During the last decade, numerous studies have identified nitric oxide (NO) as a molecule involved in different homeostatic control mechanisms, including the mechanisms of temperature regulation (14). Soszynski et al. (15) have demonstrated that pretreatment with L-NAME (nonselective inhibitor of nitric oxide synthase, nitro-L-arginine-methyl ester) markedly inhibited the fever induced by LPS. These results are consistent with a recent report by Kozak et al. (16), who demonstrated that iNOS and nNOS deficiency resulted in reduction of fever induced by endotoxin. Moreover, it has been shown that L-NAME given ip was able to inhibit open field-induced rise in Tb in rat (15). Since L-NAME is capable of crossing the blood-brain barrier (17), and because there are three NOS isoforms, in this study we have further tested the involvement of central NOS in psychological stress-induced fever. Our working hypothesis is that the elevation in Tb due to exposure to psychological stress is related to activation of NOS inside the brain. To test this hypothesis we investigated the effects of selective inhibitors of neuronal (nNOS) and inducible NOS (iNOS) given into the lateral ventricle on changes in Tb and locomotor activity in freely moving biotelemetered rats exposed to an open field.
Animals
Pathogen-free male Wistar rats weighing 300-320 g were used throughout the study. They were housed individually in plastic cages in a temperature-controlled room at 21 ± 1°C on 12:12-hour light-dark cycle with lights on at 06:00 h. Rodent laboratory chow and water were provided ad lib. All procedures were approved by our University Committee on the Use and Care of Laboratory Animals.
Surgery
Rats were anesthetized with a mixture of ketamine hydrochloride (87 mg/kg intramuscularly, im) and xylazine hydrochloride (13 mg/kg im). A guide cannula (5 mm, 26 G, Plastic Products Co., Roanoke, Va., USA) was stereotaxically implanted into the right lateral ventricle (1.0 mm posterior to the bregma and 0.5 mm lateral to the midline; coordinates according to the atlas of Pellegrino et al., ref. 18) and affixed to the skull with dental cement. The guide cannula was closed with a dummy cannula that extended from the tip of the guide cannula by no more than 0.2 mm.
Deep Tb (± 0.1°C) and activity of rats were measured using intraperitoneally implanted telemetry units (model VMFH, MiniMitter, Bend, Oreg., USA). Immediately following the cannulation of the lateral ventricle while the rats were still anesthetized, a 1 cm abdominal incision was made for placement of the transmitter. After the transmitter was inserted into the peritoneal cavity, the wound was sutured and swabbed with antibiotic ointment (Neomycinum).
Animals were allowed to recover for at least 7 days prior experimentation. After the completion of experiments, the animals were sacrificed and then dye was injected through the cannula to mark the ventricular space. The brain section was visually examined to verify that the tip of the stainless steel cannula was located in the right lateral ventricle.
Measurements of Tb and motor activity
Temperature and locomotor activity of rats were measured with a peripheral processor (VitalView3000, MiniMitter, Inc, Bend, Oreg. USA), connected to an IBM personal computer.
Nitric oxide synthase inhibitors
Vinyl-L-NIO (N5 - (1-Imino-3-butenyl) - ornithine; Alexis Biochemicals, USA), a neural nitric oxide synthase (nNOS) inhibitor, and aminoguanidine hydrochloride (Sigma, USA), an inducible nitric oxide synthase (iNOS) inhibitor, were dissolved in pyrogen-free water. Both inhibitors were prepared freshly on the day of experiment. For icv injections, rats were restrained in a towel while the dummy cannula was removed and the injection cannula inserted into the guide cannula. Injections were made using a Hamilton syringe. After injection, the injection cannula was removed and the dummy cannula replaced. Control rats were injected icv with an equal volume of pyrogen-free water.
General procedure
Rats were exposed to an open field (60 x 30 x 30 cm, plastic aquarium) for 30 min at an ambient temperature of 21 ± 1°C. vL-NIO at a dose of 5 µg/rat, and aminoguanidine at a dose of 10 µg/rat were injected icv in final volume of 5 µl just before the exposure to open field. The rationale for the icv dose of inhibitors was based on our earlier observations indicating that both inhibitors at selected doses did not influence the normal body temperature (data not shown). The control animals were given the same volume of appropriate vehicle solution and then exposed to open field. Unstressed rats (not exposed to open field) also received injections of inhibitors or control vehicle solution and then returned to their home cages. To minimize possible circadian variability, exposure to the open field occurred between 8 am and 1 pm. Temperature as well as motor activity (A) in all experiments were monitored and recorded in 5-min intervals beginning 30 min before the injection and continued until the end of the 30-min exposure to the open field. The 5 min temperature and activity readings during the exposure to the open field were averaged and used to calculate the mean change in Tb and A due to exposure to an open field.
Statistical analysis
All data are presented as mean ± SE. All comparisons between groups were made using 1-factor ANOVA, followed by pair-wise comparisons tested by Fisher's Protected Least Significant Difference (PLSD). A value of p<0.05 was considered to be significant.
Effect of icv injection of vL-NIO, a nNOS inhibitor, on open field-induced rise in Tb and locomotor activity
The results of vL-NIO injection on the open field-induced rise in Tb and motor activity are shown in Fig. 1. As for all experiments in this part of the study, handling due to injection, independent of the nature of the injected substance, caused a transient increase in Tb as well as in locomotor activity. In the group of animals not exposed to the open field, where each rat was simply picked up, injected and immediately returned to his own cage, the handling-induced rise in Tb and activity was followed by a gradual fall in Tb and activity to the initial preinjection level. As shown in Fig. 1, all vehicle-injected rats exposed to the open field responded with rapid rise in Tb which started within 5 min and reached a maximum temperature at 25 min after start of exposure to psychological stress (38,27 ± 0,13°C), whereas the body temperature of rats not exposed to open field at the same time-point was 37,39 ± 0,10°C. The mean Tb for 30-min period of exposure to the open field was significantly higher in vehicle-treated rats exposed to the open field than that of the vehicle-treated and non-exposed group of rats: 37.91 ±0.08 vs. 37.41 ± 0.05°C (p<0.0001, Fig. 1A). Exposure to open field for 30 min caused increase in locomotor activity (Fig. 1B). There was significant difference in motor activity between vehicle-injected rats exposed or not exposed to the open field (83.16 ± 16.09 counts vs. 16.39 ± 5.43 counts, p<0.0001).
 |
Fig. 1. Effect of vL-NIO, a selective inhibitor of nNOS, injected icv at a dose of 5 µg/animal on body temperature (A) and locomotor activity (B) of control rats (not exposed to open field), or rats exposed to open field for 30 min. vL-NIO or appropriate vehicle was injected just before the start of open field (Time 0). Arrowhead indicates time of injection. Black horizontal bar indicates the period of exposure to an open field. Values are means ± SE. |
The 5-µg dose of vL-NIO delayed and attenuated the open field-induced rise in Tb . Compared to vehicle-injected rats exposed to open field, the body temperature measured at 25 min after start of exposure to open field was significantly lower in v-LNIO pretreated rats (37,89 ± 0,26°C). The mean Tb for the 30-min period of exposure to the open field was 37.56 ± 0.15°C for 5 µg of vL-NIO vs. 37.91 ± 0.08°C for vehicle-treated rats (p<0.0001). The 5 µg dose of this inhibitor delayed the handling-induced increase in Tb within the first 10 min in rats not exposed to open field. Animals treated with vL-NIO and exposed to the open field had a smaller increase in locomotor activity than that of vehicle-injected and open field-exposed rats. However, there was no significant difference between both groups of animals in an elevation of locomotor activity due to exposure to psychological stress (83.16 ± 16,09 counts for vehicle-injected rats vs. 73,27 ± 11.05 counts for vL-NIO-treated rats, p=0.169). Moreover, there was still significant difference in motor activity between unstressed and stressed rats treated with vL-NIO (31.86 ± 11.29 counts vs. 73.27 ± 11.05 counts, p<0.0001).
Effect of icv injection of aminoguanidine, an iNOS inhibitor, on open field-induced rise in Tb and locomotor activity
The results of aminoguanidine injection on the open field-induced rise in Tb and motor activity are shown in Fig. 2. Exposure of rats to open field induced stress-induced rise in Tb with temperature peak occurring at 25 min after start of exposure to psychological stress (38.29 ± 0,08°C vs. 37,37 ± 0,06°C, for rats exposed and not exposed to open field, respectively). Aminoguanidine blocked the open field-induced rise in Tb (Fig. 2A). By comparison, at 25 min after start of exposure to open field the Tb of aminoguanidine-treated and open field exposed rats reached 37,64 ± 0,26°C. Moreover, 10 µg dose of this inhibitor caused a transient fall in Tb even below baseline lasting 10 min in rats either exposed or not exposed to open field. This drop was followed by a gradual increase in Tb . The mean Tb for 30-min period of exposure to the open field was 37.34 ± 0.22°C for 10 µg of aminoguanidine vs. 37.82 ± 0.08°C for vehicle-injected rats (p< 0.001). It is important to note, that there was no significant difference in handling-induced increase in Tb in rats not exposed to open field (37.38 ± 0.05°C for vehicle-injected rats vs. 37.33 ± 0.24°C for aminoguanidine-treated animals, p=0.479). Aminoguanidine at a dose of 10 µg was able to suppress the rise in locomotor activity in open field-exposed rats (Fig. 2B). The mean motor activity for 30-min exposure to open field were 74.25 ± 14.17 counts for vehicle-treated rats and 43.24 ± 5.72 counts for aminoguanidine-injected rats (p<0.0001). However, aminoguanidine at a dose of 10 µg had no effect on locomotor activity in rats not exposed to open field. The mean motor activity for 30-min postinjection period in vehicle-treated rats not exposed to the open field was 9.27 ± 2.55 counts and 13.67 ± 2.86 counts for aminoguanidine-injected rats (p=0.319).
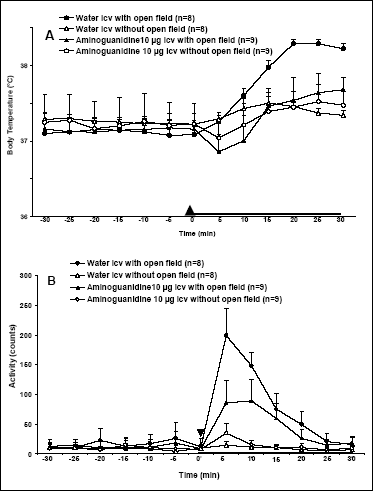 |
Fig. 2. Effect of aminoguanidine, a selective inhibitor of iNOS, injected icv at a dose of 10 µg/animal on body temperature (A) and locomotor activity (B) of control rats (not exposed to open field), or rats exposed to open field for 30 min. Aminoguanidine or appropriate vehicle was injected just before the start of open field (Time 0). Arrowhead indicates time of injection. Black horizontal bar indicates the period of exposure to an open field. Values are means ± SE. |
These results support the hypothesis that activation of nitric oxide synthase within the central nervous system may play an important role in the stress-induced rise in Tb .
Two selective NOS inhibitors (vL-NIO for nNOS and aminoguanidine for iNOS) injected icv reduced stress fever. Aminoguanidine blocked stress fever at 10 µg (Fig. 2A). vL-NIO blocked stress fever at even lower dose of 5 µg icv (Fig.1A) and had no any effect on the rise in locomotor activity due to exposure to open field (Fig. 1B). Since aminoguanidine at a selected dose caused a transient fall in Tb below the baseline in rats exposed to open field, it is not certain whether its effect is solely due to inhibition of iNOS. This dose of aminoguanidine had a hypothermic effect in rats that were not exposed to the open field, and therefore the inhibition of a stress-induced rise in Tb by this substance may have resulted not from specific inhibiting properties but rather from nonspecific influence on temperature regulation system. Moreover, aminoguanidine was able to attenuate open field-induced rise in locomotor activity (Fig. 2B). Thus, another mechanism by which aminoguanidine prevent stress fever, may be by inducing sedation and preventing the perception of stress.
The three isoforms of nitric oxide synthase, neuronal (nNOS), endothelial (eNOS), and inducible (iNOS), are found in the central nervous system (19). eNOS isoform is primarily localized in endothelial cells and plays a substantial role in blood pressure control (20). An inducible isoform (iNOS) becomes expressed in brain in response to cytokines and exogenous pyrogens (14). However, a recent study tends to indicate that activation of iNOS may participate in development and maintenance of sleep/wake cycle of the rat (21). nNOS has been localized in structures involved in thermoregulation (hypothalamic preoptic nuclei; 22) and perception of psychological stress (structures belong to the limbic system; 23). Taken together, these data along with our results suggest the hypothesis that among the three known NOS isoforms, nNOS expression inside the brain is critically involved in the rise in Tb due to exposure to psychological stress.
The mechanisms underlying the NOS inhibitor-induced suppression of fever response due to exposure to psychological stress are far to be understand. According to current model of emotional fever, this response appears to be PGE2-dependent (6, 7). Recent investigations have shown that blockade of NO production promotes down-regulation of COX2 and decrease PGE2 biosynthesis (24). Thus, it is tempting to speculate that the inhibition of stress fever by NOS inhibitors is due to the lack of stimulatory effect of nitric oxide on COX2 activity and, consequently, on PGE2 production. Another possibility comes from studies showing that inhibition of NOS attenuated the brain catecholaminergic response to restrain in rodents (25). Since stress-induced rise in Tb is centrally mediated by ß2-adrenoceptors (12, 13), it is possible that NOS expression in the brain is important for activation of catecholaminergic system during development of stress fever.
In summary, the presented data provide clear evidence that nNOS activation in the brain is essential for development of fever response due to exposure to psychological stress. However, further research is needed to identify the relationship between expression of nNOS and possible pathways responsible for induction of stress fever.
Acknowledgments: Research was supported by the Polish Committee for Scientific Research (KBN) grant 3PO4C02625.
- Long NC, Vander AJ, Kluger MJ. Stress-induced rise of body temperature in rats is the same in warm and cool environments. Physiol Behav 1990; 47: 773-775.
- Briese E, Cabanac M. Stress hyperthermia: physiological arguments that it is a fever. Physiol Behav 1991; 49: 1153-1157.
- Caputa M. Selective brain cooling: a multiple regulatory mechanism. J Therm Biol 2004; 29: 691-702.
- Kluger MJ, Kozak W, Leon LR, Soszynski D, Conn CA. Cytokines and fever. Neuroimmunomodulation 1995; 2:216-223.
- Milton AS. Prostaglandins and fever. Prog Brain Res 1998; 115: 129-139.
- Kluger MJ, O'Reilly B, Shope TR, Vander AJ. Further evidence that stress hyperthermia is a fever. Physiol Behav 1995; 2: 216-223.
- Singer R, Harker CT, Vander AJ, Kluger MJ. Hyperthermia induced by open-field stress is blocked by salicylate. Physiol Behav 1986; 36: 1179-1182.
- Long NC, Kluger MJ, Vander AJ. Antiserum against mouse IL-1 alpha does not block stress hyperthermia or LPS fever in the rat. In Thermoregulation Research and Clinical Application, P Lomax, E Schonbaum (eds). Basel, Karger, 1989, pp. 78-84.
- Kozak W, Zheng H, Conn CA, Soszynski D, Van der Ploeg LHT, Kluger MJ. Thermal and behavioral effects of lipopolisaccharide and influenza in interleukin-1b-deficient mice. Am J Physiol 1995; 269: R969-R977.
- Kozak W, Poli V, Soszynski D, Conn CA, Leon LR, Kluger MJ. Sickness behavior in mice deficient in interleukin-6 during turpentine abscess and influenza pneumonitis. Am J Physiol 1997; 272: R1298-R1307.
- Soszynski D, Kozak W, Kluger MJ. Endotoxin tolerancje does not alter open field-induced feler in rats. Physiol Behav 1998; 63: 689-692.
- Soszynski D, Kozak W, Conn CA, Rudolph K, Kluger MJ. Beta-adrenoceptor antagonists suppress elevation in body temperature and increase in plasma IL-6 in rats expose to open field. Neuroendocrinology 1996; 63: 459-467.
- Mayfield KP, Soszynski D, Kozak W, Kozak A, Rudolph K, Kluger MJ. b-adrenergic receptor subtype effects on stress fever and thermoregulation. Neuroimmunomodulation 1999; 6: 305-317.
- Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev 1991; 43: 109-142.
- Soszynski D. The inhibition of of nitric oxide synthase suppresses LPS- and psychological stress-induced fever in rats. Physiol Behav 2001; 72: 65-72.
- Kozak W, Kozak A. Differential role of nitric oxide synthase isoforms in fever of different etiologiest: studies using NOS gene-deficient mice. J Appl Physiol 2003; 94: 2534-2544.
- Dwyer MA, Bredt DS, Snyder SH. Nitric oxide synthase: irreversible inhibition by L-NG-nitroarginine in brain in vitro and in vivo. Biophys Res Commun 1991; 176: 1136-1141.
- Pellegrino LJ, Pellegrino AS, Cushman AJ. A stereotaxic atlas of the rat brain. New York, Plenum Press, 1979.
- Chabrier PE, Demerle-Pallardy C, Auguet M. Nitric oxide synthases: targets for therapeutic strategies in neurological disesases. Cell Mol Life Sci 1999; 55: 1029-1035.
- Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Ann Rev Physiol 1995; 57: 707-736.
- Monti JM, Jantos H. Microinjection of the nitric oxide synthase inhibitor l-NAME into the lateral basal forebrain alters the sleep/wake cycle of the rat. Prog Neuro-Psychopharmacol Biol Psych 2004; 28: 239-247.
- Gath I, Benamar K, Jurat G, Roth J, Simon E, Schmid HA. Expression of nitric oxide synthase (NOS) isoforms in rat brain during lipopolysaccharide-induced fever. Pflugers Arch 1999; 437: R143.
- Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience 1992; 46: 755-784.
- Perkins DJ, Kniss DA. Blockade of nitric oxide formation down-regulates cyclooxygenase-2 and decrease PGE2 biosynthesis in macrophages. J Leukocyte Biol 1999; 65: 792-799.
- Dunn AJ. Brain catecholaminergic and tryptophan responses to restraint are attenuated by nitric oxide synthase inhibition. Neurochem Int 1999; 33: 551-557.