Since the lung has been exposed to microorganisms and foreign substances from the environment for millions of years, during the evolution process a complex defense system has been developed protecting the respiratory tract from the nostrils down to the alveoli. The defense mechanisms of upper airways and the bronchi consist of anatomic barriers, cough, mucociliary apparatus, airway epithelium, secretory immunoglobulin A (IgA), dendritic cell network and lymphoid structure (6). About 90% of inhaled particles with diameters larger than 2 to 3 µm are deposited in the central airways on the mucus overlying the cilial epithelium (2, 3, 6). After deposition they are rapidly transported to the trachea by means of the mucociliary escalator and swallowed into the gastrointestinal tract (Fig. 1). Furthermore, the thickness of mucus layer and respiratory epithelium as well as peroxidases reduce the absorption of biomolecules deposited in the central airways.
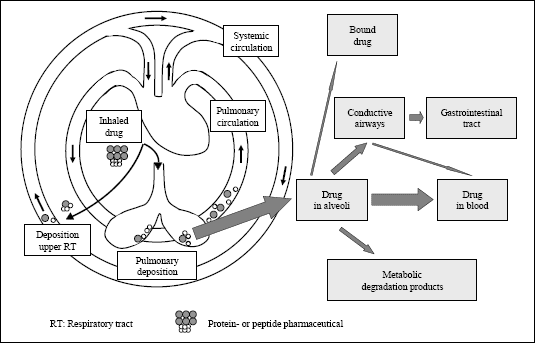 |
| Fig. 1. Uptake of inhaled drugs after peripheral/alveolar deposition (modified according to (1)). |
Much better conditions for absorption of inhaled macromolecules are found in the lung periphery making the lung an important target for inhalant administration of pharmaceuticals for systemic treatment. Firstly, the size of the alveolar surface depends on the distension of the lung and varies between 80 and 140 m2, which is about the half of a tennis court (132 m2) and much larger than that of the nose (about 180 cm2) (4, 7, 8). Another advantage of the lung is the thin alveolar epithelium. The thickness of this epithelium in most regions is between 0.1 and 0.2 µm (9, 10) resulting in a total distance between epithelial surface and blood between 0.5 and 1.0 µm (8), which is much less than that in the bronchial tract, where the deposited substances have to pass a distance of 30-40 µm, and more between mucus surface and blood (Fig. 2) (8, 9). Furthermore, the lung is perfused with a blood volume of about 5 l/min at rest (11) without a first-pass effect, which plays a large role for orally administered drugs even though some metabolism takes also place in the lung (1, 4, 8, 10, 12, 13). However, even in the lung periphery a number of defense mechanisms exist that inhibit the absorption of biomolecules, e.g., macrophage uptake.
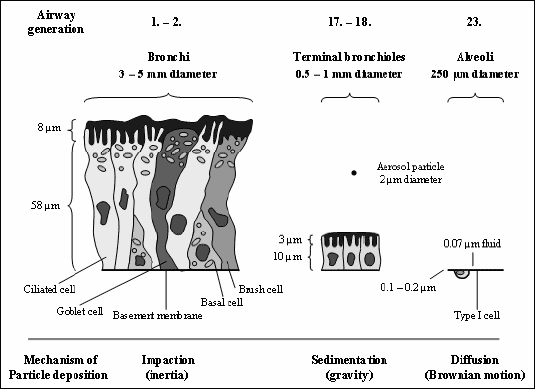 |
| Fig. 2. Lung epithelium and mechanisms of particle deposition at different sites within the lungs [modified from (9)]. Lung epithelial cells of the different lung regions are drawn at their relative sizes. The higher the number of the airway generation the deeper the particle is inspired into the lung (0: Trachea, 1-2: Bronchi, 3-5: Bronchioles, 17-18: Terminal bronchioles, 19-20: Respiratory bronchioles, 21-22: Alveolar ducts, 23: Alveolar sacs). Mechanisms of particle deposition depending on the aerodynamic particle diameters (dae) are impaction (inertia), sedimentation (gravity) and diffusion (Brownian motion) in bronchi, terminal bronchioles and alveoli, respectively. A typical aerosol particle (dae: 2 µm) contains tens to hundreds of millions of insulin molecules or hundreds of millions/billions small molecules depending on its physical character (liquid or solid). Solid aerosol particles are too large to be absorbed in total and must dissolve to release their drugs for absorption. The deeper an aerosol particle penetrates into the lung the thinner becomes the airway epithelium and the larger becomes the lung surface. In consequence, the function of the epithelial absorption barrier decreases and the absorption increases as a function of the particle penetration depth into the lung. Typical cells in the bronchi are basal cells, which serve as the stem or progenitor cells for the other epithelial cells in case of injury or apoptosis, ciliated cells, which provide the mechanism for moving the mucus blanket, goblet cells, which secrete the mucus and brush cells, which are involved in drug metabolism. The same cells and the mucus layer are also found in the smaller airways, but not as tall. The thinnest absorption barrier is found in lung alveoli. The basement membrane is not a membrane, but an extracellular matrix of different biopolymers to which epithelial cells attach. |
Physical methods for aerosol administration
Different types of nebulizers, metered dose inhalers (MDI), and powder inhalers have been developed for aerosol therapy. Requirements for this type of administration are high efficiency of drug delivery, reproducible dosing, targeted delivery of the inhaled drug to the site of action, ease of device operation, short duration of treatment, minimized risk to the patient and the medical personnel, environmental protection, and cost-effectiveness (14). However, the various products differ strongly in respect to their suitability for nebulization and administration of the different compounds. In the past, low rates of pulmonary drug absorption were observed, because the used nebulizers were not qualified for production of an adequate aerosol particle spectrum (5, 8) and did not take care on the breathing patterns of the patients.
Nebulizers
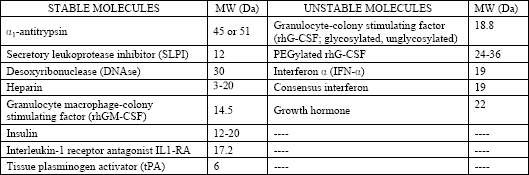
The appropriateness of nebulizers for administration of macromolecular compounds depends on the performance of them (e.g., aerosol output, distribution width, and variability of the aerosol particle spectrum) as well as the stability of the biochemical compounds used for nebulization. Within the nebulization process in air-jet nebulizers, protein structure and function can be compromised independently from the molecular weight of the protein (Table 1) by surface denaturation, shear-stress induced denaturation, and desiccation of the aerosol droplets (4). The role of these degenerative processes in aerosol production is enhanced by the operating mode of air-jet nebulizers, because just 1% of the produced droplets leaves the nebulizer, whereas the other 99% remain inside and undergoes the nebulization process at least 10-15 times (1, 4). Several additives, such as lipids, surfactant, amino acids, albumin, polyols, and packing into liposomes result in an increased protein stability and additional absorption enhancement (4, 5, 15-17).
| Table 1. Stability of selected biomolecules in air-jet nebulizers (from (4)). Molecular weight for some compounds not provided for monomers. |
 |
Ultrasonic nebulizers act by disruption of liquid surfaces by means of ultrasound and, therefore, allow production of high concentration aerosols (16). That requires a supply of high energy doses, which especially in viscous liquids may cause formation of large surfaces with cavitation and distinct heat development (4, 16). However, most of the clinically used pharmaceuticals have a sufficient stability and are not affected by these denaturating processes. In contrast, peptides and proteins (e.g., insulin,
Another approach for nebulization is the vibrating plate technology. In this type of aerosol devices, a liquid aerosol is produced by means of a vibrating mesh or plate with multiple apertures. Devices of this type allow the generation of aerosols with a high fine-particle fraction. The aerosols are generated as a fine mist without requirement of an internal baffling system (14, 16). Compared with conventional jet nebulizers and ultrasonic nebulisers, they have a higher efficiency for the delivery of drugs to the respiratory tract. Some other advantages are that these devices effectively aerosolize solutions, have only a minimal residual volume of medication left in the device (cost sparing effect), and might be breath-actuated, thereby limiting the release of aerosolized drugs into the environment (14). These devices sometimes fail when liposomal formulations should be aerosolized and it usually is difficult to aerosolize suspensions (exception: nanosuspensions).
Powder aerosols
Powder aerosols are produced by disaggregation of preformed (e.g., milled or spray-dried) micronized particles. The energy required for disaggregation is supplied by the inhalation maneuver or alternatively by means of an external energy (4, 18). Advantages of dry powder inhalers are their environmental sustainability due to a propellant-free design, the ease to use, because not much patient coordination is needed, and the formulation stability. On the other hand, typical disadvantages are the dependency of the deposition efficiency on the patients inspiratory airflow, their potential for dose uniformity problems, and their relative high complexity and costs for development and manufacture. The use of dry powder aerosols is established for treatment of asthma and chronic obstructive pulmonary disease (COPD), e.g., by means of ß-mimetics, anticholinergics, or steroids. However, up to now there is little experience on inhalant administration of biomolecules except insulin (Exubera®) for systemic treatment (1, 17, 18). This is caused by specific problems for the use of proteins or peptides occurring in the processes of lyophilization or spray drying, micronisation, completeness of dispersion and disaggregation, and the surveillance of the latter.
For passive systems, the inspiratory air flow of the patient is an essential parameter. If this air flow is insufficient for complete disaggregation, large aggregates will be inhaled and cannot reach the alveolar region. Humidity can also be a large problem, because it impairs the stability of proteins and peptides, and also affects disaggregation and dispersion (4, 16, 18, 19). However, if the underlying problems, especially in particle engineering, are solved by novel techniques (20), the inhalation of dry powder aerosols may be an interesting tool for inhalant treatment of systemic diseases by inhaled biomolecules deposited in the alveolar region. In this case it should additionally be considered that high powder doses (over a few milligram) may cause cough and in that way influence deep lung deposition significantly.
Metered dose inhalers (MDI)
In metered dose inhalers compounds are dissolved or suspended in a pressurized propellant that should be nontoxic, noninflammable, compatible with drugs formulated, as suspensions or solutions, and to have appropriate boiling points and densities. For consistent dosing the vapor pressure must remain constant throughout the product´s life. These requirements are typically fulfilled by chlorofluorocarbons (e.g., dichlorodifluoromethane, dichlorotetrafluoroethane, and trichlorofluoromethane), but not by pressurized carbon dioxide. After its release with high velocity, the mixture rapidly expands forming an aerosol. Because of the high velocity of the aerosol directly after its release, different types of spacers are often required for optimization of the aerosol deposition (4, 21). Aerosols from metered dose inhalers are established in clinical treatment of patients with asthma or COPD from about 50 years, and many different types of metered dose inhalers have been developed (4, 16, 21). Unfortunately, these devices, up to now, cannot be used for treatment with macromolecules (e.g., peptides and proteins), because a number of prerequisites (stability of the compound within storage in the inhaler, no denaturation of the compound within the nebulization process, production of an aerosol with appropriate particle distribution pattern) are not sufficiently fulfilled.
General and specific factors affecting the absorption
Proteins with lower molecular weight are absorbed more rapidly after alveolar deposition than those with higher molecular weight (5, 8, 22-25). Numerous studies have shown that the bioavailability of proteins with molecular weights up to 30 kDa (which includes the vast majority of proteins used in clinical therapy) is between 20 and 50% (Fig. 3) (8, 25). However, the bioavailability of some proteins is much smaller, because they are subject of proteolytic degradation (8, 22). Other variables affecting the absorption are pH-value, electrical charge, surface activity, solubility and stability in the alveolar environment (4, 10, 22). Pharmacokinetics of the different macromolecules also depends on their molecular weight. For example, the half-life time of the alveolar absorption of hydrophilic compounds increases with their molecular weight (sucrose: MW: 342 Da, t0.5: 87 min; inulin: MW: 5250 Da, t0.5: 225 min; dextran: MW: 20000 Da, t0.5: 688 min; dextran: MW: 75000 Da, t0.5: 1670 min) (22). Accordingly, the time to reach the maximum serum concentration (tmax.) is also increasing as a function of the molecular weight of peptides and proteins (Fig. 4) (5, 22, 24, 25).
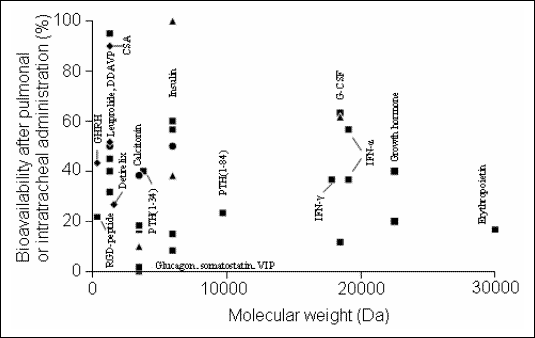 |
| Fig.
3. Bioavailability of peptides and proteins after pulmonary deposition
or intratracheal adminstration (from (8, 25)). Data were obtained in rodents
( |
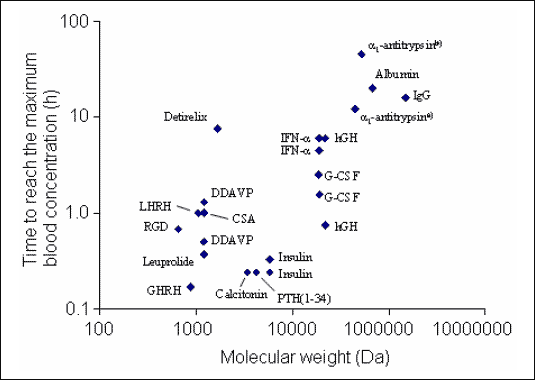 |
| Fig.
4. Time to reach the maximum blood concentration (tmax.)
after pulmonary administration as function of the molecular weight of
various peptides and proteins [modified from (5, 22, 24, 25)]. Most of
the biomolecules were administered intratracheally in rats, few in other
species (e.g. dogs). Data of some biomolecules show a large variability.
The variability can be caused by differences in the experimental settings
(e.g., animal species and mode of administration (substances administered
by aerosol peak more rapidly than those administered intratracheally))
and by the glycosylation of a protein. Abbreviations: AAT: |
Proteins deposited on the mucociliar epithelium of the conducting airways are poorly absorbed and show a small bioavailability, because they are transported to the pharynx by mucociliary transport and degraded in the intestinal tract. In contrast, proteins deposited in the alveoli can be absorbed by four distinct mechanisms: phagocytosis by alveolar macrophages, paracellular diffusion via tight junctions, vesicular endocytosis or pinocytosis, and receptor dependent transcytosis (4, 8, 10). The functional role of barriers and transport mechanisms is very different and underlies control by physiological and pharmacological factors (4, 8, 10, 15). For example, absorption enhancing substances (15) and cigarette smoking cause an inflammation of the lower respiratory tract followed by an increased epithelial permeability (8, 10). In consequence, inhaled insulin is more rapidly absorbed in smokers than in nonsmokers (5, 8, 10, 16, 17, 22, 26, 27). On the other hand, an alveolar inflammation, which can be even induced by the inhalation therapy itself (e.g., by absorption enhancers), can result in a reduction of the bioavailability (15). However, an immunization against the administered peptides and proteins, which might cause an incompatibility or an inactivation, obviously plays no relevant role (8, 12, 28). Finally, pulmonary diseases affecting convective gas transport, size of the alveolar surface or alveolar permeability (asthma, COPD, smoking) can preclude or hamper a pulmonary drug therapy (8, 11, 17).
Physiological absorption barriers
A number of physiological barriers inhibit the absorption of inhaled proteins after their pulmonary deposition (Fig. 5) (4, 8, 10). The first barriers after contact are the mucus layer and the alveolar lining fluid. The mucus layer consists of a complex mixture of lipids and glycoproteins, but also surfactant from the lower respiratory tract. The amount, composition and thickness of the mucus layer depend on their localization in the respiratory tract and are also influenced by local inflammatory and neuronal factors. Pulmonary diseases, local inflammation, and administered drugs cause a variation of the mucus volume and composition and of the airway diameters, all of which affects deposition and absorption. In consequence, patients with pulmonary diseases must be thoroughly investigated prior to inhalation therapy for treatment of systemic diseases, because aerosol deposition and absorption differ from those in healthy individuals and data obtained in individuals with normal lung function cannot be extrapolated to these patients (4). The alveolar lining fluid includes a large amount of surfactant with phospholipids and surfactant apolipoproteins acting as a surface active substance. Hyperventilation causes a release of surfactant from the type II pneumocytes located in the alveoli. However, numerous other endogenous and exogenous factors (pharmaceuticals) modulate cellular surfactant synthesis. Pulmonary surfactant interacts with the deposited substances affecting their stability and solubility, e.g., by formation of liposomes (Fig. 5).
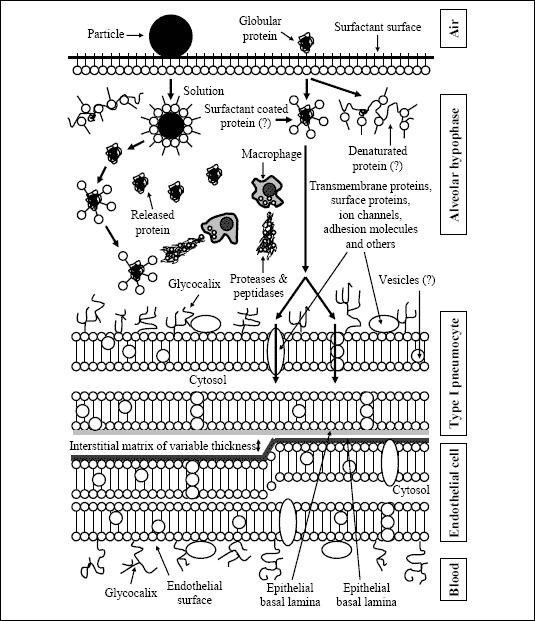 |
| Fig. 5. Barriers for absorption of peptides and proteins after peripheral/alveolar deposition [modified from (4)]. |
Cells located in the respiratory tract also counteract the absorption of inhaled substances after their alveolar deposition. Macrophages represent about 85% of the cells retrieved by bronchoalveolar lavages and are normally the only type of phagocytic cells within the lower respiratory tract (6). They play a predominant role in this process for absorption inhibition, which serves as an unspecific defense mechanism of the lung against bacteria and inhaled particles. Lombry et al (29) demonstrated that alveolar macrophages serve as a primary barrier to the pulmonary absorption of macromolecules, as a depletion of alveolar macrophages was followed by an improved absorption of proteins into circulation after intratracheal instillation even though there seems to be differences regarding the administered type of protein (e.g., IgG and hCG). Macrophages are differentiated from blood monocytes after they have emigrated into the tissues and they occur in the respiratory tract, the alveoli, and the interstitial matrix, and their number can rapidly increase in case of an inflammation (4, 6). Furthermore, they can rapidly incorporate particles deposited in the lung alveoli, secrete reactive oxygen species (ROS) by means of respiratory burst and release mediators of inflammation (e.g., granulocyte macrophage-colony stimulating factor (GM-CSF)), cytokines (e.g., IL-1ß/IL-1ra, IL-6 and tumor necrosis factor a (TNF-
The largest proportion of substances deposited in lung alveoli achieves the surface of the alveolar type I pneumocytes. These cells cover about 97% of the alveolar surface and serve for pulmonary gas exchange. The remaining area consists of the type II pneumocytes producing lung surfactant. Type I pneumocytes express carboxypeptidase on their membrane, which degrades a number of peptides and proteins. The total distance between respiratory tract and circulation is only 0.5 µm facilitating the diffusion of gasses and penetration and transport of fluids and (inhaled) macromolecules (4). Inhaled macromolecules can pass alveolar epithelium via different transport mechanisms, which are intracellular tight junctions, membrane pores, and vesicular transport by type I and type II pneumocytes (4). Tight junctions are located between epithelial barriers, have a radius between about 0.8 and 1.0 nm and regulate the transport of small soluble substances, fluids, and ions. In the normal lung they play obviously no relevant role in the transport of proteins. In contrast, in cases of cellular damage the size selectivity is lost allowing permeation of larger molecules and fluid volumes. Furthermore, the permeability of chemical compounds like bile acids and calcium chelators is also increased. However, there are structural differences between epithelial tight junctions and endothelial tight junctions. The latter allow a permeation of molecules with molecular weights more than 12 kDa into the interstitium. In cases of hydrostatic or oncotic pressure gradients larger molecules can also permeate (4).
Membrane pores are discussed as another transport mechanism allowing the exchange of fluids and macromolecules. It is assumed that pores of different sizes exist, which can increase their diameter in case of an existing hydrostatic pressure gradient (4). In pneumocytes types I and II another mechanism of vesicular transport has been described, which is comparable with that in epithelial and endothelial cells. This transport mechanism is of larger relevance in type I pneumocytes, because they line a much larger proportion of the alveolar surface than type II pneumocytes. In detail, the vesicular transport mechanism of type I pneumocytes is pressure independent and allows the transcellular transport of fluids and macromolecules. The vesicles have a diameter of about 35.5 nm allowing the transport of even larger macromolecules. For example, the hydrodynamic radii of lysozyme (MW: 14.1 kDa) and catalase (MW: 230 kDa) are 2.1 and 5.2 nm, respectively. However, an estimation of the functional capacity of this transport mechanism is difficult, because (1) the number of vesicles increases in liquid filled lung indicating their role in the transport of fluids, (2) the glycocalix affects the uptake of proteins via specific or unspecific binding mechanisms and a number of receptors and binding proteins were identified on capillary endothelia, (3) the definite processing of the vesicles inside the cells and the mechanisms for their movement (e.g., Brownian movement) are not conclusively identified, (4) the energetic mechanisms of membrane displacement and fusion of the vesicles are not yet conclusively elucidated, and (5) different types of vesicles (e.g., clathrin-coated and clathrin-uncoated) exist, which both play a role in transcytosis, but differ in respect to their characteristics of protein uptake (e.g.,
In contrast to the type I pneumocytes described before, type II pneumocytes cover only a small area of the alveolar surface and produce pulmonary surfactant. The latter together with proteins plays an important role in the clearance of macromolecules by means of the alveolar lining fluid. Further cellular processing can take place with or without binding of the macromolecules on the cellular surface and depends strongly on the charge of the molecules. For example, cationic ferritin is absorbed much better than uncharged or anionic molecules. A large proportion of the material absorbed by endocytosis from the type II pneumocytes is deposited in lamellar bodies. In addition, transcellular transport represents another mechanism for absorption of macromolecules.
The basal lamina has a thickness of about 20 to 25 nm and is placed below the epithelium. It predominantly consists of glycoproteins (laminin, heparan sulphate, proteoglycan, fibronectin, and collagen) and has an anionic charge on its outer surface. Presumably, the latter regulates the permeation dependent on the size and charge of molecules. However, the mechanism of the permeation inhibition is not yet fully elucidated. After their passage through the alveolar wall and alveolar basal lamina inhaled substances reach the interstitium, where proteins can be bound by macromolecules or inactivated or phagocytosed by macrophages or transported to the lymphatic system. In the latter case, proteins can be detected after some hours in the circulation. The endothelial basal lamina and endothelium are also barriers for the absorption of macromolecules. However, compared with the other barriers described before they act only as a minor barrier for inhaled biomolecules before entering the circulation (4).
Methods for absorption improvement
A number of physiological barriers inhibit the absorption of macromolecules via the gastrointestinal tract and other mucosal surfaces, the respiratory tract, and the skin. In addition, various enzymes, especially peptidases and proteases, degrade macromolecules, especially peptides and proteins, by proteolysis. Addition of absorption enhancers to the pharmacological compound considerably increases the transdermal (30), gastrointestinal (31), and respiratory absorption (5, 15, 31, 32). Prevention of proteolysis by addition of protease inhibitors or packing of the macromolecules into particles can further increase the bioavailability. Packing into microparticles can also be used for the development of sustained release pharmaceuticals. However, it should be considered that all these substances for absorption enhancement do not only affect the pharmacological properties of the administered macromolecules (e.g., bioavailability, time to reach the maximum plasma concentration (tmax.), and maximum plasma concentration (Cmax.)), but also have an own active profile and toxicity (1, 15, 22).
Enzyme inhibitors
The activity of proteases and peptidases in the alveolar region of the respiratory tract is much lower than in the gastrointestinal tract (13, 22). However, proteolytic degradation, especially of susceptible peptides and proteins, cause a relevant reduction of the bioavailability even after pulmonary administration of these macromolecules. The bioavailability and pharmacological activity of inhaled peptides and proteins can be improved by addition of protease inhibitors preventing the inactivation of these biomolecules by proteolytic cleavage (1, 5, 15). Examples of protease inhibitors are nafamostat mesilate (doubling the insulin bioavailability) and aprotinin and (p-amidinophenyl)- methanesulfonylfluorideHCl (p-AMF) (increase the bioavailability of rhG-CSF 1.5-times and 3-times, respectively) (Table 2) (15).
| Table 2. Substances tested for promoting pulmonary protein absorption of pharmaceuticals for systemic treatment (modified from (15, 34-36)). Most substances were tested in animals only and the absorption enhancing effect differs strongly between the various compounds and the administered doses. Note that liposomes and microparticles differ strongly regarding their composition. |
 |
Surface active substances
This group includes compounds, which are very different in their molecular structure (bile acids, fatty acids, nonionic detergents). The mode of action is not yet completely understood, and it is assumed that the increase of the alveolo-capillary transport is caused by an interaction with the cell membrane resulting in a liquefaction followed by an increased permeability and/or a modulation of cellular tight junctions followed by an increased paracellular permeability (15, 33). Presumably, bile acids increase the absorption by alteration of the mucus layer, protection of proteins against enzymatic degradation, disaggregation of protein multimers, opening of epithelial tight junctions, and solubilization of phospholipids and proteins out of the cell membrane, followed by formation of micelles. However, the strong absorption enhancing effect (e.g., of bile acids for insulin) (5) can result in a damage to the epithelial surfaces after treatment for longer periods. The absorption can also be increased by fatty acids (or their sodium salts) or nonionic detergents. For example, beside other fatty acids (or their sodium salts), oleic acid and linoleic acid and polyoxyethylene cause a distinct increase of calcitonin absorption. Lauryl ether enhances the absorption of rhG-CSF and Span 85 increases the absorption of inhaled insulin aerosol without lung damage (Table 2) (5, 15).
Cyclodextrins
Cyclodextrins are cyclic polymers of glucose that form complexes with molecules fitting into their lipophilic inner structure. An absorption enhancing effect of cyclodextrins was observed for luteinic hormone releasing hormone (LH-RH), granulocyte-colony stimulating factor (G-CSF), calcitonin, and analogs of the adrenocorticotrophic hormone (ACTH). However, inhalation of insulin with different compounds of this group demonstrated that the intensity of the absorption enhancing effect of cyclodextrins, but also their toxicity, depends on their structure (1, 15). In detail, the toxicity increases with the intensity of absorption enhancement (15). The underlying modes of action are solubilization and complexation of membrane lipids and proteins of epithelial cells, inhibition of proteolytic enzymes, and modification of the physicochemical properties (e.g., solubility and partition coefficient) of the administered substances. The latter is important for hydrophilic peptides and proteins with high molecular weight that can only partially be incorporated into complexes and are subject of changes of their conformity (15) (Table 2).
Other substances
Other very different compounds also serve as absorption enhancers for pharmaceuticals after inhalant administration and pulmonary deposition. For example, salts of different lanthanides (CeCl3, GdCl3, LaCl3, LuCl3) interact with membrane components and cause a conformational change of membrane proteins resulting in a distinct increase of insulin absorption depending in its intensity on the type of the lanthanide salt (15). Ethylene diamine tetraacetic acid (EDTA) and salicylates increase the paracellular transport by a calcium regulated modification of cellular tight junctions (15). Polyethylene glycol (PEG) also increases the bioavailability of inhaled macromolecules (e.g., rhG-CSF) after alveolar deposition (15). As shown for insulin, hydroxyl methyl amino propionic acid (HMAP, an amino acid) increases absorption and bioavailability of the inhaled peptide. However, the inhalation of HMAP is followed by a temporary alveolar inflammation (15). In another study, the bioavailability of leuprolide acetate was enhanced by additionally administered alcohol. However, repeated administration resulted in an inflammation followed by a reduced effect (Table 2) (15).
Another, recently described approach is the modification of therapeutic proteins by fusion to the Fc domain of an IgG1 (IgG subtype 1). The Fc fusion proteins can be efficiently administered as liquid aerosols (38). Compared to the other absorption enhancers described before, the function mode of this absorption enhancing process is more physiological. First described in the intestine of rodents, the neonatal constant region fragment (Fc) receptor (FcRn) transports maternal immunoglobulin (IgG) from milk into the circulation of newborns providing immunity in the first life span. The transport is based on the interactions between the Fc fragment of IgG and FcRn. In rodents FcRn expression in gut epithelium rapidly decreases after weaning and remains low in epithelial tissues of adult animals. In contrast, FcRn in humans is also expressed in adulthood, where it can be found in the placenta and serves for the transport of IgG from the mother to the fetus, and in several absorptive tissues (lung, kidney, and intestine) (38, 39). Physiologically, IgG is taken up into epithelial cells by pinocytosis. In detail, a coated vesicle is formed by invagination of the plasma membrane entrapping IgG and other solutes in its lumen. Obviously, only a small proportion of IgG binds to FcRn at the plasma membrane, whereas most of the binding takes place intracellularly, because the majority of FcRn is localized in acidic endosomal vesicles inside the cell. The transport vesicles containing IgG bound to FcRn do not fuse with lysosomes, but rather pass unidirectionally through the epithelial cell, driven by the pH gradient between luminal and serosal exposures of the epithelial cells. As the binding of IgG to FcRn is pH-dependent (tight binding at slightly acidic pH), there is release of IgG from FcRn after fusion of the transport vesicles with the plasma membrane at the basolateral site of the epithelial cells, because of the neutral to slightly alkaline pH value of the interstitial space. Passage of IgG into the circulation is most likely primarily paracellular because of the absence of tight junctions between endothelial cells. The FcRn receptor is also responsible for the long half-life time of IgG in the bloodstream, because it protects IgG from degradation. As in epithelial cells, IgG is taken up from vascular endothelial cells by pinocytosis. However, in contrast to epithelial cells, IgG there is not subject of transcytosis, because the endocytic vesicles containing IgG bound to FcRn return to the plasma membrane of the endothelial cells, so that IgG is released back into the bloodstream. This results in a recycling process for IgG protecting IgG from lysosomal degradation (39). Both, the enhanced uptake via alveolar epithelium and the endothelial recycling process, makes the administration of Fc fusion proteins an interesting tool for inhalant application of some proteins. Fc fusion proteins with erythropoietin, interferon-a, interferon-b, and follicle-stimulating hormone (FSH) have been evaluated in animals or humans, demonstrating a good tolerability, a high bioavailability even of this large proteins, and an increased half-life time in the circulation (Table 2) (38-40, 44).
Liposomes and phospholipids
Liposomes are particles ranging in size from nanometers up to few micrometers and consist of hydrophobic lipids and phospholipids forming a closed, concentric, bilayer membrane vesicle with a hydrophilic aqueous centre (14). In their structure they have some similarities to the biological cell membrane (Fig. 6). Each phospholipid molecule is characterised by a polar (i.e., hydrophilic) head group and two hydrophobic tails. Hydration of phospholipid molecules under low-shear stress conditions results in a spontaneous arrangement of the phospholipids in heads-up and tails-down orientation followed by a join in a tail-to-tail array with formation of a concentric bilayer membrane enclosing some water in an aqueous center (Fig. 6). According to this structure both hydrophobic and hydrophilic compounds can be packed into liposomes prior to transportation into the lung. Hydrophilic compounds (e.g., pharmaceuticals and larger biomolecules) are entrapped into the vesicle inside the liposome, whereas lipophilic (hydrophobic) compounds are encapsulated into the membrane bilayer. Small liposomes are unilamellar bodies with a hydrophilic core, whereas larger multilamellar liposomes have an onion-like structure with several layers of phospholipids and aqueous compartments. Because of their strong chemical and structural similarity liposomes merge with cell membranes and facilitate drug delivery into the interior of the cell (Fig. 6). In the lung, the cellular absorption can also be influenced by the pulmonary surfactant that lines the alveolar surface, because surfactant proteins A, B, and C are subject to an intensive recycling process, which is further increased by the deposited liposomes resulting in an enhanced protein absorption (15). One more mechanism for liposome absorption is cellular phagocytosis, which seems to play a role for small liposomes only (14). Depending on their structure liposomes have a high transport capacity and allow the transport of a large number of very different lipophilic and hydrophobic compounds. One more characteristic is the sustained release of the compounds transported by liposomes (5, 14-16). The majority of studies revealed no relevant toxicity of liposomes after pulmonary deposition. However, liposomes not only enhance absorption of drugs and biomolecules, but may also damage pulmonary epithelium. Both effects, absorption enhancement and lung toxicity, depend on the physicochemical properties of liposomes (concentration, charge, chain length, and molecular weight of phospholipids) (1, 14, 15). After pulmonary deposition, soluble compounds are rapidly cleared from the lung, whereas lipids and phospholipids remain much longer in the lung because of their chemical properties and structural homology to cell membranes. Human studies demonstrated that more than 80% and 52-73% of inhaled liposomal formulation remained in the lung 8 and 24 hours after inhalation, respectively (14). Examples for the successful administration of macromolecules for systemic treatment via liposomes are the inhalation of interleukin-2 (IL-2) in patients with advanced kidney cancer, the inhalation of the immunosuppressive drug cyclosporine A in lung graft recipients and even the inhalant administration of insulin (Table 2) (1, 4, 15, 16, 17, 45).
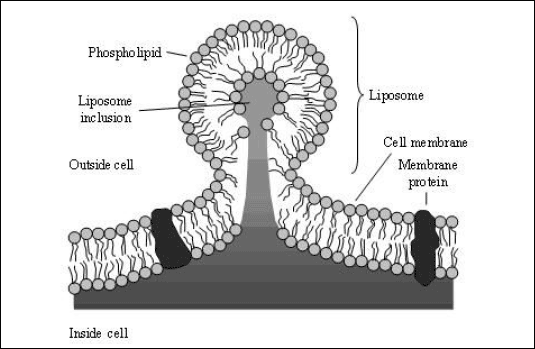 |
| Fig. 6. Acceptance of a liposome into a cell. Liposomes consist of lipids and phospholipids (from (14)). Each phospholipid has a polar hydrophilic head group and two hydrophobic tails. When phospholipid molecules are hydrated under low-shear conditions, they spontaneously arrange themselves in sheets with their heads up and tails down. These sheets then join tails-to-tails and form a bilayer membrane that encloses water and if added water soluble compounds (e.g., pharmaceuticals and larger biomolecules) in the center of the sphere. If liposomes come into contact with phospholipid cell membranes, the liposome membrane fuses with the cell membrane facilitating the entry of the encapsulated drug into the interior of the cell. |
Microparticles
In 1992, Rudt and Muller published their observation that smaller particles are more rapidly phagocytosed than larger ones (46). Based on these results, methods were developed to bind macromolecules to microparticles (1, 22, 43). For this purpose proteins are packed into biologically degradable polymers or lipids. That results in a reduction of physiological clearance in the alveolar region and proteolytic degradation of peptides and proteins after phagocytosis by alveolar macrophages. In addition, there is also a variation of the pharmacokinetics of the administered pharmaceuticals, because of a sustained release of the compounds from the microparticles (5, 22, 43). Microparticles for drug administration can be classified into porous particles and liposomes (1, 5, 15, 22, 43). The pharmacological properties of porous particles depend on the used material, particle size, porosity, and surface structure, whereas those of liposomes depend on particle size and chemical properties (charge, molecular weight) of the consisting phospholipids (1, 15, 43). For example, inhaled insulin linked to large porous particles shows a higher bioavailability than insulin from small nonporous particles (47). The same applies to insulin administered via liposomes and rhG-CSF linked to polyethylene glycol (PEGylated CSF) (1, 17). However, it cannot be excluded that microparticles can damage pulmonary tissue under specific conditions (Table 2) (1).
Examples of systemic treatment with inhaled macromolecules
Recently, the number of studies investigating the feasibility of macromolecules for systemic treatment has continuously increased (Table 3). Studies in this field focussed on hormones (insulin, calcitonin, growth hormones, somatostatin, thyroid-stimulating hormone (TSH), and follicle-stimulating hormone (FSH)), growth factors (granulocyte-colony stimulating factor (G-CSF) and granulocyte monocyte-colony stimulating factor (GM-CSF)), different interleukins and heparin (unfractionated and low molecular weight heparin (LMWH)) (1, 4, 16, 17). Most data are available for insulin, which was introduced in the market for pulmonary delivery, heparin and interleukin-2 (IL-2) (4, 5, 8, 16, 22, 27, 35, 48-50).
| Table 3. Examples of aerosol inhalation for systemic treatment (from (4, 16, 22, 49, 51)). Note that the inhalant application of most substances is experimental in animals or clinical studies or off-label use and not approved for human use. Some cytokines tested in clinical studies failed to show a sufficient antitumour effect even though there was a proven systemic effect of the cytokine (51). Furthermore, for some substances an additional local mode of action after inhalation has been described, which is not considered in this table (4, 16, 51, 52). The table is not complete, but it demonstrates the large variety of drugs, which have been administered by pulmonary instillation or aerosol inhalation in clinical investigations or experimental studies. |
 |
| #)Approximated values; data in part for non-glycosylated monomers of peptides and proteins; ##) BAY 41-2272, BAY 41-8543, BAY 58-2667; ###) Phosphodiesterase inhibitors; ####) Proteins with an arginine-glycine-aspartate sequence |
Safety of the inhalation of peptides and proteins
An analysis of safety and tolerability of pulmonary administered compounds includes their activity after inhalation, which can be largely different compared with subcutaneous administration. For example, inhaled insulin causes a more rapid decrease of the blood glucose concentration than subcutaneously administered insulin (8, 11, 12, 26-28). Pulmonary diseases may complicate or prevent inhalant drug therapy under some circumstances (8, 11, 17). Inhaled pharmaceuticals and additives may induce incompatibility. For example, peptides and proteins can cause immunisation (8, 10, 11, 28), but also can have specific effects on the target organ lung (e.g., growth stimulating effect of insulin) (12, 28). In addition, a chronic administration of bile acids, cyclodextrins, and other absorption enhancers can damage alveolar epithelium (15, 28). Finally, administration of compounds by means of microparticles and liposomes can harm the lung (15). The latter, even though often a safe type of therapy, can damage the lung via production of reactive oxygen species (ROS) in case of cationic liposomes (1).
A large number of studies have demonstrated the feasibility and safety of pulmonary administration of drugs and biomolecules for systemic treatment (8, 11, 16, 53). However, there are few data regarding the long-term effects of inhaled macromolecules, except insulin and heparin (1, 8, 11, 12, 16, 27, 28, 48, 50, 53, 82). The effects of inhaled macromolecules should be thoroughly investigated in future studies to ensure the safety of this pharmaceutical form for therapy. Packing of the macromolecules into microparticles and liposomes and addition of stabilisers or absorption enhancers can improve the bioavailability and reduce the required drug doses and therapy costs. Such compounds can strongly affect safety and tolerability of inhalant drug therapy. Therefore, they also should be subject of intensive studies, including lung function diagnostics for detection of therapy-induced untoward effects. In summary, advances in aerosol therapy in the last decades will allow the introduction of inhalation based methods for drug administration for treatment of systemic diseases as an alternative of subcutaneous injection and will improve convenience and compliance of the treated patients.
Conficts of interest: Dr. RUdiger Siekmeier has no conflicts of interest in relation to this article. Dr. Gerhard Scheuch does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. Dr. Scheuch is a consultant for several pharmaceutical companies in the field of aerosol medicine and pulmonary drug delivery (e.g., Bayer-Schering, Boehringer/Ingelheim, GSK, Novartis, Talecris, Sandoz).
- Agu RU, Ugwoke MI, Armand M, Kinget R, Verbeke N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir Res 2001; 2: 198-209.
- Scheuch G, Kohlhaeufl MJ, Brand P, Siekmeier R. Clinical perspectives on pulmonary systemic and macromolecular delivery. Adv Drug Deliv Rev 2006; 58: 996-1008.
- Scheuch G, Siekmeier R. Novel approaches to enhance pulmonary delivery of proteins and peptides. J Physiol Pharmacol 2007; 58 (Suppl 5): 615-625.
- Niven RW. Delivery of biotherapeutics by inhalation aerosol. Crit Rev Ther Drug Carrier Syst 1995; 12: 151-231.
- Yu J, Chien YW. Pulmonary drug delivery: Physiologic and mechanistic aspects. Crit Rev Ther Drug Carrier Syst 1997; 14: 395-453.
- Nicod LP. Pulmonary defense mechanisms. Respiration 1999; 66: 2-11.
- Weibel ER. Morphometry of the Human Lung. Springer-Verlag, Berlin, 1963.
- Wolff RK. Safety of inhaled proteins for therapeutic use. J Aerosol Med 1998; 11: 197-219.
- Patton JS, Byron PR. Inhaling medicines: Delivering drugs to the body through the lungs. Nat Rev Drug Discov 2007; 6: 67-74.
- Patton JS, Platz RM. Routes of delivery: Case studies. (2) Pulmonary delivery of peptides and proteins for systemic action. Adv Drug Deliv Rev 1992; 8: 179-196.
- Valente AXCN, Langner R, Stone HA, Edwards DA. Recent advances in the development of an inhaled insulin product. Biodrugs 2003; 17: 9-17.
- Cefalu WT. Concept, strategies, and feasibility of non-invasive insulin delivery. Diabetes Care 2004; 27: 239-246.
- Weibel ER. Morphometry of the human lung: The state of the art after two decades. Bull Eur Physiopathol Respir 1979; 15: 999-1013.
- Dhand R. New frontiers in aerosol delivery during mechanical ventilation. Respir Care 2004; 49: 666-677.
- Hussain A, Arnold JJ, Khan MA, Ahsan F. Absorption enhancers in pulmonary protein delivery. J Control Release 2004; 94: 15-24.
- Kohler D, Fleischer W. Medikamente. In Theorie und Praxis der Inhalationstherapie, D Köhler, W Fleischer (eds). Arcis Verlag, München, 2000, pp. 71-99.
- Siekmeier R, Scheuch G. Systemische Therapie mit Aerosolen. Beispiele zur pulmonalen Verabreichung von Makromolekülen zur systemischen Therapie. Atemw-Lungenkrkh 2005; 31: 391-410.
- Telko MJ, Hickey AJ. Dry powder inhaler formulation. Respir Care 2005; 50: 1209-1227.
- Irngartinger M, Camuglia V, Damm M, Goede J, Frijlink HW. Pulmonary delivery of therapeutic peptides via dry powder inhalation: Effects of micronisation and manufacturing. Eur J Pharm Biopharm 2004; 58: 7-14.
- Shoyele SA, Cawthorne S. Particle engineering techniques for inhaled biopharmaceuticals. Adv Drug Deliv Rev 2006; 58: 1009-1029.
- Newman SP. Principles of metered-dose inhaler design. Respir Care 2005; 50: 1177-1190.
- Byron PR, Patton JS. Drug delivery via the respiratory tract. J Aerosol Med 1994; 7: 49-75.
- Kobayashi S, Kondo S, Juni K. Critical factors on pulmonary absorption of peptides and proteins (diffusional barrier and metabolic barrier). Eur J Pharm Sci 1996; 4: 367-372.
- Patton JS, Trinchero P, Platz RM. Bioavailability of pulmonary delivered peptides and proteins: a-interferon, calcitonins and parathyroid hormones. J Control Release 1994; 28: 79-85.
- Patton JS, Fishburn CS, Weers JG. The lungs as a portal of entry for systemic drug delivery. Proc Am Thorac Soc 2004; 1: 338-344.
- Barnett AH. Exubera inhaled insulin: A review. Int J Clin Pract 2004; 58: 394-401.
- Mastrandrea LD, Quattrin T. Clinical evaluation of inhaled insulin. Adv Drug Deliv Rev 2006; 58; 1061-1075.
- Patton JS, Bukar JG, Eldon MA. Clinical pharmacokinetics and pharmacodynamics of inhaled insulin. Clin Pharmacokinet 2004; 43: 781-801.
- Lombry C, Edwards DA, Preat V, Vanbever R. Alveolar macrophages are a primary barrier to pulmonary absorption of macromolecules. Am J Physiol Lung Cell Mol Physiol 2004; 286: L1002-L1008.
- Sinha VR, Kaur MP. Permeation enhancers for transdermal drug delivery. Drug Dev Ind Pharm 2000; 26: 1131-1140.
- Song Y, Wang Y, Thakur R, Meidan VM, Michniak B. Mucosal drug delivery: Membranes methodologies, and applications. Crit Rev Ther Drug Carrier Syst 2004; 21: 195-256.
- Davis SS, Illum L. Absorption enhancers for nasal drug delivery. Clin Pharmacokinet 2003; 42: 1107-1128.
- Edwards DA, Dunbar C. Bioengineering of therapeutic aerosols. Annu Rev Biomed Eng 2002; 4: 93-107.
- Kobayashi S, Kondo S, Juni K. Study on pulmonary delivery of salmon calcitonin in rats: Effects of protease inhibitors and absorption enhancers. Pharm Res 1994; 11: 1239-1243.
- Okamoto H, Todo H, Iida K, Danjo K. Dry powders for pulmonary delivery of peptides and proteins. Kona 2002; 20: 71-83.
- Sakagami M, Byron PR. Respirable microspheres for inhalation. The potential of manipulating pulmonary disposition for improved therapeutic efficacy. Clin Pharmacokinet 2005; 44: 263-277.
- Kobayashi S, Kondo S, Juni K. Pulmonary delivery of salmon calcitonin dry powders containing absorption enhancers in rats. Pharm Res 1996; 13: 80-83.
- Bitonti AJ, Dumont JA, Low SC et al. Pulmonary delivery of an erythropoietin Fc fusion protein in non-human primates through an immunoglobulin transport pathway. Proc Natl Acad Sci USA 2004; 101: 9763-9768.
- Bitonti AJ, Dumont JA. Pulmonary administration of therapeutic proteins using an immunoglobulin transport pathway. Adv Drug Deliv Rev 2006; 58: 1106-1118.
- Dumont JA, Bitonti AJ, Clark D, Evans S, Pickford M, Newman SP. Delivery of an erythropoietin-Fc fusion protein by inhalation in humans through an immunoglobulin transport pathway. J Aerosol Med 2005; 18: 294-303.
- Grenha A, Remunan-Lopez C, Carvalho ELS, Seijo B. Microspheres containing lipid/chitosan nanoparticles complexes for pulmonary delivery of therapeutic proteins. Eur J Pharm Biopharm 2008; 69: 83-93.
- Koshkina NV, Waldrep JC, Roberts LE, Golunski E, Melton S, Knight V. Paclitaxel liposome aerosol treatment induces inhibition of pulmonary metastases in murine renal carcinoma model. Clin Cancer Res 2001; 7: 3258-3262.
- Verschraegen CF, Gilbert BE, Loyer E et al. Clinical evaluation of the delivery and safety of aerosolized liposomal 9-nitro-20(S)-camptothecin in patients with advanced pulmonary malignancies. Clin Cancer Res 2004; 10: 2319-2326.
- Low SC, Nunes SL, Bitonti AJ, Dumont JA. Oral and pulmonary delivery of FSH-Fc fusion proteins via neonatal Fc receptor-mediated transcytosis. Hum Reprod 2005; 20: 1805-1813.
- Huang YY, Wang CH. Pulmonary delivery of insulin by liposomal carriers. J Control Release 2006; 113: 9-14.
- Rudt S, Muller R. in vitro phagocytosis assay of nanoparticles and microparticles by chemiluminescence. 1. Effect of analytical parameters, particle size and particle concentration. J Control Release 1992; 22: 263-271.
- Edwards DA, Hanes J, Caponetti G et al. Large porous particles for pulmonary drug delivery. Science 1997; 276: 1868-1871.
- Guntur VP, Dhand R. Inhaled insulin: Extending the horizons of inhalation therapy. Respir Care 2007; 52, 911-922.
- Kohler D. Aerosols for systemic treatment. Lung 1990; 168 (Suppl.): 677-684.
- Laube BL. The expanding role of aerosols in systemic drug delivery, gene therapy, and vaccination. Respir Care 2005; 50: 1161-1176.
- Thipphawong J. Inhaled cytokines and cytokine antagonists. Adv Drug Delivery Rev 2006; 58: 1089-1105.
- Wylam ME, Ten R, Prakash UBS, Nadrous HF, Clawson ML, Anderson PM. Aerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis. Eur Respir J 2006; 27: 585-593.
- Kohler D. Aerosolized heparin. J Aerosol Med 1994; 7: 307-314.
- Scheuch G, Brand P, Meyer T et al. Anticoagulative effects of the inhaled low molecular weight heparin certoparin in healthy subjects. J Physiol Pharmacol 2007; 58 (Suppl 5): 603-614.
- Ryszka F, Dolinska B. Initial studies on the administration route of prolactin. Boll Chim Farm 2001; 140: 169-171.
- Anderson PM, Markovic SN, Sloan JA et al. Aerosol granulocyte macrophage-colony stimulating factor: A low toxicity, lung-specific biological therapy in patients with lung metastases. Clin Cancer Res 1999; 5: 2316-2323.
- Okumu FW, Lee RY, Blanchard JD et al. Evaluation of the AERx pulmonary delivery system for systemic delivery of a poorly soluble selective D-1 agonist, ABT-431. Pharm Res 2002; 19: 1009-1012.
- Davison S, Thippawong J, Blanchard J et al. Pharmacokinetics and acute safety of inhaled testosterone in postmenopausal women. J Clin Pharmacol 2005; 45: 177-184.
- Corcoran TE. Inhaled delivery of aerosolized cyclosporine. Adv Drug Deliv Rev 2006; 58: 1119-1127.
- Iacono AT, Johnson BA, Grgurich WF et al. A randomized trial of inhaled cyclosporine in lung-transplant recipients. N Engl J Med 2006; 354: 141-150.
- Deshpande D, Blanchard J, Srinivasan S et al. Aerosolization of lipoplexes using AERx Pulmonary Delivery System. AAPS PharmSci 2002; 4: E13.
- Farr SJ, Otulana BA. Pulmonary delivery of opioids as pain therapeutics. Adv Drug Deliv Rev 2006; 58: 1076-1088.
- Evgenov OV, Kohane DS, Bloch KD et al. Inhaled agonists of soluble guanylate cyclase induce selective pulmonary vasodilation. Am J Respir Crit Care Med 2007: 176; 1138-1145.
- Heinzer H, Mir TS, Huland E, Huland H. Subjective and objective prospective, long-term analysis of quality of life during inhaled interleukin-2 immunotherapy. J Clin Oncol 1999; 17: 3612-3620.
- Heinzer H, Huland E, Huland H. Regionale Immuntherapie beim metastasierten Nierenzellkarzinom. Urologie (A) 2002; 41: 239-248.
- Huland E, Heinzer H. Renal cell carcinoma - innovative medical treatments. Curr Opin Urol 2004; 14: 239-244.
- Skubitz KM, Anderson PM. Inhalational interleukin-2 liposomes for pulmonary metastases: A phase I clinical trial. Anticancer Drugs 2000; 11: 555-563.
- Shahiwala A, Misra A. A preliminary pharmacokinetic study of liposomal leuprolide dry powder inhaler: A technical note. AAPS PharmSciTech 2005; 6: E482-E486.
- http://www.solvaypharmaceuticals-us.com/newsroom/pressreleases/0,,28488-2-0,00.htm 2005.
- Schermuly RT, Krupnik E, Tenor H et al. Coaerosolization of phosphodiesterase inhibitors markedly enhances the pulmonary vasodilatory response to inhaled iloprost in experimental pulmonary hypertension. Maintenance of lung selectivity. Am J Respir Crit Care Med 2001; 164: 1694-1700.
- Lauterbach R, Szymura-Oleksiak J, Pawlik D, Warchol J, Lisowska-Miszczyk I, Rytlewski K. Nebulized pentoxifylline for prevention of bronchopulmonary dysplasia in very low birth weight infants: A pilot clinical study. J Matern Fetal Neonatal Med 2006; 19: 433-438.
- Aubin MC, Laurendeau S, Mommerot A et al. Differential effects of inhaled and intravenous sildenafil in the prevention of the pulmonary endothelial dysfunction due to cardiopulmonary bypass. J Cardiovasc Pharmacol 2008; 51: 11-17.
- Ichinose F, Erana-Garcia J, Hromi J et al. Nebulized sildenafil is a selective pulmonary vasodilator in lambs with acute pulmonary hypertension. Crit Care Med 2001; 29: 1000-1005.
- Pullamsetti S, Krick S, Yilmaz H et al. Inhaled tolafentrine reverses pulmonary vascular remodeling via inhibition of smooth muscle cell migration. Respir Res 2005; 6: 128.
- Ichinose F, Adrie C, Hurford WE, Bloch KD, Zapol WM. Selective pulmonary vasodilation induced by aerosolized zaprinast. Anesthesiology 1998, 88: 410-416.
- Gupta S, Moussy F, Dalby RN, Miekka SI, Bruley DF. Pulmonary delivery of human protein C and factor IX. Adv Exp Med Biol 1997; 411: 429-435.
- Beyer S, Speich R, Fischler M, Maggiorini M, Ulrich S. Long-term experience with oral or inhaled vasodilator combination therapy in patients with pulmonary hypertension. Swiss Med Wkly 2006; 136: 114-118.
- Gessler T, Seeger W, Schmehl T. Inhaled prostanoids in the therapy of pulmonary hypertension. J Aerosol Med 2008; Jan 16 [Epub ahead of print].
- Onen ZP, Akkoca Yildiz O, Eris Gulbay B, Karabiyikoglu G. Inhaled iloprost as a long-term additional therapy to oral sildenafil in severe idiopathic pulmonary arterial hypertension. Tuberk Toraks 2006; 54: 177-181.
- Olschewski H, Rohde B, Behr J et al. Pharmacodynamics and pharmacokinetics of inhaled iloprost, aerosolized by three different devices, in severe pulmonary hypertension. Chest 2003; 124: 1294-1304.
- Schermuly RT, Inholte C, Ghofrani HA et al. Lung vasodilatory response to inhaled iloprost in experimental pulmonary hypertension: Amplification by different type phosphodiesterase inhibitors. Respir Res 2005; 6: 76.
- Ceglia L, Lau J, Pittas AG. Meta-analysis: Efficacy and safety of inhaled insulin therapy in adults with diabetes mellitus. Ann Intern Med 2006; 145: 665-675.