < 6 ml/kg b.w.) for an additional 4 hours after administration of the treatment. Blood gases and respiratory parameters were measured at 0.5, 1, 2, 3, and 4 hours of the treatment. At the end of experiment, animals were overdosed by anesthetics.
Counting of cells in the bronchoalveolar lavage fluid
After sacrificing the animal, lungs and trachea were excised. The left lung was lavaged 3-times by saline (0.9% NaCl, 37°C, dose of 10 ml/kg b.w.). Total number of cells in the BAL fluid was determined by an automated cell analyzer (Countess, Invitrogen, USA). Bronchoalveolar lavage (BAL) fluid was then centrifuged at 1500 rpm for 15 min. Differential count of cells in the BAL fluid sediment was evaluated microscopically after staining by May-Grunwald/Giemsa-Romanowski.
Markers of inflammation and oxidative damage in the lung homogenate
Concentrations of cytokines, oxidative modification products and nitrite/nitrate (NOx) were determined in 10% (weight/volume) lung homogenate prepared using 0.1 M ice-cold phosphate buffer (PBS, pH 7.4) by Polytron homogenizer PT 1200 E (5-times for 25 s, 1200 rpm; Kinematica AG, Switzerland). Homogenates were then 3-times freezed and centrifuged (12000 rpm, 15 min, 4°C). Final supernatants were then stored at –70°C until the analysis was performed.
Concentrations of TNF-α, interleukin (IL)-8, and IL-6 were quantified using rabbit-specific ELISA kits (USCN Life Science Inc., China) according to the manufacturer’s instructions. Data were expressed in pg/ml. All ELISA analyses were performed in duplicates.
Protein oxidative damage was determined using OxiSelectTM nitrotyrosine ELISA Kit (Cell Biolabs, Inc., USA). Data were expressed in 3-nitrotyrosine nanomole concentration (nM, 3NT). Lipid oxidative damage expressed by concentration of thiobarbituric acid-reacting substances (TBARS) was determined by OxiSelectTM TBARS Assay Kit (Cell Biolabs Inc., USA). Data were expressed as malondialdehyde in micromole concentration (µM MDA).
The nitrate/nitrite (NOx) concentrations were determined in duplicate by using the Cayman’s Nitrate/Nitrite Colorimetric Assay Kit (Alexis Corp., USA), according to the manufacturer’s instruction. Data were expressed as nitrite/nitrate in micromole concentration (µM NOx).
Lung edema formation expressed as wet/dry lung weight ratio and protein content in the bronchoalveolar lavage fluid
For determination of wet/dry lung weight ratio, strips of the right lung tissue were weighed, dried at 60°C for 72 h and then the wet/dry weight ratio was determined, whereas the higher value indicates higher accumulation of liquid in the lung. Total protein content in the BAL fluid was determined by colorimetric method according to Bradford (29) standardized with bovine serum albumin (BSA). Results were expressed in mg/ml.
Apoptosis assays
In situ labeling of DNA strand breaks by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) method
The lungs were immersed in 4 % formaldehyde solution. After paraffin embedding, 4 µm thick slides were cut on microtome followed by deparaffinization and pretreatment with proteinase K. The tissue sections were further processed by DeadEndTM Colorimetric TUNEL System (Promega, USA). This assay labels fragmented DNA of apoptotic cells. Biotinylated nucleotide is incorporated at 3‘-OH DNA ends using the Terminal Deoxynucleotidyl Transferase, Recombinant, (rTdT) enzyme. Horseradish peroxidase-labeled streptavidin (Streptavidin HRP) was then bound to these biotinylated nucleotides. For detection of nucleotides and blocking endogenous peroxidases, the sections were incubated with 0.3% H2O2 solution. Colour of sections was developed after incubation with diaminobenzidine, DAB-chromogen solution. The sections were then counterstained with Mayer´s hematoxylin and mounted with Permount (Fisher, USA). The slides were viewed with Olympus BX41 microscope (Olympus, Japan), magnification × 400. The image capture was performed with Quick Photo Micro software, version 2.2 (Olympus). The apoptotic index was evaluated by blinded histopathologist and was calculated as a percentage of TUNEL immunoreactive (TUNEL-IR) dark brown stained nuclei in a total of 100 nuclei randomly counted from three sites within each section, 282 pictures in total, 56 per animal.
Immunohistochemistry for activated caspase-3
After deparaffinization, revitalisation and rehydratation, the tissue slides were treated with 3% H2O2 solution for 10 min for blocking endogenous peroxidases. Washing with Tris buffer was used after each handling step. The sections were incubated with the primary antibody rabbit anti-caspase 3 (1:500; Bioss, USA) for 30 min at a room temperature. The specimen was then incubated by sequential 10 min incubation with biotinylated anti-rabbit secondary antibody and pexodidase-labelled streptavidin conjugated to HRP (DAKO LSAB®2 System-HRP; Dako, Danmark). Colour of sections was developed after incubation with DAB-chromogen solution (Dako, Danmark). The sections were then counterstained with Mayer´s hematoxylin and mounted with Entellan (Merck, USA). The slides were viewed with Olympus BX41 microscope (Olympus, Japan), magnification × 400. The image capture was performed with Quick Photo Micro software, version 2.2 (Olympus). The density of activated caspase 3 immunoreactive cells (dark-brown cytoplasm; caspase 3-IR) was evaluated by blinded histopathologist and was measured randomly from three sites within each section and was calculated as a total number of caspase 3-IR cells in the field, 314 pictures in total, 62 per animal.
Measurement of respiratory parameters
During experiment, animals were ventilated with ventilator Aura V (Chirana, Slovakia). Ventilatory parameters, such as FiO2, VT, minute ventilation, f, Ti, ventilatory pressures (mean airway pressure (MAP), PIP and PEEP), dynamic (Cdyn) and static (Cst) lung compliance, and airway resistance (Raw) were automatically measured by in-build sensors and software and were displayed on the screen of ventilator. Partial pressures of oxygen and carbon dioxide (PaO2 and PaCO2), arterial pH, arterial oxygen saturation (SatO2), and parameters of acid-base balance were measured with blood gas analyzer (Rapidlab 348, Siemens, Germany). Ventilation efficiency index was calculated as VEI = 3800/[(PIP-PEEP) × frequency × PaCO2]. The oxygenation index (OI) was calculated as: OI = [MAP (kPa) × FiO2 (%)]/PaO2 (kPa). The PaO2/FiO2 ratio was calculated as: PaO2/FiO2 ratio = [PaO2 (mmHg) × 100]/FiO2 (%). Alveolar partial pressure of O2 (PAO2 mmHg) was expressed as: PAO2 = {[FiO2 (%)/ 100] × (Patm – PH2O)} –[PaCO2 (mmHg)/ RQ], whereas atmospheric pressure (Patm) at a sea level is 760 mmHg, PH2O is 47 mmHg, and RQ (respiratory quotient) is 0.8. Alveolar-arterial (A-a) gradient was calculated as: A-a gradient = PAO2 (mmHg) – PaO2 (mmHg). Right-to-left pulmonary shunts (Qs/Qt) were calculated as described elsewhere (30) and expressed in %.
Statistical analysis
GraphPad Prism (Version 6.01) software was used for statistical analyses. Statistical differences between the groups were determined by analysis of variance (ANOVA) with Bonferroni post-hoc test or Kruskal-Wallis test. A P-value below 0.05 was considered to be statistically significant. Results are presented as average with error bars indicating standard error of the mean (means ± SEM).
RESULTS
Body weights of animals and initial values of the parameters before induction of the ALI model were comparable between the groups (all P > 0.05). Average numbers of lung lavages in ALI and ALI + Sil groups were 7 times, without significant differences between the groups (P > 0.05, data not shown).
Cells in the bronchoalveolar lavage fluid
Repetitive lung lavage used for induction of the ALI model significantly elevated a total number of cells in the BAL fluid compared to controls (P < 0.01 for ALI versus Control), while sildenafil inhibited infiltration of cells into the lungs (P < 0.05 for ALI + Sil versus ALI) (Table 1). As shown in Table 1, induction of the ALI model significantly increased the percentage of neutrophils (P < 0.001 for ALI versus Control) and decreased relative number of monocytes-macrophages (P < 0.001 for ALI versus Control). Sildenafil therapy lowered number of neutrophils (P < 0.001 for ALI + Sil versus ALI) and elevated relative count of monocytes and macrophages (P < 0.001 for ALI + Sil versus ALI).

Markers of inflammation and oxidative damage
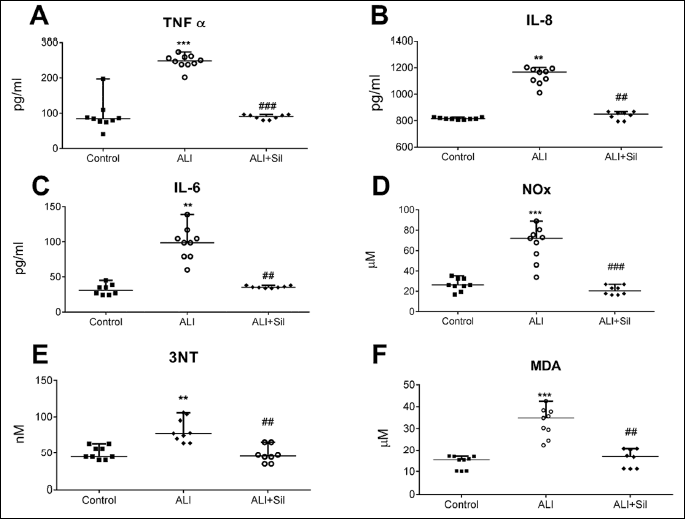
Lung lavage with saline increased all observed markers of inflammation and oxidative stress in the lung tissue. All proinflammatory cytokines (TNF-α, P < 0.001; IL-8, and IL-6; for both P < 0.01), nitrite/nitrate (NOx, P < 0.001), marker of protein oxidation 3-nitrotyrosine (3NT, P < 0.01), and marker of lipid peroxidation thiobarbituric acid-reacting substances (TBARS, P < 0.001) expressed as malondialdehyde significantly increased in untreated animals compared to controls (ALI versus Control). Sildenafil therapy decreased concentrations of the mentioned markers compared to ALI group: for TNF-α (ALI + Sil versus ALI, P < 0.001), IL-8 and IL-6 (ALI + Sil versus ALI, P < 0.01), NOx (ALI + Sil versus ALI, P < 0.001), 3NT and TBARS (ALI + Sil versus ALI, both P < 0.01) (Fig. 1).
Lung edema formation
Formation of the lung edema was expressed as wet-dry lung weight ratio (W/D ratio) and protein content in the BAL fluid. W/D ratio increased after induction of the lavage-induced lung injury compared to controls (ALI versus Control P < 0.001), what represented evident fluid accumulation in the lung tissue. Sildenafil therapy decreased W/D ratio (ALI + Sil versus ALI, P < 0.01) (Fig. 2A).
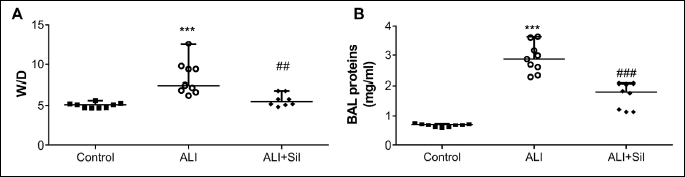
In animals with ALI, increased protein content in the BAL fluid compared to healthy controls was observed, what underlined massive deterioration of the alveolar-capillary membrane and leaking fluid into the alveolar space (ALI versus Control, P < 0.001). Similarly to W/D ratio, sildenafil therapy significantly decreased protein concentration in the BAL fluid compared to untreated animals (ALI + Sil versus ALI, P < 0.001) (Fig. 2B).
Apoptosis in the lung tissue
Apoptotic index expressed as a percentage of TUNEL-positive immunoreactive cells significantly elevated in saline-lavaged and untreated group compared to control group (ALI versus Control, P < 0.001) and decreased in sildenafil-treated animals (ALI + Sil versus ALI, P < 0.01; Fig. 3).
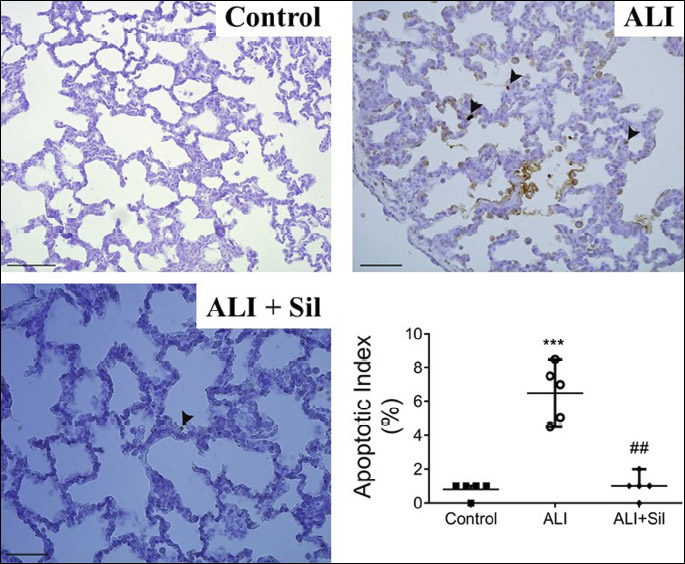
Number of caspase-3 immunoreactive cells in the lung increased in ALI group compared to control group (ALI versus Control, P < 0.01). In sildenafil-treated animals, number of caspase-3 immunoreactive cells decreased compared to untreated ALI animals (P < 0.01; Fig. 4).
Respiratory parameters
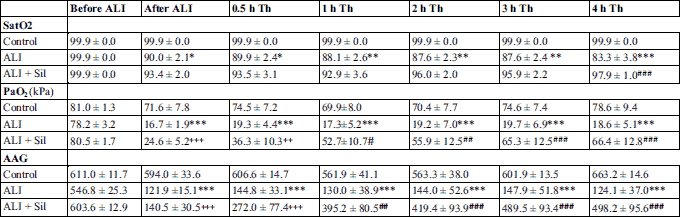
All observed respiratory parameters (PaO2/FiO2, OI, VEI, MAP, PaCO2, pH, Cdyn, Qs/Qt, PaO2, AAG) had been severely altered 15 min after the lung lavage by saline compared to the baseline values (time sequence ALI versus basal value, BV; all P < 0.01).
Repetitive lung lavage induced acute respiratory insufficiency characterized with a decrease in lung compliance, an increase in right-to-left pulmonary shunts with subsequent hypoxemia, hypercapnia and acidemia compared to non-lavaged controls (all P < 0.001, Table 2 and 3; Fig. 5). Lavage-induced deterioration of the respiratory parameters persisted in the untreated animals (ALI group) till the end of experiment.
| Fig. 5. The ratio of arterial oxygen partial pressure to fraction of inspired oxygen (PaO2/FiO2, in kPa) (A); oxygenation index (OI) (B) and ventilation efficiency index (VEI) (C) before (basal value, BV) and after lung lavage (acute lung injury, ALI) and within 4 h after administration of the therapy (Th) in healthy ventilated and non-treated animals (Control group, n = 9), in non-treated animals with ALI (ALI group, n = 9) and in animals with ALI treated with sildenafil (ALI + Sil group, n = 9). Statistical comparisons: for ALI versus Control *P < 0.05, **P < 0.01, ***P < 0.001; for ALI + Sil versus ALI #P < 0.05, ##P < 0.01, ###P < 0.001; for ALI + Sil versus Control ++P < 0.01, +++P < 0.001. Data are presented as means ± SEM. |
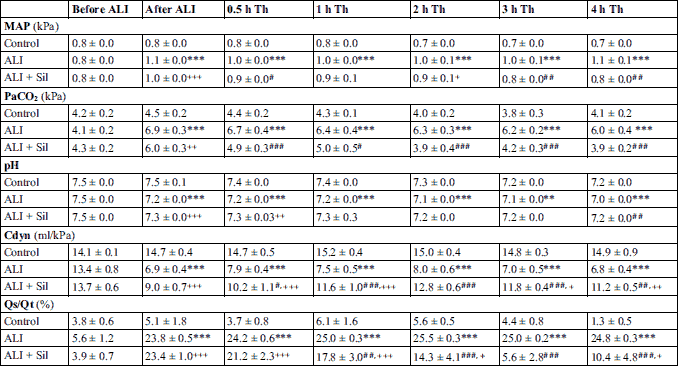
Sildenafil administration significantly increased PaO2 from 1 hour (P < 0.05, Table 3) and decreased PaCO2 from 30 min (P < 0.01, Table 2). Sildenafil influenced also other respiratory parameters compared to non-treated ALI group, i.e. decreased MAP (P < 0.01), PaCO2 (P < 0.01), and OI (P < 0.001), and elevated Cdyn (P < 0.05) from 30 min of the therapy; reduced Qs/Qt (P < 0.01) and increased PaO2 (P < 0.01), PaO2/FiO2 ratio (P < 0.01), AAG (P < 0.01), and VEI (P < 0.01) from 1 hour of the therapy, and also increased arterial pH (P < 0.01) and SatO2 (P < 0.001) compared to ALI group at the end of the experiment (Table 2 and 3; Fig. 5).
DISCUSSION
ALI is characterized by an extensive inflammatory reaction within the lung tissue along with disruption of alveolar-capillary membrane leading to severe impairment of gas exchange (31). Since no pharmacological approaches were clearly shown to be effective for management of ALI, development of therapeutic strategies for ALI is urgently needed. The present study demonstrated that repetitive lung lavage by saline induced severe changes in the respiratory parameters, elevated levels of oxidative stress markers, inflammatory cytokines, and NO metabolites, and increased permeability of alveolar-capillary membrane and epithelial lung cell apoptosis. Intravenous administration of 1 mg/kg b.w. of sildenafil improved gas exchange and other respiratory parameters, reduced transmigration of cells, particularly of neutrophils, into the lung, decreased levels of nitrite/nitrate, proinflammatory cytokines and markers of protein and lipid oxidation in the lung, and reduced formation of lung edema and lung cell apoptosis.
Inflammatory cell infiltration and accumulation in the lung tissue is one of the characteristic pathological hallmarks of ALI (32). PMNs are essential for clearance of debris and pathogens from the alveolar space. However, excessive and persistent sequestration of PMNs may result in additional damage to the lung tissue through release of numerous toxic mediators including reactive oxygen species, proteases, proinflammatory cytokines and changed apoptosis which exacerbate ALI (33, 34). During ALI, neutrophils are the first immune cells recruited into the inflammation site. Activated neutrophils extravasate and migrate into the alveolar space where they secrete chemoattractants and recruit more leukocytes to amplify the inflammation response (35, 36).
Our data suggest that the protective activity of sildenafil in ALI may be mediated through inhibition of inflammatory cell influx, mainly of neutrophils, into the lung. During ALI, large amounts of ROS are generated which cause cell membrane lipid peroxidation leading to destruction of lung parenchymal cells. Overproduction of ROS and proinflammatory substances deteriorates the alveolar epithelium and surfactant function (37, 38). The present study confirmed elevation of MDA, a lipid peroxidation parameter, and 3NT, an indirect marker of nitrosative stress, in the lung tissue of ALI animals while sildenafil therapy decreased oxidative stress. However, the role of inflammation in the pathogenesis of ALI is not limited to oxidative stress. Combined action of inflammation and oxidative stress increases a capillary permeability leading to lung edema and exacerbation of tissue injury.
Activated macrophages produce large amounts of proinflammatory cytokines, such as TNF-α and interleukins, which promote lung inflammation. TNF-α is the first multifunctional cytokine released from stimulated alveolar macrophages. It elicits the inflammatory cascade as it stimulates other cells to secrete more cytokines and chemokines such as IL-6, monocyte chemotactic protein, and macrophage inflammatory protein which subsequently mediate the recruitment of PMNs, and increase the severity of lung injury (39, 40). In our experiments, significantly elevated levels of circulatory TNF-α and IL-6 in the plasma come in agreement with previous report (34). In this study, sildenafil therapy markedly suppressed the elevation of inflammatory cytokines what suggests that sildenafil might suppress the inflammatory cell infiltration into the lung via inhibition of the release of inflammatory cytokines.
The effect of selective phosphodiesterase-5 (PDE-5) inhibitor sildenafil is mediated through increasing the tissue level of the second messenger cGMP what leads to relaxation of the smooth muscle (41) through reduction of intracellular [Ca2+] and downregulation of contractile apparatus. PDE-5 is found in high concentrations in the pulmonary artery smooth muscle, and therefore its inhibition leads to a decrease in pulmonary vascular resistance (42). Pulmonary vasodilation effect of sildenafil was also demonstrated in our study where it decreased pulmonary right-to-left pulmonary shunts. Level of cGMP in the smooth muscle is also increased by nitric oxide (NO), which is formed from l-arginine through the actions of different types of NO synthase. NO acts as a vasodilator, neurotransmitter and inflammatory mediator in the airways, and decreases a methacholine-induced bronchoconstriction in the experimental animals (43). Sildenafil elevates cellular cGMP levels through competition for the PDE binding site with cGMP and in that way inhibits degradation of cGMP to GMP. cGMP through protein kinase G (PKG) plays an important role in the regulation of activity of various cell populations including immune cells. The level of cGMP is also regulated by NO. NO exerts multiple modulating effects on inflammation and plays a key role in the regulation of immune responses. Both the inducible NO synthase (iNOS) and the PDEs enzymes are expressed in numerous cell types including macrophages, dendritic cells, T cells, and neutrophils (44). NO is the main activator of soluble guanylate cyclase (sGC), an enzyme which synthesizes cGMP. Inhibition of cGMP catabolism by selective PDE5 inhibitors increases NO levels as confirmed by increasednitrite/nitrate (NOx) (45). Circulating nitrite/nitrate (NOx) levels not only reflect the level of NO synthesis but also act as a reservoir of alternative substrate for NO synthesis. NO metabolites can be chemically reduced to NO in vivo by haem proteins, such as xanthine oxidoreductase and aldehyde oxidase which exhibit nitrite reductase activity (43). Thus, measurement of circulating NOx could serve as an index of vascular NO synthesis and/or could reflect an ability to synthesize NO via this alternative pathway (46). Physiologic levels of NO exert antiinflammatory effects through suppression of leukocyte activation and microvascular leakage (47, 48). Excessive NO derived from iNOS favors inflammation through the development of Th2- lymphocyte responses (e.g., eosinophilia and IgE ‘priming’ of mast cells) and microvascular leakage. Inflammation may be also exacerbated via formation of cytotoxic peroxynitrite from excess of NO and inflammatory-derived superoxide (47). In our study, concentrations of NO metabolites in the lungs of ALI animals were elevated, probably due to increased expression of iNOS induced by proinflammatory mediators. Sildenafil significantly reduced their levels, suggesting direct involvement of cGMP in inhibition of NO overproduction (47). Resident macrophages are likely to orchestrate neutrophil recruitment through chemotactic factors such as TNF-α and IL-8 (49). TNF-α also stimulates the transcription of iNOS, responsible for NO overproduction (48).
Activation and infiltration of neutrophils and macrophages into the lung tissue participate in increased permeability of the alveolar-capillary membrane, as well. These changes result into generation of the lung edema which can be clearly distinguished from cardiogenic pulmonary edema (50, 51). Integrity of the alveolar-capillary membrane and extent of the lung edema formation can be estimated by lung W/D ratio and protein content in the BAL fluid (52). In the present study, repetitive saline lung lavage used to evoke the ALI model caused a development of lung edema which was expressed by elevation in the lung W/D ratio and total protein content in the BAL fluid whereas sildenafil therapy significantly reduced these parameters.
Apoptosis of alveolar cells is an additional mechanism involved in the pathogenesis of ALI in response to various stimuli via induction of endothelial/epithelial barrier dysfunction. Apoptosis is an important mechanism which maintains normal cellular homeostasis through the removal of damaged or potentially harmful cells. Apoptotic cells which are not quickly removed undergo a secondary necrosis resulting in leakage of cellular content, inflammation and severe tissue injury (53). In the regulation of apoptosis, Bcl2 proteins have an essential role. Bcl-2 family proteins include the anti-apoptotic proteins such as Bcl-2 and Bcl-XL, and pro-apoptotic proteins, such as Bax and BH3 interacting-domain death agonist (Bid). The imbalance between proapoptotic and anti-apoptotic proteins promotes the response to apoptotic stimuli leading to apoptosis and lung injury (54, 55). In addition to Bcl2, caspases, a family of proteases, also have a pivotal role in apoptotic and inflammatory processes. Caspases activate pro-apoptotic pathways leading to DNA degradation and cell death (55) whereas caspase-3 is known as a key executor of apoptosis. Induction of cellular apoptosis is closely associated with ALI. Previous studies showed that intratracheal instillation of LPS elevated apoptotic factors such as caspases and Bax in the lung what caused the progression of ALI (56). In our present study, finding of increased levels of pro-apoptotic markers, caspase-3, and increased apoptotic index in the lung of animals with ALI clearly confirmed the role of apoptosis in the pathogenesis of ALI. DNA damage in the lung tissue was also published in similar model of surfactant depletion in rabbits (26).
Our results showed that sildenafil therapy significantly counteracted elevation of apoptosis markers in the lung of saline lavage-injured animals. Similar results were recently published by Song et al. (22) who found that sildenafil attenuated apoptosis in the lung of neonatal rats with other form of ALI induced by instillation of meconium. There are several mechanisms by which sildenafil can influence the apoptosis. Sildenafil is known to enhance the effect of NO. NO-cGMP pathway has neuroprotective and antiapoptotic effects by increasing the level cGMP. Intracellular accumulation of cGMP in different models of inflammation reduces production of proinflammatory cytokines and reduces oxidative stress (57). We may presume that influencing inflammation and oxidative stress sildenafil could provide anti-apoptotic action also in our experiments.
As a result of complex changes following repetitive saline lung lavage leading to surfactant depletion we could observe an obvious and stable decrease in arterial PaO2 in the ALI group. Whereas the values of PaO2 remained below 20 kPa and index of blood oxygenation (PaO2/FiO2) below 200 mmHg, our results indicate that the used model was suitable to mimic the changes related to the surfactant depletion-associated lung damage as it correlates well with the clinical criteria of acute respiratory distress syndrome (ARDS) in patients (58). Our data are consistent with findings of earlier studies (26, 27, 39) which used the saline lavage-induced surfactant-depleted ALI model. Administration of sildenafil in the present study led to significant improvement of blood gases and other measured respiratory parameters and calculated indexes compared to non-treated animals with ALI, whereas the changes become significant already within 1 hour after sildenafil administration. Similarly, in a piglet model of pulmonary hypertension induced by meconium sildenafil reversed an increase in pulmonary vascular resistance within 1 hour after the infusion, and improved an cardiac output that was not accompanied by deterioration in oxygenation (21). We presume that this rapid improvement in oxygenation can be related to a decrease in the right-to-left pulmonary shunts what is in accordance with the previous study (58).
Of course, we are aware of some limitations of direct applicability of our results. Surfactant depletion is rather rare situation in patients with ARDS, except of near-drowning or RDS in preterm neonates which has some similar features. Repetitive saline lung lavage induces the model of ALI with predominant signs of surfactant depletion, such as immediate hypoxemia, decrease in lung compliance and lung edema formation. Despite several literature sources declared only negligible recruitment and activation of neutrophils and pulmonary vasoconstriction (59, 60), our results showed significant accumulation of neutrophils in the lungs, increased production of cytokines and markers of oxidative stress and increased right-to-left pulmonary shunts. Positive response to sildenafil in this animal model widens future perspectives of PDE5 inhibitors also for situations with prominent surfactant depletion and edema formation.
According to the above mentioned results we can conclude that sildenafil intravenous administration reduced PMNs migration into the lung, mitigated lung injury, decreased concentrations of proinflammatory cytokines, lung edema formation and concentration of proteins in the BAL fluid, prevented apoptosis of lung epithelial cells and improved ventilation in a rabbit model of ALI induced by repetitive saline lung lavage. Taken together, this study provides an experimental evidence of protective activity of sildenafil against the lung injury due to surfactant depletion.
Acknowledgements: Authors thank M. Petraskova, M. Hutko, D. Kuliskova, Z. Remisova, M. Kondekova, M. Letrichova, and A. Resetarova for technical assistance.
This work was supported by projects „Biomedical Center Martin“, ITMS code: 26220120187; „Center of Experimental and Clinical Respirology II“, ITMS code: 26220120034; and by projects APVV-15-0075, and VEGA 1/0356/18.
Conflict of interests: None declared.
REFERENCES
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med 2000; 342: 1334-1349.
- Fanelli V, Vlachou A, Ghannadian S, Simonetti U, Slutsky AS, Zhang H. Acute respiratory distress syndrome: new definition, current and future therapeutic options. J Thorac Dis 2013; 5: 326-334.
- Sweeney RM, McAuley DF. Acute respiratory distress syndrome. Lancet 2016; 388: 2416-2430.
- Cornet AD, Hofstra JJ, Swart EL, Girbes AR, Juffermans NP. Sildenafil attenuates pulmonary arterial pressure but does not improve oxygenation during ARDS. Intensive Care Med 2010; 36: 758-764.
- Cowburn AS, Condliffe AM, Farahi N, Summers C, Chilvers ER. Advances in neutrophil biology: clinical implications. Chest 2008; 134: 606-612.
- Frank JA, Parsons PE, Matthay MA. Pathogenetic significance of biological markers of ventilator-associated lung injury in experimental and clinical studies. Chest 2006; 130: 1906-1914.
- Summers C, Singh NR, White JF, et al. Pulmonary retention of primed neutrophils: a novel protective host response, which is impaired in the acute respiratory distress syndrome. Thorax 2014; 69: 623-629.
- Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol 2004; 202: 145-156.
- Andersson KE. PDE5 inhibitors - pharmacology and clinical applications 20 years after sildenafil discovery. Br J Pharmacol 2018; 175: 2554-2565.
- Simonca L, Tulloh R. Sildenafil in infants and children. Children (Basel) 2017; 4(7): E60. doi: 10.3390/ children4070060
- Ahluwalia A, Foster P, Scotland RS, et al. Antiinflammatory activity of soluble guanylate cyclase: cGMP-dependent down-regulation of P-selectin expression and leukocyte recruitment. Proc Natl Acad Sci USA 2004; 101: 1386-1391.
- von Bulow V, Rink L, Haase H. Zinc-mediated inhibition of cyclic nucleotide phosphodiesterase activity and expression suppresses TNF-α and IL-1b production in monocytes by elevation of guanosine 3‘,5‘-cyclic monophosphate. J Immunol 2005; 175: 4697-4705.
- Kiemer AK, Hartung T, Vollmar AM. cGMP-mediated inhibition of TNF-α production by the atrial natriuretic peptide in murine macrophages. J Immunol 2000; 165: 175-181.
- Wu BN, Chen CW, Liou SF, Yeh JL, Chung HH, Chen IJ. Inhibition of proinflammatory tumor necrosis factor- induced inducible nitric-oxide synthase by xanthine-based 7-[2-[4-(2-chlorobenzene)piperazinyl]ethyl]-1,3-dimethyl- xanthine (KMUP-1) and 7-[2-[4-(4 -nitrobenzene)piperazinyl]- ethyl]-1,3-dimethylxanthine (KMUP-3) in rat trachea: the involvement of soluble guanylate cyclase and protein kinase G. Mol Pharmacol 2006; 70: 977-985.
- Gerassimou C, Kotanidou A, Zhou Z, Simoes DC, Roussos C, Papapetropoulos A. Regulation of the expression of soluble guanylyl cyclase by reactive oxygen species. Br J Pharmacol 2007; 150: 1084-1091.
- Filippov G, Bloch DB, Bloch KD. Nitric oxide decreases stability of mRNAs encoding soluble guanylate cyclase subunits in rat pulmonary artery smooth muscle cells. J Clin Invest 1997; 15: 942-948.
- Farrow KN, Groh BS, Schumacker PT, et al. Hyperoxia increases phosphodiesterase 5 expression and activity in ovine fetal pulmonary artery smooth muscle cells. Circ Res 2008; 102: 226-233.
- Witwicka H, Kobialka M, Siednienko J, Mitkiewicz M, Gorczyca WA. Expression and activity of cGMP-dependent phosphodiesterases is up-regulated by lipopolysaccharide (LPS) in rat peritoneal macrophages. Biochim Biophys Acta 2007; 1773: 209-218.
- Corbin JD, Beasley A, Blount MA, Francis SH. High lung PDE5: a strong basis for treating pulmonary hypertensionwith PDE5 inhibitors. Biochem Biophys Res Commun 2005; 334: 930-938.
- Toward TJ, Smith N, Broadley KJ. Effect of phosphodiesterase-5 inhibitor, sildenafil (Viagra), in animal models of airways disease. Am J Respir Crit Care Med 2004; 169: 227-234.
- Shekerdemian LS, Ravn HB, Penny DJ. Intravenous sildenafil lowers pulmonary vascular resistance in a model of neonatal pulmonary hypertension. Am J Respir Crit Care Med 2002; 165: 1098-1102.
- Song J, Wang YY, Song P, Li LP, Fan YF, Wang YL. Sildenafil ameliorated meconium-induced acute lung injury in a neonatal rat model. Int J Clin Exp Med 2016; 9: 10238-10246.
- Yildirim A, Ersoy Y, Ercan F, et al. Phosphodiesterase-5 inhibition by sildenafil citrate in a rat model of bleomycin-induced lung fibrosis. Pulm Pharmacol Ther 2010; 23: 215-221.
- Cadirci E, Halici Z, Odabasoglu F, et al. Sildenafil treatment attenuates lung and kidney injury due to overproduction of oxidant activity in a rat model of sepsis: a biochemical and histopathological study. Clin Exp Immunol 2011; 166: 374-384.
- Gokakin AK, Deveci K, Kurt A, et al. The protective effects of sildenafil in acute lung injury in a rat model of severe scald burn: A biochemical and histopathological study. Burns 2013; 39: 1193-1199.
- Ronchi CF, Fioretto JR, Ferreira AL, et al. Biomarkers for oxidative stress in acute lung injury induced in rabbits submitted to different strategies of mechanical ventilation. J Appl Physiol (1985) 2012; 112: 1184-1190.
- Fioretto JR, Campos FJ, Ronchi CF, et al. Effects of inhaled nitric oxide on oxidative stress and histopathological and inflammatory lung injury in a saline-lavaged rabbit model of acute lung injury. Respir Care 2012; 57: 273-281.
- Jung SY, Seo YG, Kim GK, Woo JS, Yong CS, Choi HG. Comparison of the solubility and pharmacokinetics of sildenafil salts. Arch Pharm Res 2011; 34: 451-454.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976; 72: 248-254.
- Vodoz JF, Cottin V, Glerant JC, et al. Right-to-left shunt with hypoxemia in pulmonary hypertension. BMC Cardiovasc Disord 2009; 9: 15. doi: 10.1186/1471-2261-9-15
- Wheeler AP, Bernard GR. Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet 2007; 369: 1553-1564.
- Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012; 122: 2731-2740.
- Bhattacharya J, Matthay MA. Regulation and repair of the alveolar-capillary barrier in acute lung injury. Annu Rev Physiol 2013; 75: 593-615.
- Mokra D, Kosutova P, Balentova S, et al. Effects of budesonide on the lung functions, inflammation and apoptosis in a saline-lavage model of acute lung injury. J Physiol Pharmacol 2016; 67: 919-932.
- Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med 2011; 17: 293-307.
- Piechota A, Goraca A. Influence of nuclear factor-kB inhibition on endothelin-1 induced lung edema and oxidative stress in rats. J Physiol Pharmacol 2011; 62: 183-188.
- Verbrugge SJ, Sorm V, Lachmann B. Mechanisms of acute respiratory distress syndrome: role of surfactant changes and mechanical ventilation. J Physiol Pharmacol 1997; 48: 537-557.
- Walski M, Pokorski M, Antosiewicz J, et al. Pulmonary surfactant: ultrastructural features and putative mechanisms of aging. J Physiol Pharmacol 2009; 60 (Suppl. 5): 121-125.
- Kamiyama J, Jesmin S, Sakuramoto H, et al. Assessment of circulatory and pulmonary endothelin-1 levels in a lavage-induced surfactant-depleted lung injury rabbit model with repeated open endotracheal suctioning and hyperinflation. Life Sci 2014; 118: 370-378.
- Kosutova P, Mikolka P, Balentova S, et al. Intravenous dexamethasone attenuated inflammation and influenced apoptosis of lung cells in an experimental model of acute lung injury. Physiol Res 2016; 65 (Suppl. 5): S663-S672.
- Birschmann I, Walter U. Physiology and pathophysiology of vascular signaling controlled by guanosine 3’,5’-cyclic monophosphate-dependent protein kinase. Acta Biochim Pol 2004; 51: 397-404.
- Sebkhi A, Strange JW, Phillips SC, Wharton J, Wilkins MR. Phosphodiesterase type 5 as a target for the treatment of hypoxia-induced pulmonary hypertension. Circulation 2003; 107: 3230-3235.
- Bueno M, Wang J, Mora AL, et al. Nitrite signaling in pulmonary hypertension: mechanisms of bioactivation, signaling, and therapeutics. Antioxid Redox Signal 2013; 18: 1797-1809.
- Kobayashi Y. The regulatory role of nitric oxide in proinflammatory cytokine expression during the induction and resolution of inflammation. J Leukoc Biol 2010; 88: 1157-1162.
- Liu XM, Peyton KJ, Wang X, Durante W. Sildenafil stimulates the expression of gaseous monoxide-generating enzymes in vascular smooth muscle cells via distinct signaling pathways. Biochem Pharmacol 2012; 84: 1045-1054.
- Klinger JR. Plasma nitrite/nitrate levels: a new biomarker for pulmonary arterial hypertension? Eur Respir J 2016; 48: 1265-1267.
- Barnes PJ, Chung KF, Page CP. Inflammatory mediators of asthma: an update. Pharmacol Rev 1999; 50: 515-575.
- Colasanti M, Suzuki H. The dual personality of NO. Trends Pharmacol Sci 2000; 21: 249-252.
- Brigham KL, Meyrick B. Endotoxin and lung injury. Am Rev Respir Dis 1986; 133: 913-927.
- Zemans RL, Matthay MA. Bench-to-bedside review: the role of the alveolar epithelium in the resolution of pulmonary edema in acute lung injury. Crit Care 2004; 8: 469-477.
- Schmickl CN, Biehl M, Wilson GA, Gajic O. Comparison of hospital mortality and long-term survival in patients with acute lung injury/ARDS vs cardiogenic pulmonary edema. Chest 2015; 147: 618-625.
- Kosutova P, Mikolka P, Kolomaznik M, Rezakova S, Calkovska A, Mokra D. Effects of roflumilast, a phosphodiesterase-4 inhibitor, on the lung functions in a saline lavage-induced model of acute lung injury. Physiol Res 2017; 66 (Suppl. 2): S237-S245.
- Yun JH, Henson PM, Tuder RM. Phagocytic clearance of apoptotic cells: role in lung disease. Expert Rev Respir Med 2008; 2: 753-765.
- Wang L, Ye Y, Su HB, Yang JP. The anesthetic agent sevoflurane attenuates pulmonary acute lung injury by modulating apoptotic pathways. Braz J Med Biol Res 2017; 50: e5747. doi: 10.1590/1414-431X20165747
- Inoue S, Browne G, Melino G, Cohen GM. Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ 2009; 16: 1053-1061.
- Lin WC, Chen CW, Huang YW, et al. Kallistatin protects against sepsis-related acute lung injury via inhibiting inflammation and apoptosis. Sci Rep 2015; 5: 12463. doi: 10.1038/srep12463
- Szczypka M, Obminska-Mrukowicz B. The effects of selective and nonselective phosphodiesterase inhibitors on phagocytic cells in mice. Immunopharmacol Immunotoxicol 2010; 32: 507-513.
- Kelly LE, Ohlsson A, Shah PS. Sildenafil for pulmonary hypertension in neonates. Cochrane Database Syst Rev 2017; 8: CD005494. doi: 10.1002/14651858.CD005494.pub4
- Matute-Bello G, Frevert CW, Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 2008; 295: L379-L399.
- Ballard-Croft C, Wang D, Sumpter LR, Zhou X, Zwischenberger JB. Large-animal models of acute respiratory distress syndrome. Ann Thorac Surg 2012; 93: 1331-1339.
A c c e p t e d : October 30, 2018