INHIBITION OF SOLUBLE EPOXIDE HYDROLASE
OFFERS PROTECTION AGAINST FRUCTOSE-INDUCED DIABETES
AND RELATED METABOLIC COMPLICATIONS IN RATS
INTRODUCTION
The 1-trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) as seen in Fig. 1, is the most potent, metabolically stable and with appreciable pharmacokinetic profile among other soluble epoxidehydrolase (sEH) enzyme inhibitors (1). sEH is the enzyme responsible for degradations of epoxyeicosatrienoic acids (EETs) to the corresponding biologically inactive dihydroxyeicosatrienoic acids (DHETs). This enzyme is expressed in different tissues and organs including kidney, liver, intestine, vascular smooth muscles, astrocytes and neuronal cells. The sEH inhibition effectively raises EETs level and decreases DHETs level, which may be translated to therapeutic potentials for several ailment indications (2). It has been reported that sEH inhibitors can treat dyslipidemia, stroke, diabetes, hypertension, pain, eye diseases, immunological disorders and neurological diseases (3). EETs are cytochrome p-450 metabolites of arachidonic acid that are produced by vascular endothelium (4). EETs are lipid mediators that are involved in regulating blood pressure, inflammatory cascades, and glucose homeostasis (5). Significant amount of EETs are produced in the pancreas of human and rat providing evidence that they may regulate b-cell function (6). Luo et al. provided the first evidence that sEH blockade promotes insulin secretion in vivo, resulting in the prevention of hyperglycemia in diabetic mice. They also showed sEH blockade by t-AUCB augmented glucose-stimulated insulin secretion (GSIS) in islets (7). Similarly, sEH blockade by t-AUCB significantly reduced islet cell apoptosis relative to that in control mice (8). These studies suggest that sEH is involved in b-cells apoptosis. Since sEH is expressed in the human pancreas and mouse β-cells (7, 9), it is possible that blockade of sEH will be an important means of increasing the bioavailability of EETs in β-cells. Extensive research has focused on whether increasing EETs, sEH blockade, or sEH gene deletion has beneficial effect against type 2 diabetes mellitus (T2DM). Xu et al. reported earlier that CYP2J3 gene delivery in vivo increased EETs levels and reversed insulin resistance in diabetic db/db mice and mice with fructose induced T2DM. Moreover, the over-expression of CYP2J3 prevented decreases in insulin receptor signaling in various organs including heart, kidney, and aorta of T2DM rats (10). Zeldin et al. published significant amount of endogenous EETs in the human and rat pancreas (6). EETs are potent mediators of insulin release in isolated rat islets (11). Literature also highlighted that sEH inhibition possesses glucose lowering effects (12), thus resulting in improvement in diabetes and glucose homeostasis probably by increasing in the size, function of β-cells and regulation of adipocytes (2, 13). On the other hand resultant EETs preservation is considered a pivotal component playing its role in homeostasis of the glycemic system. EETs and sEHIs do not directly modify insulin levels, insulin sensitivity or blood glucose levels of normal animals. These mediate a positive interference in dysregulated glucose metabolism. Keeping all these in consideration, it has been hypothesized that sEHIs inhibition to preserve EETs with a novel inhibitor results in improvement of experimentally-induced metabolic alteration in animals.
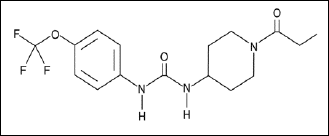 |
Fig. 1. Structure of 1-trifluoromethoxyphenyl-3-(1-propionylpiperidine-4-yl) urea (TPPU). |
Although the potential of sEH inhibitors for the treatment of diabetes appears promising, however, it has not been fully established. Hence, this study was aimed at exploring the role of 1-trifluoromethoxyphenyle-3-(1-propionylpiperididin-4-yl) urea (TPPU), a sEH inhibitor, as a preventive and/or therapeutic agent in fructose induced diabetic rat’s model.
MATERIALS AND METHODS
Chemicals
Fructose, hydrogen peroxide, hematoxylin, xylene and ethanol were procured from Sigma Aldrich (Germany). TPPU was procured from Synthia laboratories Davis, California, USA. TPPU was solubilized in 2% Tween 80 and 1.5% ethanol to prepare a stock solution of 20 ml for further dosing. Similarly vehicle was prepared by excluding TPPU in same volume. Sodium bicarbonate, potassium dihydrogen phosphate, sodium chloride, isoflurane, formaldehyde solution and calcium chloride were purchased from Merck (Darmstadt, Germany). The chemicals used in this study were of analytical grades and their solutions were prepared in distilled water.
Animals
Sprague-Dawley (SD) rats (150 – 200 g) of both sexes were housed at 23 – 25°C in Animal House of Aga Khan University, Karachi, Pakistan. The animals were housed under standard laboratory conditions with 12-h light/dark cycle with access to food and water ad libitum.
Experimental protocol was reviewed and approved by the Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council (National Research Council, 1996) and Institutional Ethics Committee.
Fructose induced diabetes
Rats were kept separately in cages (3 in each cage) of each group. Diabetes was induced in normal SD by adding 25% fructose in drinking water that was prepared every day and fed for 12 weeks (14).
Treatment protocol
SD rats (150 – 200 g) were divided into four groups, each containing 10 animals. Animals in group-1 received standard diet and tap water. Animals of the group-2 received standard diet, 25% fructose in drinking water and were treated with vehicle only. The group-3 animals received standard diet, 25% fructose in drinking water and TPPU as a test drug orally at the dose of 2 mg/kg body. The group-4 animals received standard diet, 25% fructose in drinking water and metformin as a reference drug orally at the dose of 150 mg/kg. The treatment of animals was continued for 12 weeks. Water intake and food of all the experimental animals were daily measured and at the end of study animals were fasted for 12 – 14 hours, anesthetized with isoflurane, dissected and blood and tissue sample were collected.
Histological studies
The thoracic aorta, liver and pancreas of the animals were isolated and fixed in 10% buffered neutral formalin solution for 24 to 48 hours. Using metallic blocks, the isolated tissues were embedded in molten paraffin followed by covering with flexible plastic molds. The samples were then kept under freezing plates for allowing solidification of molten paraffin. The fixed tissues were subjected to cross sectioning into 5 µm thick sections. Staining of the sections was achieved with eosin and hematoxyline (H & E) and the sections were mounted on light microscope and microscopic architectures of all the tissues were studied (15).
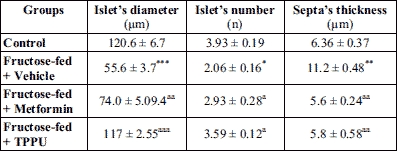
Histomorphometric analysis was conducted using Olympus research microscope (OlympusWF10X, Japan) with a linear scale-ocular micrometer and an area-measuring ocular grid inserted in the eyepiece. Different slides form each group were studied for morphometric measurements for (a) the diameter of the islets and (b) the number of islets in pancreas sections in each pancreatic section at 400 × magnification using ocular grid in the eyepiece (×10). The numbers of islets were quantified in these sections at ×40. The numbers of islets were expressed as N/10 mm2 of the pancreatic parenchyma. The major axis (a) and minor axis (b) at right angles to the major axis of the islets were measured. The diameter of the islets (DI) was calculated using following equation DI = √ab. Septa thickness was measured using the major axis and minor axis (16). The bar scale was 50 µm.
Immunohistochemistry
Imunohistochemistry was performed on 5 µm thick sections of pancreas. Xylene was used for the deparaffinizing of paraffin fixed sections followed by their rehydration with descending grades of alcohol. Treatment with hydrogen peroxide (0.3%) was used for suppressing the non-specific binding sites for antibodies of the sections. This was followed by their incubation with primary antibodies and then secondary antibodies. Staining of the nuclei was achieved with hematoxylin followed by their dehydration with graded alcohol. The nuclei were cleared with xylene and then were mounted on dibutylphthalate polystyrene xylene (17). Insulin immunohistochemical localization was visualized and studied by keeping the slides under light microscope.
Measurement of biochemical parameters
Automated Analyzer, Roche Cobas c111 was used for the analysis of serum samples. Optics, reagents and calibrators sourced from Cobas Integra compatible with Roche Cobas c111 were used for the determination of total serum cholesterol (TC), triglycerides (TG), fasting glucose, total bilirubin, low-density lipoprotein (LDL), high-density lipoprotein (HDL), direct bilirubin, alkaline phosphatase (ALP), uric acid, alanine aminotransferase (ALT), creatinine and urea levels in serum.
Measurement of serum insulin level
The serum insulin levels were assessed by means of an enzyme-linked immunosorbent assay (ELISA) by using commercially available kits. Samples were assayed as per manufacturer’s recommendation. Commercially available 96 well insulin ELISA kit (Rat/Mouse Insulin ELISA EZRMI-13K- Merck Millipore, Germany) was used for quantitative determination of serum insulin concentrations. The insulin containing in the samples reacts with both peroxidase-conjugated anti-insulin antibodies and anti-insulin antibodies attached to titration well. Quenching of reaction was achieved by the addition of acid indicated by colorimetric endpoint that is read spectrophotometrically at 450 nm with the help of a microplate reader (18). All reagents and solutions were prepared at the day of assay as per manufacturer instructions. The serum samples were set out and allowed to thaw at room temperature. All incubations were carried out at room temperature. All standards, controls and unknown samples were run in triplicate for validity of results.
Statistical analysis
Results are expressed as mean ± SEM. Unpaired student’s t-test and one-way ANOVA followed by Dunnett’s test were used for statistical analysis of data. P < 0.05, was considered statically significant.
RESULTS
Effect on the body weight
Initial body weights (g) of normal, fructose-fed (FF), FF + metformin (150 mg/kg) and FF + TPPU (2 mg/kg) treated groups were (mean ± SEM, n = 10/group) 180 ± 12, 200 ± 14, 190 ± 7 and 183 ± 16 g, respectively. Fructose feeding resulted in a significant (P < 0.001) increase in the body weight gain compared to normal rats with respective values of 310 ± 17 versus 200 ± 14 g. At 12th week of the study, administration of TPPU to the fructose-fed animals caused a marked (P < 0.05) decrease in the body weight gain compared to the body weight of only fructose fed rats (249 ± 11 versus 310 ± 17 g) similar to the effect of metformin (270 ± 16 versus 310 ± 17 g, P < 0.05).
Biochemical parameters
As shown in Table 1, the vehicle treated animals receiving fructose in drinking water for 12 weeks showed a significant increase (P < 0.001) in the serum levels of glucose, insulin, lipid serum markers (TC, TG, LDL), renal markers (uric acid and urea) and hepatic markers (ALT and ALP) compared to vehicle treated control animals receiving drinking water only. Treatment of animals with TPPU (2 mg/kg p.o.) significantly (P < 0.001) reduced serum glucose, insulin, lipid parameters (TC, TG, LDL), renal markers (uric acid and urea) and hepatic markers (ALT and ALP) compared to only fructose-fed vehicle treated animals as shown in Table 1. Metformin used as reference compound caused similitude significant decrease in the biochemical parameters in rats as seen with TPPU treatment (Table 1). No changes were observed in the serum levels of direct bilirubin, total bilirubin and creatinine.
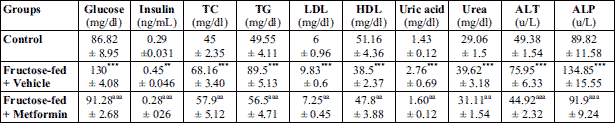
TC, total cholesterol; TG, triglycerides; LDL, low-density lipoprotein HDL, high-density lipoprotein.
As shown in Fig. 2, homeostatic model assessment of insulin resistance (HOMA-IR) was markedly increased in fructose-fed untreated rats compared to the normal rats. Treatment of animals with TPPU or metformin significantly decreased (P < 0.01) insulin resistance compared vehicle treated and fructose-fed animals.
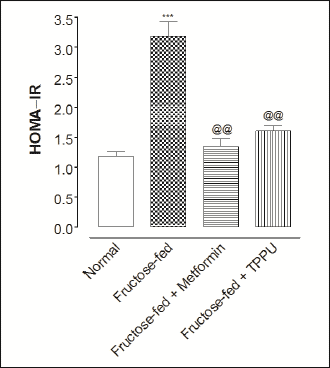 |
Fig. 2. Effect of TPPU treatment on HOMA-IR index in animal model of fructose induced diabetes. Animals were fed on fructose for 12 weeks. Metformin (150 mg/kg) or TPPU (2 mg/kg) were administrated orally for 12 weeks. Animal either received normal food and water (Normal) or fructose rich feed and vehicle (Fructose-fed) or metformin (Metformin) or TPPU (TPPU). Data represent mean ± SEM of 10 determinants. Different superscript letters indicate significant differences between groups @@P < 0.01 and ***P < 0.001; * shows a comparison of normal versus fructose-fed control group (unpaired Student’s t-test), while @ shows a comparison of fructose-fed control group versus TPPU or metformin treated and fructose fed animals (one-way ANOVA followed by Dunnett’s test). |
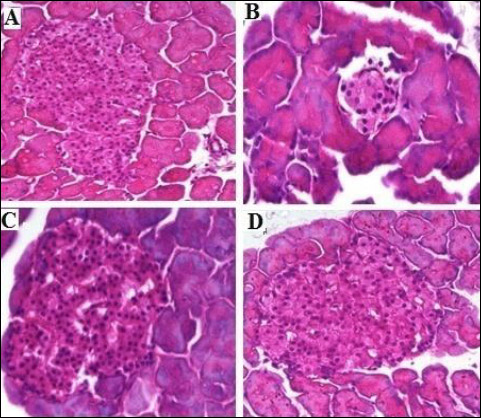 |
Fig. 3. Photomicrographs of histopathology of the pancreatic tissues showing the effect of TPPU treatment in fructose-fed rats. (A): normal rats, (B): fructose-fed rats, (C): metformin treated fructose-fed rats and (D): TPPU treated fructose-fed rats. Magnification × 400, scale bar displays 50 µm. |
Microscopic observations of pancreatic tissues
The H&E stained tissue sections of pancreas, microscopic examination of the normal rats showed typical histological structures with normal islets. In contrast, fructose fed untreated group showed marked shrinkage in islets cells, decreased number of islets as well as thickening of the septa were clearly seen when compared with normal rats. Treatment with TPPU (2 mg/kg) showed expansion of islets, significantly increased the numbers of islets cells and reduced septa thickness near to normal, similar to the effect of metformin (150 mg/kg) as seen in Figs. 3-5.
As shown in Table 2 and Fig. 3 and 5, the mean values of islet diameter of different groups were measured. There was significant difference (P < 0.001) in islet diameter of normal control and fructose-fed vehicle treated animals. Treatment of animals with TPPU or metformin caused marked (P < 0.001) increase in islet diameter.
The mean numbers of islets of different groups were counted and compared. The results revealed a significant reduction (P < 0.05) in the number of islet in the vehicle treated diabetic animals. However, the numbers of islets were significantly increased in the TPPU or metformin treated animals (Table 2). There was a significant difference (P < 0.05) between the septa of normal and untreated group (Fig. 4). The TPPU treatment reduced septa thickness near to the normal (P < 0.01), similar to the effect of metformin administered animals, micrometry measurements are shown in Table 2.
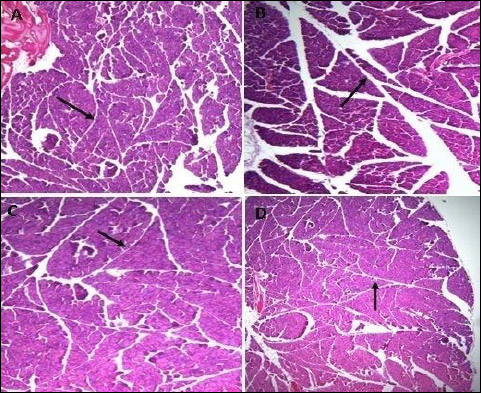 |
Fig. 4. Photomicrographs of pancreatic septa, indicated by arrows, showing the effect of TPPU treatment in fructose-fed rats. (A): normal rats, (B): fructose-fed rats, (C): metformin treated fructose-fed rats and (D): TPPU treated fructose-fed rats. Magnification × 400, scale bar displays 50 µm. |
Microscopic observations of hepatic tissues
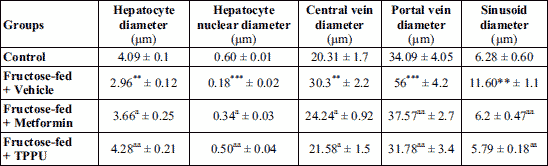
As shown in Figs. 5-7, in normal control animals, the hepatic stained specimen showed normal cellular patterns. Clear portal vein and central vein were seen. The hepatocytes were normally arranged in plates or cords, radiating from central vein toward portal areas. The sinusoidal spaces between plates or cords were normal. Mostly cells with large round nucleolus were identified. H&E stained sections of diseased untreated group of hepatic sections revealed changed cellular architecture, hepatocyte shrinkage, central and portal vein dilatation were seen at various fields.
The liver specimens of TPPU or metformin treated group revealed normal hepatic pattern and cellular architecture patterns. No major structural variations of the liver features were seen. The hepatic architectures were similar to normal group as shown in Figs. 6-8.
The mean values of portal vein diameter of hepatic tissues of various groups were measured. Diseased and TPPU or metformin treated animals with respect to portal vein diameter revealed significant mean difference with P values of each P < 0.01. There was significant difference (P < 0.01) between the normal and diseased vehicle treated group.
The sinusoid diameters of rat’s liver of various groups were measured. Significant mean difference (P < 0.01) vehicle treated and fructose fed animals compared to TPPU or metformin treated and fructose fed animals with respect to sinusoidal diameter. There was significant difference between the control and fructose fed and vehicle treated animals with P value ***P < 0.001. Hepatic micrometry measurements are shown in Table 3. Treatment of animals with TPPU or metformin attenuated fructose-induced deleterious hepatic changes (Figs. 6-8 and Table 3).
Immunohistochemistry
The immunohistochemical staining with anti-insulin antibody confirmed a marked reduction in beta cell of islet of Langerhans in vehicle treated fructose-fed rats. The pancreas sections from the TPPU treated group showed marked improvement when compared with untreated fructose-fed group. Marked difference was observed between beta cell of untreated fructose-fed rats and healthy control rats as shown in Fig. 5.
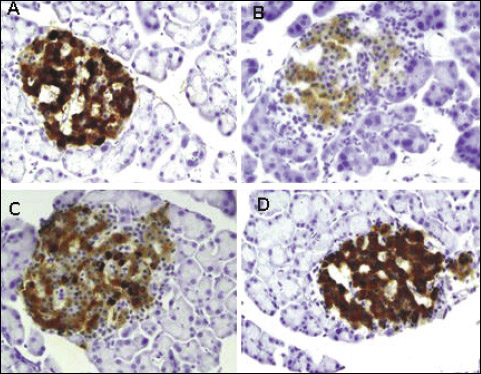 |
Fig. 5. Immunohistochemical staining with anti-insulin antibody of the pancreatic tissues showing the effect of TPPU treatment in fructose-fed rats. (A): normal rats, (B): fructose-fed rats, (C): metformin treated fructose-fed rats and (D): TPPU treated fructose-fed rats. Magnification × 400, scale bar displays 50 µm. |
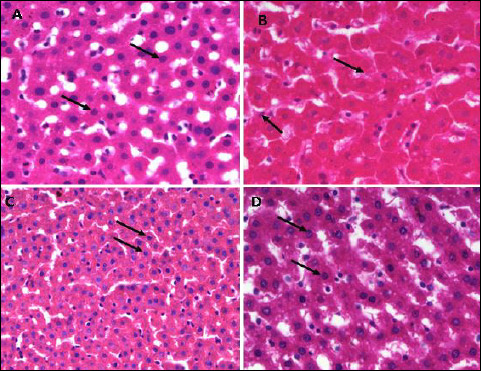 |
Fig. 6. Photomicrographs of the liver tissues showing the effect of TPPU treatment on hepatocytes, indicated by arrows, in fructose-fed rats. (A): normal rats, (B): fructose-fed rats, (C): metformin treated fructose-fed rats and (D): TPPU treated fructose-fed rats. Magnification × 400, scale bar displays 50 µm. |
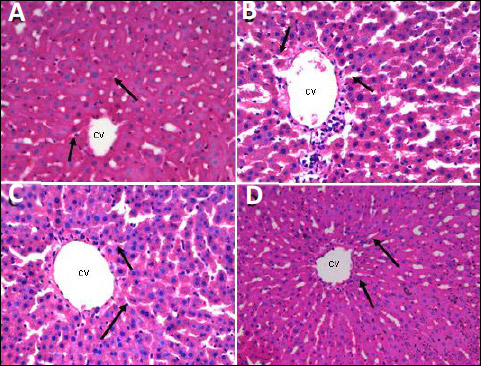 |
Fig. 7. Photomicrographs of central vein (CV) and sinusoid, indicated by arrows, of liver tissues showing the effect of TPPU treatment in fructose-fed rats. (A): normal rats, (B): fructose-fed rats, (C): metformin treated fructose-fed rats and (D): TPPU treated fructose-fed rats. Magnification × 400, scale bar displays 50 µm. |
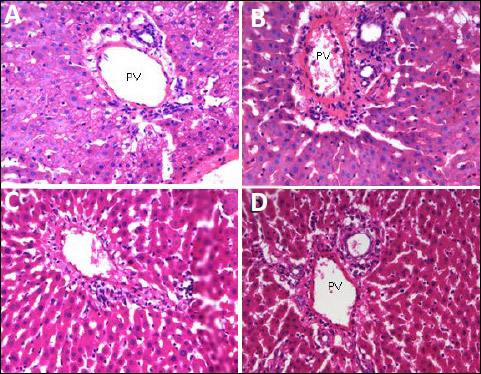 |
Fig. 8. Photomicrographs of portal vein (PV) of the liver tissues showing the effect of TPPU treatment in fructose-fed rats. (A): normal rats, (B): fructose-fed rats, (C): metformin treated fructose-fed rats and (D): TPPU treated fructose-fed rats. Magnification × 400, scale bar displays 50 µm. |
DISCUSSION
Data from our current study revealed that rats receiving fructose in drinking water for 12 weeks developed classic symptoms of metabolic such as weight gain, hyperglycemia, hyperinsulinemia, and hyperlipidemia and insulin resistance, while intake of low caloric diet is known for its cardiometabolic protective effects (19). There was significant increase of ALT, ALP, urea and uric acid. Together, these findings are consistent with previous studies (20). TPPU prevented these changes most likely causing down regulation of insulin receptors due to decrease in mRNA expression in skeletal muscle (21). Fructose over consumption results in increased body weight, decreased insulin secretion, reduced leptin production and increased levels of circulating nonesterified fatty acids. The higher proportions of nonesterified fatty acids may reduce insulin sensitivity by increasing the intramyocellular lipid content and also pose deleterious effects on β-cell functions. Increased portal delivery of nonesterified fatty acids, particularly from visceral adipose tissue, could also lead to impaired carbohydrate metabolism. As it is known that elevated portal nonesterified fatty acid concentrations causes increase in hepatic glucose, VLDL and triacylglycerol production. High fructose consumption is known to develop hypertriacylglycerolemia. Also, insulin resistance and reduced insulin binding have been reported in hypertriacylglycerolemic condition which may in part explains the development of insulin resistance in fructose-fed animal model (22). Moreover, fructose intake can also cause impaired endothelial function through over production of uric acid. Uric acid prevents the nitric oxide vasodilation ability (23) and decreasing the insulin potentials for glucose uptake into the tissues. This may leads to hyperglycemia (24). EETs and sEHIs have a beneficial effect in insulin resistance hyperglycemia states, they do not modify insulin levels, insulin sensitivity or blood glucose levels of normal animals. However, EETs and sEHIs effectively correct dysregulated glycemic condition (25). Data from animal studies have shown that sEH inhibition has proven glucose lowering effects (12). sEH inhibitors have been shown to improve diabetes and glucose homeostasis probably by increasing the size, function of β-cells and regulation of adipocytes (13).
The results of the current study suggest that in contrast to untreated animals, TPPU or metformin treated animals exhibited improved insulin levels, indicating sEH inhibition a potential target of insulin regulation in diabetic model (26), metformin is known for its beneficial effects in metabolic syndrome (27). Increasing EETs concentrations improves insulin resistance in fructose fed rats through prevention of fructose-induced decline in phosphorylation of AMP-activated protein kinase (AMPK) and insulin receptor signaling in muscles, liver, kidneys, aorta and heart (10).
Diet rich in fructose contents increases the triglycerides levels by decreasing clearance of VLDL triacylglycerol and hepatic de novo lipogenesis stimulation (28). Moreover, fructose also increases the activity of sterol regulatory element binding protein (SREBP). SREBP is a transcription factor which initiates the biosynthesis of cholesterol and regulating fatty acid (29). EETs also appear to contribute in the regulation of lipid homeostasis (30), enhancing EETs level through inhibition of sEH was found to lower total cholesterol, LDL cholesterol and increase of HDL cholesterol, indicating their beneficial effects on atherosclerotic process (31). In the current study, the TPPU treatment lowered serum cholesterol, triglycerides and LDL, while HDL levels were elevated in rats similar to earlier reports, having a protective role in cardiometabolic complications (32).
sEH inhibitors improve diabetes probably by increasing the size and function of β-cells (13). In the current investigation, the histopathological and immunohistochemistry analysis revealed that similar to metformin, TPPU displayed regenerative effect on β-cells of the pancreas in diabetic rats. Moreover, there was an effective increase in the number of islet of Langerhan’s in the pancreas of diabetic rats treated with TPPU compared with untreated diabetic animals. Immunohistochemical staining with anti-insulin antibody confirmed a marked reduction in insulin secreting cells in diabetic vehicle treated animals which were markedly improved with TPPU pre-treatment. The results of the present study revealed regeneration in the pancreatic β-cells in diabetic rats treated with the TPPU. The islet cell regeneration in diabetic rats could be due TPPU as a sEH blocker and EETS enhancer. EETs stimulate cell angiogenesis and proliferation mainly by stimulation of the EGF receptor which results in the activation of protein kinase B (Akt) and an up-regulation in cyclin D1 expression (33), activation of a cyclic adenosine monophosphate (cAMP) dependent pathway by EETs, especially 11,12-EET (34) and activation of p38 mitogen-activated protein kinase (p38 MAPK) pathway by 8,9-EET and 11,12-EET (35, 36). The beneficial effect of TPPU observed in our study may have occurred via regeneration by stimulating the angiogenesis, endothelial cell proliferation and neovascularization through EETs enhancement.
Fructose intake is coupled with increased production of reactive oxygen species leading to increased oxidative stress in the body. Fructose induced oxidative stress is implicated to cause deep and chronic liver injuries (37). Fructose and its metabolites directly and indirectly cause oxidative stress, chronic inflammation and may lead to tissues damages (38). Liver enzymes such as alanine aminotransferase (ALT) and alkaline phosphatase (ALP) are known markers for the assessment of the functional integrity of the liver cells (39, 40). The deleterious liver changes in diabetic vehicle treated animals were evident in our study. The histopathological assessment of hepatic tissue specimen fructose induced diabetic rats and treated with TPPU showed tremendous improvement in the structural component of hepatic tissues comparable to control vehicle treated animals. Endoplasmic reticulum stress leads to the induction of inflammatory responses and eventually cell damage. TPPU attenuates ER stress and is capable of preventing liver damage and helps to maintain structural integrity of liver (41).
These data show that TPPU possesses protective effect against fructose-induced hyperglycemia, hyperlipidemia and impaired renal and hepatic serum markers. These effects are possibly achieved by inhibition of sEH activity, thus preserving EETs levels. Current investigation reveals TPPU as a novel potential drug candidate for the management of diabetes and its associated complications.
Acknowledgement: The authors extend their appreciation to the Deanship of Scientific Research at King Saud University Riyadh for funding this work through research group No (RG 1439-002).
Conflict of interests: None declared.
REFERENCES
- Liu J-Y, Lin Y-P, Qiu H, et al. Substituted phenyl groups improve the pharmacokinetic profile and anti-inflammatory effect of urea-based soluble epoxide hydrolase inhibitors in murine models. Eur J Pharm Sci 2013; 48: 619-627.
- Morisseau C, Hammock BD. Epoxide hydrolases: mechanisms, inhibitor designs, and biological roles. Annu Rev Pharmacol Toxicol 2005; 45: 311-333.
- Shen HC. Soluble epoxide hydrolase inhibitors: a patent review. Expert Opin Ther Pat 2010; 20: 941-956.
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Res 1996; 78: 415-423.
- Huang H, Weng J, Wang M-H. EETs/sEH in diabetes and obesity-induced cardiovascular diseases. Prostaglandins Other Lipid Mediat 2016; 125: 80-89.
- Zeldin DC, Foley J, Boyle JE, et al. Predominant expression of an arachidonate epoxygenase in islets of Langerhans cells in human and rat pancreas. Endocrinology 1997; 138: 1338-1346.
- Luo P, Chang H-H, Zhou Y, et al. Inhibition or deletion of soluble epoxide hydrolase prevents hyperglycemia, promotes insulin secretion, and reduces islet apoptosis. J Pharmacol Exp Ther 2010; 334: 430-438.
- Chen L, Fan C, Zhang Y, et al. Beneficial effects of inhibition of soluble epoxide hydrolase on glucose homeostasis and islet damage in a streptozotocin-induced diabetic mouse model. Prostaglandins other Lipid Mediat 2013; 104: 42-48.
- Enayetallah AE, French RA, Barber M, Grant DF. Cell-specific subcellular localization of soluble epoxide hydrolase in human tissues. J Histochem Cytochem 2006; 54: 329-335.
- Xu X, Zhao CX, Wang L, et al. Increased CYP2J3 expression reduces insulin resistance in fructose-treated rats and db/db mice. Diabetes 2010; 59: 997-1005.
- Falck JR, Manna S, Moltz J, Chacos N, Capdevila J. Epoxyeicosatrienoic acids stimulate glucagon and insulin release from isolated rat pancreatic islets. Biochem Biophys Res Commun 1983; 114: 743-749.
- Shen HC, Hammock BD. Discovery of inhibitors of soluble epoxide hydrolase: a target with multiple potential therapeutic indications. J Med Chem 2012; 55: 1789-1808.
- Henquin J-C. Pathways in beta-cell stimulus-secretion coupling as targets for therapeutic insulin secretagogues. Diabetes 2004; 53: S48-S58.
- Mamikutty N, Thent ZC, Sapri SR, et al. The establishment of metabolic syndrome model by induction of fructose drinking water in male Wistar rats. BioMed Res Int 2014; 2014: 263897. doi: 10.1155/2014/263897
- Suman RK, Ray Mohanty I, Borde MK, Maheshwari U, Deshmukh Y. Development of an experimental model of diabetes co-existing with metabolic syndrome in rats. Adv Pharmacol Sci 2016; 2016: 9463476. doi: 10.1155/2016/9463476
- Adeyemi DO, Komolafe OA, Adewole OS, Obuotor EM, Abiodun AA, Adenowo TK. Histomorphological and morphometric studies of the pancreatic islet cells of diabetic rats treated with extracts of Annona muricata. Folia Morphol (Warsz) 2010; 69: 92-100.
- Abdul-Hamid M, Moustafa N. Protective effect of curcumin on histopathology and ultrastructure of pancreas in the alloxan treated rats for induction of diabetes. J Basic Appl Zool 2013; 66: 169-179.
- Csont T. Determination of serum insulin level by ELISA. Practical Course: Basic Biochemical Methods and Ischemic Heart Models Hungary-Romania Cross Border Co-operation Programme 2007; 2013: 3-4.
- Zubrzycki A, Cierpka-Kmiec K, Kmiec Z, Wronska A. The role of low-calorie diets and intermittent fasting in the treatment of obesity and type-2 diabetes. J Physiol Pharmacol 2018; 69: 663-683.
- Mahmoud AA, Elshazly SM. Ursodeoxycholic acid ameliorates fructose-induced metabolic syndrome in rats. PLoS One 2014; 9: e106993. doi: 10.1371/journal.pone.0106993
- Catena C, Cavarape A, Novello M, Giacchetti G, Sechi LA. Insulin receptors and renal sodium handling in hypertensive fructose-fed rats. Kidney Int 2003; 64: 2163-2171.
- Elliott SS, Keim NL, Stern JS, Teff K, Havel PJ. Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr 2002; 76: 911-922.
- Gromotowicz-Poplawska, A, Kloza M, Aleksiejczuk M, et al. Nitric oxide as a modulator in platelet- and endothelium-dependent antithrombotic effect of eplerenone in diabetic rats. J Physiol Pharmacol 2019; 70: 187-198.
- Tappy L, Le K-A. Metabolic effects of fructose and the worldwide increase in obesity. Physiological Rev 2010; 90: 23-46.
- Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol 2013; 53: 37-58.
- Luria A, Bettaieb A, Xi Y, et al. Soluble epoxide hydrolase deficiency alters pancreatic islet size and improves glucose homeostasis in a model of insulin resistance. Proc Natl Acad Sci USA 2011; 108: 9038-9043.
- Zawiejska A, Wender-Ozegowska E, Grewling-Szmit K, Brazert M, Brazert J. Short-term antidiabetic treatment with insulin or metformin has a similar impact on the components of metabolic syndrome in women with gestational diabetes mellitus requiring antidiabetic agents: results of a prospective, randomised study. J Physiol Pharmacol 2016; 67: 227-233.
- Tappy L, Le KA, Tran C, Paquot N. Fructose and metabolic diseases: new findings, new questions. Nutrition 2010; 26: 1044-1049.
- Basciano H, Federico L, Adeli K. Fructose, insulin resistance, and metabolic dyslipidemia. Nutr Metab (Lond) 2005; 2: 5. doi: 10.1186/1743-7075-2-5.
- Roche C, Besnier M, Cassel R, et al. Soluble epoxide hydrolase inhibition improves coronary endothelial function and prevents the development of cardiac alterations in obese insulin-resistant mice. Am J Physiol Heart Circ Physiol 2015: 308: H1020-H1029.
- Zhang L-N, Vincelette J, Cheng Y, et al. Inhibition of soluble epoxide hydrolase attenuated atherosclerosis, abdominal aortic aneurysm formation, and dyslipidemia. Arterioscler Thromb Vasc Biol 2009; 29: 1265-1270.
- EnayetAllah AE, Luria A, Luo B, et al. Opposite regulation of cholesterol levels by the phosphatase and hydrolase domains of soluble epoxide hydrolase. J Biol Chem 2008; 283: 36592-36598.
- Michaelis UR, Fisslthaler B, Medhora M, Harder D, Fleming I, Busse R. Cytochrome P450 2C9-derived epoxyeicosatrienoic acids induce angiogenesis via cross-talk with the epidermal growth factor receptor (EGFR). FASEB J 2003; 17: 770-772.
- Michaelis UR, Falck JR, Schmidt R, Busse R, Fleming I. Cytochrome P4502C9-derived epoxyeicosatrienoic acids induce the expression of cyclooxygenase-2 in endothelial cells. Arterioscler Thromb Vasc Biol 2005; 25: 321-326.
- Pozzi A, Macias-Perez I, Abair T, et al. Characterization of 5, 6-and 8, 9-epoxyeicosatrienoic acids (5, 6-and 8, 9-EET) as potent in vivo angiogenic lipids. J Biol Chem 2005; 280: 27138-27146.
- Potente M, Michaelis UR, Fisslthaler B, Busse R, Fleming I. Cytochrome P450 2C9-induced endothelial cell proliferation involves induction of mitogen-activated protein (MAP) kinase phosphatase-1, inhibition of the c-Jun N-terminal kinase, and up-regulation of cyclin D1. J Biol Chem 2002; 277: 15671-15676.
- Li S, Tan H-Y, Wang N, et al. The role of oxidative stress and antioxidants in liver diseases. Int J Mol Sci 2015; 16: 26087-26124.
- Zhang D-M, Jiao R-Q, Kong L-D. High dietary fructose: direct or indirect dangerous factors disturbing tissue and organ functions. Nutrients 2017; 9: 335. doi: 10.3390/nu904033
- Adaramoye OA, Osaimoje DO, Akinsanya AM, Nneji CM, Fafunso MA, Ademowo OG. Changes in antioxidant status and biochemical indices after acute administration of artemether, artemether-lumefantrine and halofantrine in rats. Basic Clin Pharmacol Toxicol 2008; 102: 412-418.
- Eze E, Dawud F, Zainab A, Jimoh A, Malgwi I, Isa A. Preliminary studies of effects of vitamin C and zinc on some liver enzymes in alloxan-induced diabetic wistar rats. Asian J Med Sci 2012; 4: 17-22.
- Harris TR, Bettaieb A, Kodani S, et al. Inhibition of soluble epoxide hydrolase attenuates hepatic fibrosis and endoplasmic reticulum stress induced by carbon tetrachloride in mice. Toxicol Appl Pharmacol 2015; 286: 1-2-111.
A c c e p t e d : October 30, 2020