However, pulmonary application of drugs by means of aerosols is influenced by a number of physical, physiological and individual factors which are described elsewhere (2-4, 6-11). A biopharmaceutical must have a sufficient physical and chemical stability to persist the process of nebulization without loss of its functional properties and without relevant aggregation within or after the nebulization process. The aerosol must be homogenous with respect to the produced particle size and the particle diameter should be optimized (aerodynamic diameter: 1-3 µm) for deposition in the alveolar region of the lung. Particles with aerodynamic diameters <1 µm are not deposited in the lung but expired. On the other hand, larger particles (>3 µm) are deposited in the tracheobronchial airways and do not reach the alveolar region. The breathing maneuver is another critical parameter for pulmonary drug application. An optimal pulmonary deposition is achieved with a slow and deep inhalation procedure. In addition, variations in lung morphology and ventilation due to diseases (e.g., asthma, chronic obstructive pulmonary disease (COPD)) and individual factors (e.g., smoking) have an influence on the alveolar deposition of inhaled particles. Finally, the absorption of the biomolecules after alveolar deposition is affected by structure and function of the physiological pulmonary defence mechanisms (e.g., proteases/peptidases, alveolar macrophages, physiological absorbance barriers) and specific properties of the biopharmaceuticals (e.g., molecular weight, lipophilicity, solubility in water and lipids).
The application of biomolecules by means of different inhalation approaches has been investigated in a large number of studies (4, 7, 8, 12, 13). In principle, some of the biomolecules can be given without additives. Other large molecules, especially peptides and proteins, require stabilisers and inhibitors of phagocytosis (e.g., protease inhibitors, microspheres, liposomes) or absorption enhancers (e.g., detergents, bile acids, cyclodextrins) which can cause tissue irritation (1, 2, 4, 8, 11, 14-16). Compared with absorption enhancers, the use of carrier-based systems (e.g., liposomes and microspheres) has some more specific advantages for sustained and targeted drug delivery as compiled in Table 1 (17).
In the last years, a large number of studies on pulmonary application of metabolically active hormones (e.g., insulin, calcitonin, growth hormone, somatostatin, thyroid-stimulating hormone (TSH) and follicle-stimulating hormone (FSH)), growth factors (e.g., granulocyte-colony stimulating factor (G-CSF) and granulocyte monocyte-colony stimulating factor (GM-CSF)), distinct interleukins (e.g., IL-2) and heparin (unfractionated and low molecular weight heparin (LMWH)) have been performed (1, 4, 8, 9, 13). However, most experience is available for the inhalation of insulin. In addition, from the large number of substances insulin is the one with the greatest relevance because of the large number of diabetic patients worldwide. In our review we describe the current status and problems of devices for pulmonary administration of insulin.
| Table 1. Advantages of carrier-based systems for sustained drug delivery; according to (17). |
 |
Insulin, a peptide hormone (MW: 6000 Da) consisting of 2 chains (a und b) linked by three disulfide bonds, has been isolated 1921 by Banting and Best and was introduced into clinical treatment on January 11th 1922 (18-20). At the beginning, it was exclusively administered by intramuscular injection. However, because of the lower traumatisation of the patient subcutaneous application was rapidly established (20). Other techniques for drug application (transdermal, ocular, oral, buccal, nasal, pulmonal, rectal, vaginal, and transuterine) were also investigated and some of them are currently under further investigation (3, 18, 20, 21) (Table 2).
| Table 2. Methods for non-invasive administration of insulin; according to (18, 20). |
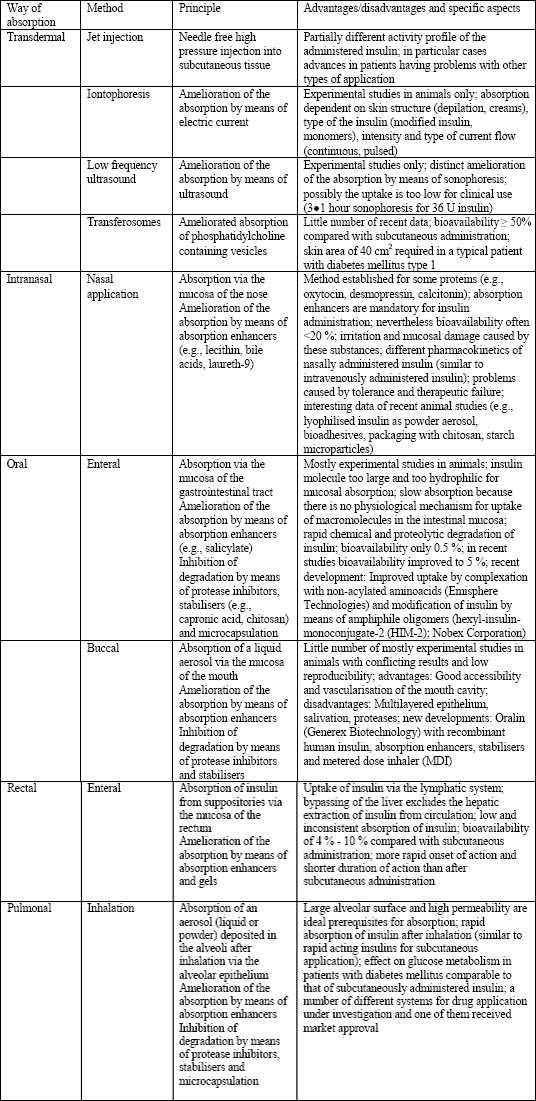 |
In 1924 and 1925 only two years after the start of the therapeutic insulin era the first studies on insulin inhalation were published. Laqueur and Grevenstuk (22) published her investigation on intratracheal administration of insulin in 1924 and reported a more rapid onset of action after intratracheal administration compared with subcutaneous administration. A first study on inhalation of insulin in patients was performed by Heubner et al (23) also in 1924. These investigators reported a dose-dependent effect of insulin inhalation on blood glucose. However, a 30-times higher dose for inhalation was required than that for subcutaneous administration and the authors assumed a problem in the requirement of high amounts of insulin, even though they also emphasised the advantage of this type of administration for the patients (23). At the same time and independently from the investigations of Heubner et al (23), Gänsslen (24) performed the investigations in patients and reported that the inhalation of insulin was well tolerated and caused a significant decrease of the blood glucose concentration, and that the amounts of insulin required for inhalation in relation to subcutaneous application were not as high as described by Heubner et al (23). However, because of the large number of unsolved problems, it took 46 years more until Wigley et al (20, 25) published their pivotal study of insulin inhalation offering the proof of principle of this therapy. They investigated three subjects without diabetes mellitus and four patients with diabetes and they were able to demonstrate that pork-beef insulin administered by a nebulizer caused a prompt increase in plasma immunoreactive insulin and that hypoglycemia showed a temporal relationship with the increase in plasma immunoreactive insulin (25). However, even after the investigation of Wigley et al (25) inhalant insulin therapy was far away from its introduction into clinical therapy and in the next two decades several studies ruled out the basics of insulin inhalation (8, 26-28). In these years, it was observed that the bioavailability of inhaled insulin in case of improved application procedures was only about 20 to 25% of that after subcutaneous administration, but also that inhalation might be an important alternative administration route (4, 8, 20). However, the methods under investigation were not able to administer sufficient drug doses in a reproducible way, because their particle spectrum was optimized for aerosol deposition in the bronchial system and not in the alveoli (20, 29).
Based on the advances in asthma therapy by means of aerosols, nebulizers, metered dose inhalers (MDIs) and dry powder inhalers (DPI) at about 1990 the scientific and technical prerequisites for the inhalant application of insulin were established and a number of studies on the inhalant application of insulin were initiated in the following years (4, 20). Several companies developed devices for inhalant administration of insulin, which are very different in respect to the technical and pharmacological principles (e.g., manual or semi-automated systems for inhalation, powder aerosol or liquid aerosol) and briefly described in Table 3 (20). The most advanced method was Exubera® from Pfizer/Nektar, which received the approval from the American and European Drug Agencies (FDA and EMEA, respectively) in early 2006 for patients with diabetes mellitus types 1 and 2 and was marketed since September 2006. Exubera® was based on recombinant human insulin which was spray-dried and supplemented with the excipients mannitol, glycine, and sodium citrate. The insulin content of the final product, a large low-density particle, packed into small blisters was 60% (30). However, in October 2007 Pfizer announced it would be dropping Exubera®, citing that the drug had failed to gain market acceptance. Another device for insulin inhalation in advanced developmental status was AERx® iDMS (Aradigm Corporation, Novo Nordisk) (Table 3) (20). Shortly after the decision of Pfizer, Novo Nordisk also stopped all investigations on inhaled insulin.
| Table 3. Devices for inhalant administration of insulin; modified according to (18, 20, 31-36). |
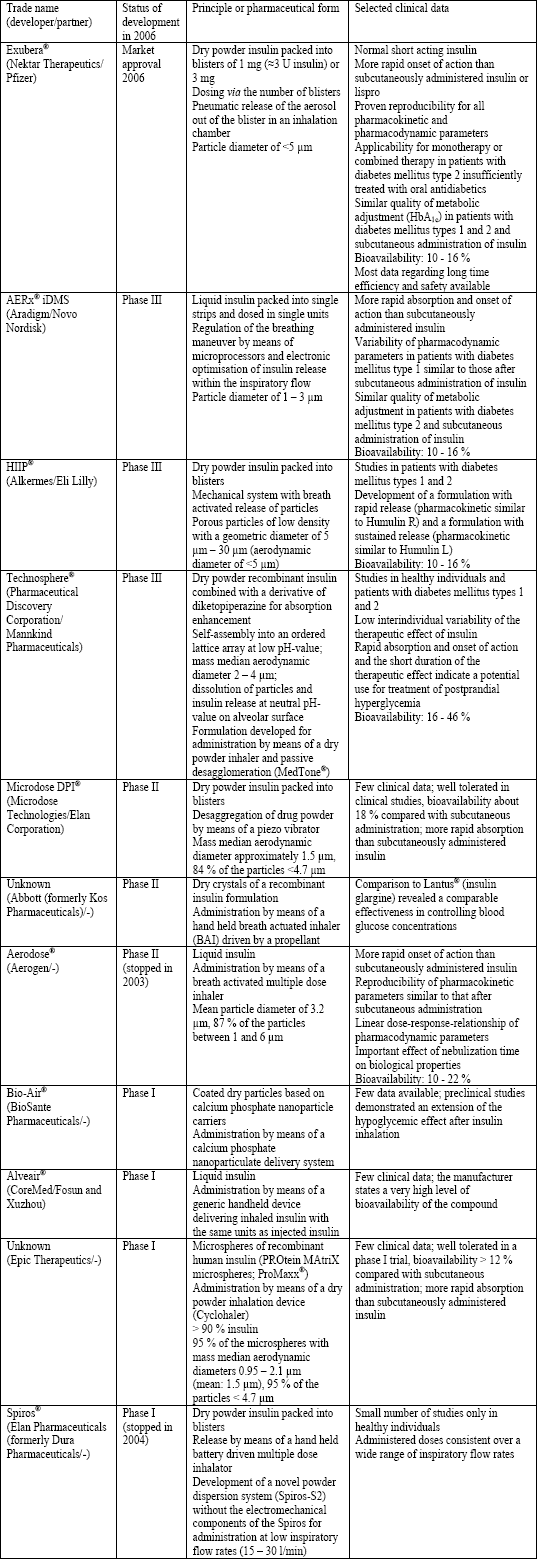 |
In some of the novel techniques for insulin inhalation the protein is formulated into microspheres (liposomes, particles, large porous particles). Even though the majority of these methods up to now was investigated in animal studies only, they may play a role for inhalant drug administration for insulin and other biomolecules in the future (Table 4) (1, 17, 37). The use of microparticles is based on the observation that smaller particles are phagocytosed more rapidly than larger ones (49). Biomolecules (e.g., insulin) can be packed into the inner part of biologically degradable polymers and lipids (microparticles and liposomes, respectively) (1, 7, 11, 15, 50). In consequence, the physiological alveolar clearance mechanism and the degradation of proteins and peptides after phagocytosis by alveolar macrophages are slowed which results in increased bioavailability. Another advantage is the alteration of the pharmacokinetic properties of the administered substances due to their slow release from these particles (7, 11, 15, 50). However, prerequisites for the use of these excipients are their rapid degradation after inhalation, readily elimination after inhalation and drug release, and immunological and toxicological inertness (37). In the last years, distinct procedures have been developed for the packing of proteins (e.g., insulin) into liposomes and solid particles (Table 4).
| Table 4. Microparticle and liposome formulations for delivery of insulin to the lungs; modified according to (17, 37). Note that some types of microparticles are also subject of clinical studies described in Table 3. |
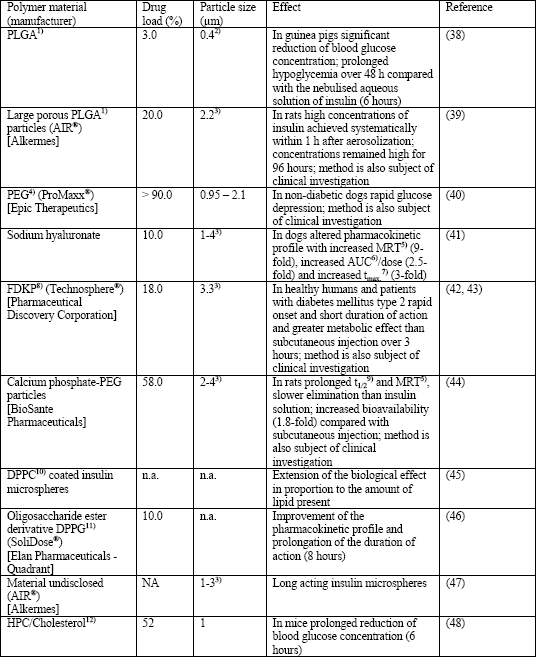 |
| 1)PLGA: Polylactic-co-glycolic acid; 2)Geometric diameter; 3)Aerodynamic diameter; 4)PEG: Polyethylene glycol; 5)MRT: Mean residence time; 6)AUC: Area under the curve; 7)tmax.: Time to reach the maximum serum concentration (Cmax.); 8)FDKP: 3,6-bis(N-fumaryl-N-(n-butyl)amino-2,5-diketopiperazine; 9)t1/2: Plasma half-life time; 10)DPPC: Dipalmitylphosphatidylcholine; 11)DPPG: Dipalmitylphosphatidylglycerol; 12)HPC: Hydrogenated egg yolk phosphatidylcholine. |
In detail, drug carrying capacity, drug release rate, toxicity, and pulmonary deposition of liposomes depend on their size, drug/lipid ratio, the properties of the used phospholipids (chain length, electrical charge, composition by neutral or anionic lipids), and the chosen method of delivery (1, 15, 17, 50). Most frequently they are made from lecithins (phosphatidylcholines), phosphatidylethanolamines, sphingomyelins, phosphatidylserines, phosphatidylglycerols and phosphatidylinositols (17). According to this structure, both hydrophobic and hydrophilic compounds can be packed into liposomes prior to the delivery to the lungs. Hydrophilic compounds (e.g., pharmaceuticals and larger biomolecules) are entrapped into the vesicle in the inner of the liposome whereas lipophilic (hydrophobic) compounds are encapsulated into the membrane bilayer. Small liposomes are unilamellar bodies with a hydrophilic core, whereas larger multilamellar liposomes have an onion-like structure with several layers of phospholipids and aqueous compartments. Because of their strong chemical and structural similarity, liposomes deposited in lung alveoli merge with cell membranes and facilitate the absorption of the carried biomolecule (e.g., insulin). Advantages of liposomes are sustained drug release, prevention of local irritation, reduced toxicity, improved stability in the large aqueous core, and the possibility for manipulation of release and targeting by variation of the bilayer constituents.
Solid particles (microspheres or large porous particles) are chemically and physically more stable than liposomes and allow higher drug loading (17). Pharmacological properties of microparticles (size range: <500 nm) depend on the used material, preparation technique, particle size, porosity, surface structure, and the delivery device (1, 15, 17, 50). Most frequently, the synthetic polymers polylactic acid (PLA) and polylactic-co-glycolic acid (PLGA) are used for their production. However, a number of other synthetic and natural polymers have been investigated (Table 4) (17). Up to now little is known about the pharmacological properties of most of the particles listed in Table 4, although some of these polymer-based systems might have toxicologically relevant effects especially after administration of high doses and/or for a longer time period (37). Microspheres can be produced by a number of distinct methods based on supercritical fluid technology, emulsion-solvent evaporation, spray-drying and phase separation. The encapsulation of peptides/proteins (e.g., insulin) by means of these techniques is affected by a number of physical and chemical properties (e.g., effect of solvents, heat, moisture, pH-value, oxygen and mechanic stress). Additionally, new techniques for production of microspheres from pure proteins have been developed (17). The release rate of the drug depends on many properties of the drug itself (concentration, solubility, molecular weight, nature of the peptide or protein) and of the polymer (e.g., nature, molecular weight, porosity, tortuosity, size, and uniformity) (17). Modification of the latter, e.g., by coating procedures, can be used to reduce the uptake by alveolar macrophages and, in consequence, to alter the pharmacological properties of the administered biomolecule (increase of pulmonary residence time and bioavailability) (17). Examples for the clinical use of microspheres for insulin inhalation are: ProMaxx®, Epic Therapeutics; Technosphere®, Pharmaceutical Discovery Corporation and calcium phosphate-polyethylene glycol particles, BioSante Pharmaceuticals (Table 4).
Large porous particles are characterized by geometric diameters >5 µm, low particle density (generally <0.1 g/ml), and aerodynamic diameters <5 µm. In consequence, these particles have good flow and aerozolization properties due to their low aerodynamic diameter and they are able to evade phagocytosis because of their large size (17). Aerosolized large porous particles deposit homogenously and reproducibly without relevant toxicity on the alveolar cell surface. However, further toxicological and pharmacological studies are required also for this excipient. Currently, only one system for insulin inhalation is based on large porous particles (AIR®, Alkermes) (Table 4).
Bioavailability of inhaled molecules after pulmonary deposition can be enhanced by a number of compounds increasing the absorption or inhibiting proteolytic degradation. Some of them are introduced into clinical treatment (e.g., Exubera®). However, most of the substances have been subject to studies in animals only and some of them can damage lung epithelium, especially after administration of higher doses and prolonged duration of treatment, necessary for patients with diabetes mellitus (2, 13, 15, 18, 36, 51).
The mode of action of absorption enhancers, which differ strongly with respect to their chemical structure and properties, is not yet completely understood. For example, bile acids presumably increase the absorption by alteration of the mucus layer, protection of proteins against enzymatic degradation, desaggregation of protein multimers, opening of epithelial tight junctions and solubilization of phospholipids and proteins out of the cell membrane followed by formation of micelles, whereas cyclodextrins, which are cyclic polymers of glucose, additionally form complexes with molecules fitting into their lipophilic inner structure (15, 50). Table 5 compiles the absorbance enhancing effect of various compounds and demonstrates that the intensity of their pharmacological effect depends on their type (e.g., different cyclodextrins and lanthanides) and their administered dose (e.g., sodium taurocholate and sodium glycocholate). However, it should be considered that the toxicity of absorption enhancers often correlates with the strength of their pharmacological effect limiting their clinical use (15, 36). The majority of data was obtained in rats only, whereas results from other mammals or human studies are available for few compounds only. For example, sodium citrate, mannitol and glycine are excipients used in Exubera® (30). The effect of bile acids was investigated in humans by Heinemann et al (36, 56), who found only a small increase in bioeffectivity if a powder aerosol of insulin was administered in combination with an endogenous bile acid in healthy individuals (12.0 ±3.5% vs. 7.6 ±2.9%). In contrast, Johansson et al (15, 22) observed a strongly increased bioavailability of insulin in dogs, if the substance was administered as a fluidic aerosol containing also taurocholate (taurocholate vs. control; 23.2 ±4.4% vs. 2.6 ±0.3%).
| Table 5. Effect of absorption enhancers on pulmonary insulin absorption; modified according to (15, 51). Note that the experiments in rats were often in situ studies. |
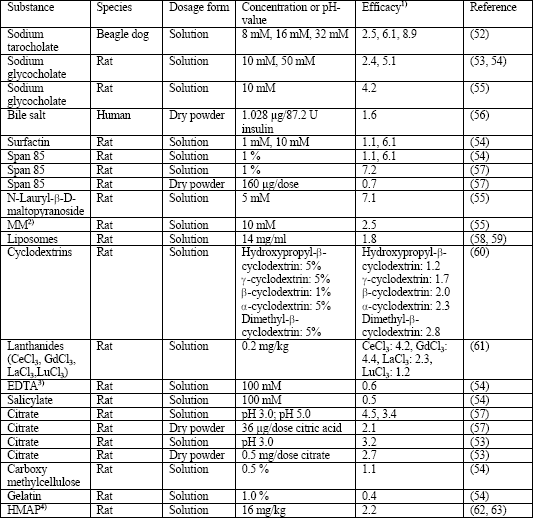 |
| 1)Efficacy: Ratio of the area under the curve (AUC) or biological response between the dosage form with absorption enhancer and that without absorption enhancer; 2)MM: Mixed micelles of linoleic acid and HCO60 (hydrogenated castor oil); 3)EDTA: Ethylene diamine tetraacetic acid; 4)HMAP: Hydroxymethyl amino propionic acid. |
Bioavailability and pharmacological activity of inhaled peptides and proteins can also be improved by addition of proteinase inhibitors preventing their inactivation by proteolytic cleavage (1, 11, 15). The effect of these compounds varies strongly depending on the type of the protease and the susceptibility of the peptide or protein. For example, an in vitro study dealing with the effect of selected protease inhibitors on the permeability of insulin across the rabbit trachea revealed peptidase efficacies in the order di-peptidylaminopeptidase IV > leu-aminopeptidase > cathepsin B > trypsin (15, 64). Another in vitro study demonstrated an inhibitory effect of the protease inhibitors bacitracin, aprotinin, soybean trypsin inhibitor, and sodium glycocholate on the degradation of insulin in lung homogenate in a descendent order (15, 65). However, the antiproteolytic properties of these compounds on insulin after tracheal or pulmonary administration were up to now only subject of few animal studies and not introduced into clinical investigations (Table 6).
| Table 6. Effect of protease inhibitors on pulmonary insulin absorption; modified according to (51). |
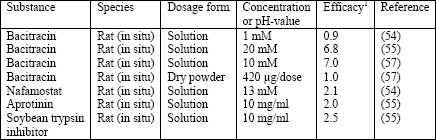 |
| 1Efficacy: Ratio of the area under the curve (AUC) or biological response between the dosage form with protease inhibitor and that without protease inhibitor. |
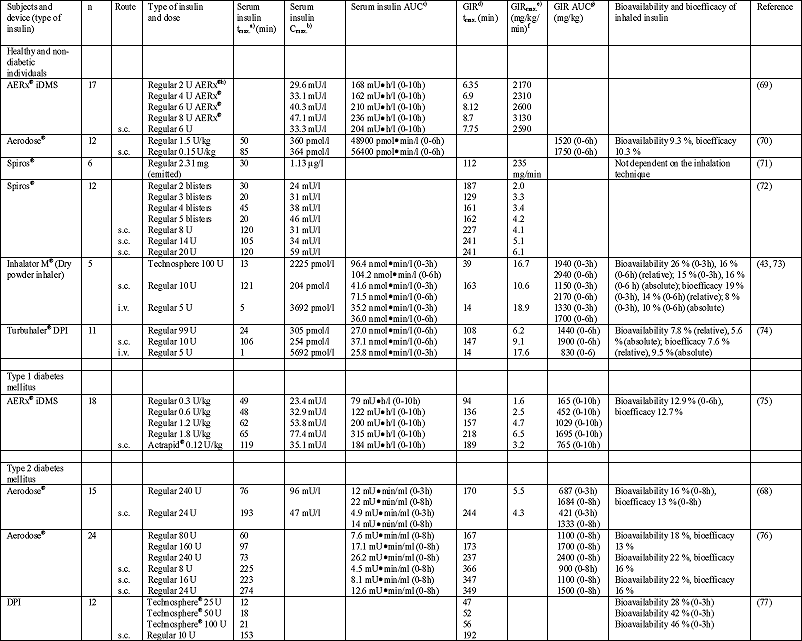
The pharmacokinetics of inhaled insulin was investigated in a large number of studies performed in healthy subjects and patients with diabetes mellitus types 1 and 2. Unfortunately, the comparison of the study results is hampered by differences of the used inhalers, administered formulations and doses of insulin, small numbers of included individuals (healthy individuals or patients), inappropriately used pharmacological models, and distinct parameters determined (36, 66). However, it has been observed that inhaled regular insulin is absorbed at least as fast as subcutaneously administered insulin (time to peak concentration in plasma (tmax.): 7-90 min vs. 42-274 min (Table 7) (4, 8, 20, 36, 66-68), an observation which has also been made in one of the first inhalation studies by Laqueur and Grevenstuk (22). The pharmacokinetics of inhaled insulin seems to be a biphasic one with a first peak rapidly after inhalation, which is followed by a slow release comparable to that after subcutaneous injection (36, 67). In the first 60 min after drug administration, the area under the concentration vs. time curve (AUC) is larger for inhaled insulin than for subcutaneously administered insulin. In contrast, subcutaneously administered insulin has a larger AUC if an observation period of 6 hours is considered (36). This suggests that inhaled insulin might have some therapeutic benefit in the treatment of prandial or postprandial hyperglycemia, when compared with conventionally administered insulin. In addition, compared with the subcutaneously administered drug, inhaled insulin shows a lower risk for postprandial hypoglycemia because of its increased clearance after inhalation (36, 78). Accordingly, determination of the glucodynamics in healthy individuals revealed a more rapid, but even shorter decrease of plasma glucose concentration than after subcutaneous administration of insulin (36).
| Table 7. Selected studies on pharmacokinetics and pharmacodynamics of inhaled insulin; according to (36). |
 |
| a)
tmax.: Time to reach Cmax. b) Cmax.: Maximum concentration of insulin in serum c) AUC: Area under the serum insulin concentration-time curve (between specified limits) d) GIR: Glucose infusion rate e) GIRmax.: Maximum GIR (peak) f) If not stated otherwise g) GIR AUC: Area under the GIR-time curve (between specified time limits) h) 1 U of AERx® |
Better conclusions on the pharmacokinetics can be obtained in studies by means of the glucose clamp technique or in patients without residual synthesis of insulin (diabetes type 1) and healthy individuals under pharmacological inhibition of insulin synthesis (Table 7) (36, 66). Different doses of inhaled insulin resulted in a widely linear dose-response relationship in patients with diabetes type 1 (36). However, although the maximum of the insulin concentration (Cmax.) increased with the administered dose, there was an increasing delay of the time to peak concentration in plasma (tmax.) indicating the existence of a dose dependent pulmonary uptake mechanism (36). In addition to the large number of studies with normal insulin, a small number of investigations were performed with lispro, an insulin derivitative modified by means of molecular biology. Compared with normal insulin lispro showed a better therapeutic effect (i.e., lower doses required to achieve the same serum concentration of insulin and more rapid onset of action). Probably, both effects are caused by a breakup of the hexamer into monomers followed by an increased bioavailability (19, 36).
In most studies, bioavailability was calculated by comparison of the AUC after inhalation to that after subcutaneous administration of insulin. In contrast, bioeffectivity describes the hypoglycemic effect of inhaled insulin compared with a defined insulin dose administered by subcutaneous injection (36). Therefore, the provision for the bioeffectivity can give further information. However, the parameters bioavailability and bioeffectivity result in an underestimation of the therapeutic effect of insulin, because only a small proportion of the administered drug is deposited in the lung periphery (i.e., alveolar region) from where it can be absorbed into circulation. In detail, the commercially available systems for pulmonary administration of insulin are characterized by bioavailabilities and bioeffectivities of 9-22 % and 8-16 %, respectively (Table 7), which is more than the 3% reported by Heubner et al (23) in 1924. In consequence, the insulin dose which is required to achieve the same therapeutic effect after inhalation is up to 11-times higher compared with subcutaneous administration (36). Between 50 and 80% of the insulin filled in the inhalation system does not reach the lung, but is remaining in the nebulizer, is deposited in the mouth, or the oropharynx, or is expired. Taking this into account, the bioavailability from the lung deposited fraction is about 2-5 times of the subcutaneously given insulin dose. However, from this dose more than 50% is deposited in the airways (bronchial system) and is removed from the lung by the mucociliary transport and/or degradation. Only about 40% is rapidly absorbed into the circulatory system. If this is also considered, it is obvious that the pulmonary extradose for insulin inhalation is 2-3-times of the dose required for injection (36).
Pulmonary deposition and, in consequence, bioavailability of inhaled aerosols (including insulin and other compounds) is mainly influenced by biological and physical parameters of the substance, the nebulizer, the breathing maneuver, and the oropharyngeal filter efficiency of the patient (9, 10, 18, 36, 66). An optimum deposition of the inhaled insulin is achieved if the aerosol is released at the beginning of a slow and deep inhalation maneuver. This enables the particles to penetrate deeply into the lung and they can be deposited in the alveolar region (9, 10, 18, 67). Farr et al (18, 67) observed in their study a later and weaker effect of insulin administered by a shallow inhalation maneuver (40% of inspiratory vital capacity (IVC)) than after a deep inspiration maneuver (80% IVC). This shows the importance of breathing pattern on alveolar deposition. The breathing maneuver does not only affect the total amount of alveolar insulin deposition, but also the intraindividual reproducibility of this therapy. However, other biological parameters (e.g., smoking, physical stress, lung perfusion) have also a strong effect on the intraindividual variability of insulin administered by inhalation. The reproducibility of pulmonary delivered insulin was investigated in several studies. In summary, these studies demonstrate a similar or even better reproducibility of insulin administration by inhalation than by subcutaneous injection (36, 68, 79-81). Obviously, the better reproducibility after inhalation is caused by the missing of some influencing parameters (e.g., physical exercise, smoking, temperature, body position and injection), which play a role after subcutaneous injection of insulin (18, 81, 82). In detail, similar intraindividual variabilities of various pharmacokinetic parameters (AUC, Cmax., tmax., blood glucose and rate of glucose infusion) were observed in the studies of Gelfand et al (36, 83) using Exubera® in patients with diabetes type 2, Hompesch et al (36, 84) using the AERx® system in patients with diabetes mellitus type 1 (36, 84) and Perera et al (68) using the Aerodose® system in patients with diabetes type 2 (coefficients of variation (CV) of the AUC0h-3h 19 and 23% after inhalation and subcutaneous injection, respectively). On the other hand, lower variabilities after inhalation than after injection were observed from Himmelmann et al (36, 80) using the AERx® system in healthy individuals (CV values of 13.7 and 16.5% in non-smokers and smokers, respectively) and Pfützner et al (36, 85) using Technosphere® in patients with diabetes mellitus type 2 (CV values 16-20%). For comparison, interindividual CV values for subcutaneous application of insulin are about 25% (79).
A number of original studies and reviews describe a higher absorption (up to 3-5 times) of inhaled insulin in smokers than in non-smokers (Table 8) (3, 8, 12, 20, 27, 36, 66, 80, 81). For example, Kohler et al (27, 66) reported a higher absorption (Cmax.) and bioavailability (65 vs. 25%) of inhaled insulin, which was accompanied by a more pronounced decrease of the glucose concentration in smokers compared with non-smokers. In another study Himmelmann et al (80) reported a higher absorption of inhaled insulin (AUC; 63.2 mUh/l vs. 40.0 mUh/l, P<0.01), a higher peak concentration (Cmax.; 42.0 mU/l vs. 13.9 mU/l, P<0.001), and a shorter time to peak (tmax.; 31.5 min vs. 53.9 min, P<0.001) in smokers compared with non-smokers. In addition, the mean residence time (MRT) in smokers was less than half of that in the non-smoker group (P<0.0001) and, accordingly, the apparent elimination rate constant of exogenous insulin was almost twice as high in smokers compared with non-smokers (P=0.0019). However, the intraindividual variability was similar in both groups (80). In another study Becker et al (87) investigated the effect of smoking cessation and subsequent resumption on the absorption of inhaled insulin. It was found that AUC and Cmax. were higher in smokers than in non-smokers, whereas tmax. was shorter. Smoking cessation resulted in a rapid change of the values obtained in smokers toward those of non-smokers. In contrast, smoking resumption completely reversed the effect of smoking cessation. In principle, this can be explained by several mechanisms. It is well established that chronic cigarette smoke inhalation increases the permeability of the alveolar-capillary barrier (91, 92). Postulated mechanisms for this increase are immunological modifications (93), an increase in the blood perfusion (94), surfactant antioxidant depletion due to an increased burden of inhaled reactive oxygen species (ROS) (95) and a disruption in surfactant function (96). On the other hand, an increased metabolism of drugs in smokers, e.g., due to an induction of drug-metabolising enzymes, has been reported (97). In consequence of augmented insulin metabolism, the metabolic activity of the hormone would be diminished. Importantly, the significantly higher values of AUC and Cmax. observed after insulin inhalation in smokers compared with non-smokers are not necessarily followed by a concomitant increase in insulin action. This apparent contradiction can be explained by the inhibition of metabolic insulin action followed by an induction of insulin resistance and glucose intolerance due to cigarette smoke inhalation (98-101). The increased epithelial permeability as a cause of the varied pharmacokinetics of insulin in smokers is reversible within a few days after the end of tobacco abuse (92), whereas the chronic bronchitis typically existing in long-time smokers is not reversible in this short period.
The effects of acute cigarette smoke inhalation on the absorption of inhaled insulin are largely different from that of chronic cigarette consumption, as cigarette consumption just before insulin inhalation significantly blunts the enhanced insulin absorption in smokers. However, there are no differences in tmax. (80). The underlying mechanisms of these effects are not understood. In principle, the well established bronchoconstrictory effect of nicotine might cause changes in ventilation and distribution which are followed by variations of the particle deposition of inhaled insulin (80). In addition, nicotine has a vasoconstrictory activity which might cause a delay of insulin absorption after subcutaneous administration (102). Likely, this vasoconstrictory effect inhibits also the absorption of insulin if the drug is administered directly after acute cigarette consumption (80). Furthermore, pulmonary neutrophils activated by components of cigarette smoke might cause an enzymatic degradation of insulin deposited in the alveoli (93). Finally, it should be noted that even acute passive cigarette smoke exposure may affect the pharmacokinetics of inhaled insulin. However, the effect of acute passive cigarette smoke exposure is just the opposite of that of active chronic smoking, because it causes a modest decrease of the bioavailability of inhaled insulin due to reduced lung permeability (103).
Inhalant insulin therapy was not approved in current smokers and individuals who quitted smoking less than 6 months before therapy until marketing was stopped by the manufacturers. In consequence, the number of diabetic patients which can be treated with insulin is strongly reduced, because about 20-25% of these patients are tobacco smokers (30, 104). By the time the inhalant therapy with insulin will be reintroduced into the market, further systematic investigations on the effect of cigarette consumption on insulin bioavailability in smokers should be performed. It might well be that insulin doses for smokers may be adapted (3, 36, 80).
The lung is a dynamic organ, strongly exposed to environmental factors and at risk for very different diseases. Patients with manifest pulmonary diseases affecting drug absorption were excluded from inhalant drug therapy in order to ensure a sufficient and reproducible deposition and bioavailability of the inhaled insulin. However, the effect of respiratory diseases on the pulmonary absorption of inhaled insulin was subject of a small number of studies. For example, infections of the upper respiratory tract have obviously no relevant effect on the bioavailability of inhaled insulin as it was shown by McElduff et al (36, 88) who observed no differences of pharmacokinetics and pharmacodynamics in otherwise healthy individuals within the period of an acute respiratory infection (Table 8). Another respiratory disease, asthma bronchiale, is characterized by hyperreactivity with bronchospasm, inflammation and airway remodelling. There are two primary concerns regarding the inhalation of insulin in asthma patients. Firstly, drug inhalation especially by means of DPI can induce bronchospasm. Secondly, in asthma exacerbation respiratory effort and bronchospasm limit the deposition of inhaled insulin in the lung alveoli. This is due to a variation of pulmonary convective gas transport, a smaller airway diameter and in consequence the higher rate of particle deposition in the central airways of these patients. Henry et al (36, 89) investigated the pharmacokinetics of inhaled insulin in asthma patients and reported a mild decrease of Cmax. and a distinct decrease of AUC (bioavailability) and plasma glucose concentration (bioeffectivity) after insulin inhalation in asthma patients compared with healthy individuals. Furthermore, patients with asthma showed a higher variability of Cmax. and AUC, but not of the glucose lowering effect than healthy controls after insulin inhalation (Table 8). Presumably, these inappropriate effects can be improved by administration of bronchodilators in these patients (105). Data regarding the pharmacokinetics of inhaled insulin in patients with chronic obstructive pulmonary disease (COPD) are limited and conflicting. COPD patients demonstrated a variable (higher or lower) absorption of insulin compared with subjects without COPD. It is not clear whether this variability is secondary to differences in inhalation devices or different study populations (105, 106). In consequence, the effect of COPD on insulin absorption should be subject of further studies prior to the reintroduction of inhalant insulin therapy into the market, because COPD is a frequent and often not or not correctly diagnosed pulmonary disease.
| Table 8. Studies investigating factors influencing the pharmacokinetics of regular human insulin inhaled by devices developed for insulin inhalation; according to (36). |
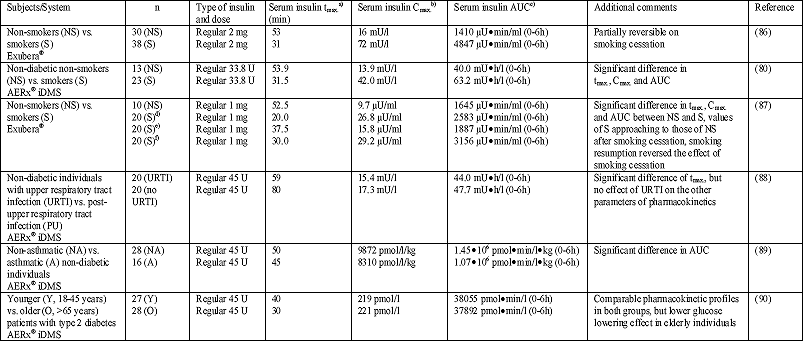 |
| a)
tmax.: Time to reach Cmax. b) Cmax.: Maximum concentration of insulin in serum c) AUC: Area under the serum insulin concentration-time curve (between specified limits) d) Before smoking cessation e) 7 days after smoking cessation f) 9-10 days after smoking resumption |
Lung morphology and function change as a function of age. Elder individuals show a decrease of the alveolar surface, a variation of lung elasticity, a decrease of the alveolar capillary volume combined with a decline of the ventilation/perfusion ratio, a decrease of the pulmonary diffusion capacity for carbon monoxide (DLCO), and an increase of the pulmonary residual volume (RV) (21). Therefore, the age is another important parameter influencing the pharmacokinetics of inhaled insulin. Henry et al (36, 90) reported similar values of Cmax. and AUC in patients with diabetes type 2 aged >65 years and young individuals of the age between 18 and 45 years. The variability in these parameters was not different between both study groups either. However, the observed decrease of plasma glucose concentrations was more pronounced in younger individuals than in elder patients indicating a requirement of higher doses in aged patients (Table 8).
The experience of the last 80 years in millions of patients has shown that the treatment of diabetes mellitus with subcutaneously administered insulin is relatively safe. However, beside the specific aspects of bioavailability and bioeffectivity discussed before, the aspects of tolerability and toxicity must be once more investigated for the inhalant therapy with insulin. In principle, not only insulin, but also absorption enhancers might cause adverse effects in the lung. The latter are variations in lung function, local inflammation, allergic reaction, formation of antibodies against insulin, pulmonary fibrosis, and lipodystrophy (3, 20, 36). Most data regarding the long-term tolerability are published for the Exubera® system and the AERx iDMS® system for study periods of up to 2 years and more in patients with diabetes types 1 than 2 (18, 33, 36, 81, 107-113).
Animal studies and investigations in diabetic patients demonstrated that diabetes and consecutive insulin treatment cause a morphological change of lung structure (e.g., thickening of the alveolar membrane and the capillary basal lamina, vascular hyalinosis, granulomas, intraseptal nodular fibrosis and emphysema-like septal obliteration) which depends on the duration and severity of the disease and on additional factors, like smoking (104, 114). Therefore, the effect of insulin inhalation on lung function has been thoroughly investigated. In most studies inhalation of insulin caused no changes of spirometric parameters of lung function (e.g., forced expiratory volume in 1 s (FEV1), forced vital capacity (FVC)) and parameters of diffusion capacity for carbon monoxide (DLCO). and blood gas analysis (3, 18, 20, 33, 36, 81, 107-120). Changes in the DLCO which were observed in some studies could not be explained by the investigators. However, based on the results of the lung function tests and the observed variations in individual patients, the manufacturer recommended spirometric measurement of lung function before treatment, after 6 months, and thereafter at least annually in the product information for Exubera® (121). In summary, the experience regarding the effect on lung function indicates that inhaled insulin is characterized by a low pulmonary toxicity, good tolerance, and good bioeffectivity (3, 18, 20, 36). This may be explained by a relatively low toxicity of insulin itself and the distribution of the inhaled doses of 4-5 mg (three times a day) on a total alveolar surface of about 80-120 m2 (3, 9, 10). The total quantity of the inhaled substance is lower than the threshold value for dust inhalation of 30 mg/day recommended by the American Council of Government Industrial Hygienists (3, 78). Furthermore, the initially high concentration of insulin in the epithelial lining fluid is rapidly decreasing due to absorption and distribution in the body fluid and proteolytic degradation. As a result, there is no evidence that pulmonary tissue is exposed to a higher insulin concentration after inhalation than after subcutaneous injection (36).
Beside its strong metabolic effect, insulin also acts as a weak growth factor (efficiency of only 1/100 of insulin-like growth factor-1(IGF-1)) after binding to the receptor for IGF-1. However, there is up to now no evidence for a relevant competitive effect of inhaled insulin at the IGF-1 receptors in the lung (36). In one of the studies on inhalant insulin application, pulmonary fibrosis had been observed, but there was obviously no relationship to the study medication (81). Recently, about 6 months after the end of the marketing of Exubera®, the American Food and Drug Administration (FDA) published a press release reporting a potentially increased risk for bronchial carcinoma in ex-smokers treated with inhalant insulin (122). In detail, there have been 6 newly diagnosed cases of primary lung malignancies in clinical trials among Exubera®-treated patients, and 1 newly diagnosed case among comparator treated patients. There has also been 1 post-marketing report of a primary lung malignancy in an Exubera®-treated patient. All these patients had a prior history of cigarette smoking. However, even though the number of cases is too small for a final risk evaluation, the potentially increased risk for lung cancer should be subject of further investigation prior to product relaunch of Exubera® or similar products of competitors.
In a number of studies, inhalation of insulin was followed by increased serum titres of non-neutralizing IgG antibodies against insulin. However, the development of these antibodies had no therapeutic relevance, i.e., there was no correlation to the metabolic control, lung function, and adverse events (18, 20, 21, 33, 36, 81, 107, 108, 110-112, 116-118, 120, 123, 124). The occurrence of these antibodies has been observed in up to one third of patients with diabetes mellitus (especially those of type 1) and long-time subcutaneous administration of insulin, more frequently in younger individuals than in elderly and with a strong increase in frequency and titre until 6 to 12 months after start of insulin inhalation, but also in patients without diabetes mellitus (e.g., patients with autoimmune diseases). It is likely, that the induction of antibodies against insulin is caused by its formulation and dose (inhaled insulin is given in higher doses and more frequently for treatment of postprandial hyperglycemia than subcutaneous insulin) and the site of delivery (presence of macrophages, dendritic cells, and lymphocytes in the lung), whereas impurity of the peptide, a structural alteration of insulin in the powder aerosol during its preparation, a modification of the molecule by storage, and immunogenicity of excipients added to insulin are less likely, or were refuted (3, 81, 123, 124).
Lipodystrophy is a phenomenon observed in up to 30% of patients treated with subcutaneous injections of insulin developing at the site of injection and was firstly described by Lawrence 1925, i.e., soon after the introduction of insulin into clinical treatment (125-127). Lipohypertrophy is caused by the anabolic effect of insulin promoting the synthesis of protein and fat, whereas lipoatrophy is caused by an inflammatory process and is nowadays rarely observed because of the use of highly purified insulins (127). Since adipocytes are also located in the lung, inhaled insulin can also affect these cells after pulmonary deposition. However, at present it is not known if and how inhaled insulin affects pulmonary adipocytes (128).
Cough is a typical symptom in clinical treatment with inhalation of dry powder aerosols which might affect patient convenience and compliance. Therefore, cough was addressed in a number of studies investigating inhaled insulin. Mild to moderate cough was reported to occur rapidly after inhalation (seconds to minutes) in up to 20-30% of patients. However, the reported symptoms were transient, settled with continuation of the therapy and seldom resulted in treatment withdrawal (107, 108, 111, 112, 117-120).
Hypoglycemia is a common problem in patients treated with antidiabetics, especially insulin. Therefore, the evaluation of hypoglycemia incidence and severity was subject of many clinical studies investigating inhaled insulin. The data obtained in these studies are conflicting, demonstrating an increased frequency of severe hypoglycemic events in patients treated with inhaled insulin compared with patients treated with subcutaneous injections in some of these studies (115-120). However, there is no, or only little, difference regarding the risk for the occurrence of hypoglycemia between inhaled and subcutaneous insulin (129), whereas the risk is expectedly higher for patients treated with inhaled insulin when compared with treatment with oral antidiabetics (107, 108, 111, 112).
In the last decades of diabetic therapy, patient convenience and compliance were improved by development of smaller and sharper needles and pen injector systems and insulin pumps for injection. However, all these systems are based on needles and, therefore, are invasive. The development of inhaled insulin was an approach for a non-invasive insulin therapy in patients with diabetes types 1 and 2. A number of studies were performed to investigate patient convenience and improvement of life quality and improvement of metabolic control (determined by measurement of glycated haemoglobin (HbA1c)). In these studies, it was found that patients welcome the non-invasive alternative for administration of insulin by means of an inhaler, even though its handling (use and cleaning) requires a large number of steps (30, 110). Especially patients with diabetes type 2, who fail on oral antidiabetic therapy, and whose switch to insulin treatment is often delayed, and patients with needle phobia representing at least 10% of the population should profit from insulin inhalation (32, 128, 130). However, these advantages are opposed by a number of other arguments. In detail, because of the small bioavailability of inhaled insulin much higher doses must be administered than in conventional therapy by means of subcutaneous injection. In addition, the high costs for the development of inhalant therapy and the device were considered in the price of the product. Furthermore, the patients must be thoroughly trained prior to inhalant therapy and require controls of lung function before and under therapy. All these factors result in an extra cost between 600 and more than 1000 (currently £1
More than 80 years after the first experiments, insulin received the approval for inhalant administration (Exubera®). In addition, several other techniques for insulin aerosolization and aerosol application have been investigated and are in a different phases of their development. Shortly after its market launch, Exubera® has been stopped by the manufacturer (Pfizer), because of unexpectedly low sales. To our information, competitors have also put their developments on hold. During a short time on the market, Exubera® was accepted by patients (although not reimbursed by most health insurance companies) and well tolerated without adverse effects. However, up to now some questions are not completely answered, e.g., effects of long-time inhalation on lung function and effects of pulmonary diseases on deposition, absorption, and pharmacokinetics of inhaled insulin, or increased risk for lung cancer in ex-smokers. Under the current circumstances, it is unlikely that inhaled insulin will be relaunched in any formulation in the near future. However, if there is a relaunch, the unanswered questions should be subject to further investigation. The experience on insulin inhalation may help develop inhalation therapies for other compounds serving for the treatment of systemic diseases, because such type of treatment seems to be a safe and reliable technique for drug application improving the patient compliance due to its non-invasive character.
Conflict of interest: Dr. Rudiger Siekmeier has no conflicts of interest in relation to this article. Dr. Gerhard Scheuch does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. Dr. Scheuch is a consultant for several pharmaceutical companies in the field of aerosol medicine and pulmonary drug delivery (e.g., Bayer-Schering, Boehringer/Ingelheim, GSK, Novartis, Talecris, Sandoz).
- Agu RU, Ugwoke MI, Armand M, Kinget R, Verbeke N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir Res 2001; 2: 198-209.
- Siekmeier R, Scheuch G. Systemische Therapie mit Aerosolen Allgemeine Grundlagen zur pulmonalen Verabreichung von Makromolekulen zur systemischen Therapie. Atemw-Lungenkrkh 2005; 31: 374-386.
- Heinemann L, Pfutzner A, Heise T: Alternative routes of administration as an approach to improve insulin therapy: Update on dermal, oral, nasal and pulmonary insulin delivery. Curr Pharm Des 2001; 7: 1327-1351.
- Niven RW. Delivery of biotherapeutics by inhalation aerosol. Crit Rev Ther Drug Carrier Syst 1995; 12: 151-231.
- Patton JS, Byron PR. Inhaling medicines: Delivering drugs to the body through the lungs. Nat Rev Drug Discov 2007; 6: 67-74.
- Wolff RK. Safety of inhaled proteins for therapeutic use. J Aerosol Med 1998; 11: 197-219.
- Byron PR, Patton JS. Drug delivery via the respiratory tract. J Aerosol Med 1994; 7: 49-75.
- Kohler D, Fleischer W. Medikamente. In Theorie und Praxis der Inhalationstherapie. Köhler D, Fleischer W (eds). Arcis Verlag, Munchen 2000, pp. 71-99.
- Scheuch G, Kohlhaeufl MJ, Brand P, Siekmeier R. Clinical perspectives on pulmonary systemic and macromolecular delivery. Adv Drug Deliv Rev 2006; 58: 996-1008.
- Scheuch G, Siekmeier R. Novel approaches to enhance pulmonary delivery of proteins and peptides. J Physiol Pharmacol 2007; 58 (Suppl 5, Pt 2): 615-625.
- Yu J, Chien YW. Pulmonary drug delivery: Physiologic and mechanistic aspects. Crit Rev Ther Drug Carrier Syst 1997; 14: 395-453.
- Kohler D. Aerosols for systemic treatment. Lung 1990; 168 (Suppl): 677-684.
- Siekmeier R, Scheuch G. Systemische Therapie mit Aerosolen. Beispiele zur pulmonalen Verabreichung von Makromolekulen zur systemischen Therapie. Atemw-Lungenkrkh 2005; 31: 391-410.
- Davis SS, Illum L. Absorption enhancers for nasal drug delivery. Clin Pharmacokinet 2003; 42: 1107-1128.
- Hussain A, Arnold JJ, Khan MA, Ahsan F. Absorption enhancers in pulmonary protein delivery. J Control Release 2004; 94: 15-24.
- Song Y, Wang Y, Thakur R, Meidan VM, Michniak B. Mucosal drug delivery: Membranes methodologies, and applications. Crit Rev Ther Drug Carrier Syst 2004; 21: 195-256.
- Cryan SA. Carrier-based strategies for targeting protein and peptide drugs to the lungs. AAPS J 2005; 7: E20-E41.
- Owens DR. New horizons Alternative routes for insulin delivery. Nat Rev Drug Discov 2002; 1: 529-540.
- Wilde MI, McTavish D. Insulin lispro: A review of its pharmacological properties and therapeutic use in the management of diabetes mellitus. Drugs 1997; 54: 597-614.
- Cefalu WT. Concept, strategies, and feasibility of non-invasive insulin delivery. Diabetes Care 2004; 27: 239-246.
- Belmin J, Valensi P. Novel drug delivery systems for insulin. Clinical potential for use in the elderly. Drugs Aging 2003; 20: 303-312.
- Laqueur E, Grevenstuk A. Uber die Wirkung intratrachealer Zuführung von Insulin. Klin Wochenschr 1924; 3: 1273-1274.
- Heubner W, de Jongh SE, Laquer E. Uber Inhalation von Insulin. Klin Wochenschr 1924; 3: 2342-2343.
- Gansslen M. Uber Inhalation von Insulin. Klin Wochenschr 1925; 4: 71.
- Wigley FW, Londono JH, Wood SH, Ship JC, Waldman RH. Insulin across respiratory mucosae by aerosol delivery. Diabetes 1971; 20: 552-556.
- Elliott RB, Edgar BW, Pilcher CC, Quested C, McMaster J. Parenteral absorption of insulin from the lung in diabetic children. Aust Paediatr J 1987; 23: 293-297.
- Kohler D, Schlüter KJ, Kerp L, Matthys H. Nicht radioaktives Verfahren zur Messung der Lungenpermeabilität. Inhalation von Insulin. Atemw-Lungenkrkh 1987; 13: 230-232.
- Laube BL, Georgopoulos A, Adams GK. Preliminary study of the efficacy of insulin aerosol delivered by oral inhalation in diabetic subjects. JAMA 1993; 269: 2106-2109.
- Gonda I. The ascent of pulmonary drug delivery. J Pharm Sci 2000; 89: 940-945.
- Guntur VP, Dhand R. Inhaled insulin: Extending the horizons of inhalation therapy. Respir Care 2007; 52: 911-922.
- Charlish P. Therapeutic focus How would you like your insulin. BioPartnering Today 2006; 6-9.
- de Galan BE, Simsek S, Tack CJ, Heine RJ. Efficacy and safety of inhaled insulin in the treatment of diabetes mellitus. Neth J Med 2006; 64: 319-325.
- Laube BL. The expanding role of aerosols in systemic drug delivery, gene therapy, and vaccination. Respir Care 2005; 50: 1161-1176.
- Mastrandrea LD, Quattrin T. Clinical evaluation of inhaled insulin. Adv Drug Deliv Rev 2006; 58: 1061-1075.
- Pearson J. Inhalation technologies A breath of fresh air. Drug Delivery Report 2006; Spring/Summer: 19-21.
- Patton JS, Bukar JG, Eldon MA. Clinical pharmacokinetics and pharmacodynamics of inhaled insulin. Clin Pharmacokinet 2004; 43: 781-801.
- Sakagami M, Byron PR. Respirable microspheres for inhalation. The potential of manipulating pulmonary disposition for improved therapeutic efficacy. Clin Pharmacokinet 2005; 44: 263-277.
- Kawashima Y, Yamamoto H, Takeuchi H, Fujioka S, Hino T. Pulmonary delivery of insulin with nebulized DL-lactide/glycolide copolymer (PLGA) nanospheres to prolong hypoglycaemic effect. J Control Release 1999; 62: 279-287.
- Edwards DA, Hanes J, Caponetti G et al. Large porous particles for pulmonary drug delivery. Science 1997; 276: 1868-1871.
- Brown L, Rashba-Step J, Scott T. Pulmonary delivery of novel insulin microspheres. In Respiratory Drug Delivery XIII. Dalby RN, Byron PR, Peart J, Farr SJ (eds), Davis Horwood International Publishing, Raleigh NC, 2002, pp. 431-433.
- Surendrakumar K, Martyn GP, Hodgers ECM, Jansen M, Blair JA. Sustained release of insulin from sodium hyaluronate based dry powder formulations after pulmonary delivery to beagle dogs. J Control Release 2003; 91: 385-394.
- Pfutzner A, Mann AE, Steiner SS. Technosphere/Insulin a new approach for effective delivery of human insulin via the pulmonary route. Diabetes Technol Ther 2002; 4: 589-594.
- Steiner S, Pfutzner A, Wilson BR, Harzer O, Heinemann L, Rave K. Technosphere/Insulin proof of concept study with a new insulin formulation for pulmonary delivery. Exp Clin Endocrinol Diabetes 2002; 110: 17-21.
- Garcia-Contreras L, Morcol T, Bell SJD, Hickey AJ. Evaluation of novel particles as pulmonary delivery systems for insulin in rats. AAPS PharmSci 2003; 5: E9.
- Bhat M. Development of a novel spray-drying technique to produce particles for aerosol delivery. In Respiratory Drug Delivery XIII. Dalby RN, Byron PR, Peart J, Farr SJ (eds), Davis Horwood International Publishing, Raleigh NC, 2002, pp. 427-429.
- Blair J, Coghlan D, Langner E, Jansen M, Askey-Sarvar A. Sustained delivery of insulin via the lung using Solidose technology. In Respiratory Drug Delivery XIII. Dalby RN, Byron PR, Peart J, Farr SJ (eds), Davis Horwood International Publishing, Raleigh NC, 2002, pp. 411-414.
- Hrkash J, Batycky R, Chen D. AIR insulin: Complete diabetes therapy via inhalation of fast-acting and slow-acting dry powder aerosols. Diabetes 2000; 49: A9 (Abstract).
- Huang YY, Wang CH. Pulmonary delivery of insulin by liposomal carriers. J Control Release 2006; 113: 9-14.
- Rudt S, Müller R. in vitro phagocytosis assay of nanoparticles and microparticles by chemiluminescence. 1. Effect of analytical parameters, particle size and particle concentration. J Control Release 1992; 22: 263-271.
- Edwards DA, Dunbar C. Bioengineering of therapeutic aerosols. Annu Rev Biomed Eng 2002; 4: 93-107.
- Okamoto H, Todo H, Iida K, Danjo K. Dry powders for pulmonary delivery of peptides and proteins. Kona 2002; 20: 71-83.
- Johansson F, Hjertberg E, Eirefelt S, Tronde A, Hultkvist Bengtsson U. Mechanisms for absorption enhancement of inhaled insulin by sodium taurocholate. Eur J Pharm Sci 2002; 17: 63-71.
- Komada F, Iwakawa S, Yamamoto N, Sakakibara H, Okumura K. Intratracheal delivery of peptide and protein agents: Absorption from solution and dry powder by rat lung. J Pharm Sci 1994; 83: 863-867.
- Okumura K, Iwakawa S, Yoshida T, Seki T, Komada F. Intratracheal delivery of insulin. Absorption from solution and aerosol by rat lung. Int J Pharm 1992; 88: 63-73.
- Yamamoto A, Umemori S, Muranishi S. Absorption enhancement of intrapulmonary administered insulin by various absorption enhancers and protease inhibitors in rats. J Pharm Pharmacol 1994; 46: 14-18.
- Heinemann L, Klappoth W, Rave H, Hompesch B, Linkeschowa R, Heise T. Intra-individual variability of the metabolic effect of inhaled insulin together with an absorption enhancer. Diabetes Care 2000; 23: 1343-1347.
- Todo H, Okamoto H, Iida K, Danjo K. Effect of additives on insulin absorption from intratracheally administered dry powders in rats. Int J Pharm 2001; 220: 101-110.
- Li Y, Mitra AK. Effects of phospholipid chain length, concentration, charge, and vesicle size on pulmonary insulin absorption. Pharm Res 1996; 13: 76-79.
- Liu F, Shao Z, Kildsig DO, Mitra AK. Pulmonary delivery of free and liposomal insulin. Pharm Res 1993; 10: 228-232.
- Shao Z, Li Y, Mitra AK. Cyclodextrins as mucosal absorption promoters of insulin. III: Pulmonary route of delivery. Eur J Pharm Biopharm 1994; 40: 283-288.
- Shen ZC, Cheng Y, Zhang Q, Wei SL, Li RC, Wang K. Lanthanides enhance pulmonary absorption of insulin. Biol Trace Elem Res 2000; 75: 215-225.
- Garcia-Contreras L, Sarubbi D, Flanders E, Smart J, Newcomer C, Hicke AJ. Immediate and short-term cellular and biochemical responses to pulmonary single-dose studies of insulin and H-MAP. Pharm Res 2001; 18: 1685-1693.
- Suarez S, Garcia-Contreras L, Sarubbi D et al. Facilitation of pulmonary insulin absorption by H-MAP: Pharmacokinetics and pharmacodynamics in rats. Pharm Res 2001; 18: 1677-1684.
- Morimoto K, Uehara Y, Iwanaga K, Kakemi M. Effects of sodium glycocholate and protease inhibitors on permeability of TRH and insulin across rabbit trachea. Pharm Acta Helv 2000; 74: 411-415.
- Fukuda Y, Tsuji T, Fujita T, Yamamoto A, Muranishi S. Susceptibility of insulin to proteolysis in rat lung homogenate and its protection from proteolysis by various protease inhibitors. Biol Pharm Bull 1995; 18: 891-894.
- Sakagami M. Insulin disposition in the lung following oral inhalation in humans. A meta-analysis of its pharmacokinetics. Clin Pharmacokinet 2004; 43: 539-552.
- Farr SJ, McElduff A, Mather LE et al. Pulmonary insulin administration using the AERx system: Physiological and physicochemical factors influencing insulin effectiveness in healthy fasting subjects. Diabetes Technol Ther 2000; 2: 185-197.
- Perera AD, Kapitza C, Nosek L et al. and metabolic effect of inhaled insulin. Intrapatient variability after inhalation via the Aerodose insulin inhaler in patients with type 2 diabetes. Diabetes Care 2002; 25: 2276-2281.
- Heise T, Scharling B, Bellaire S. Dose-response of pulmonary insulin with the AERx insulin diabetes management system in healthy subjects. Diabetologia 2001; 44 (Suppl 2): A212 (Abstract).
- Fishman RS, Guinta D, Chambers F. Insulin administration via the AerodoseTM inhaler: Comparison to subcutaneously injected insulin. Diabetes 2000; 49 (Suppl 1): A9 (Abstract).
- Chien JY, Wise SD, Sathirakul K. Time action profile of inhaled insulin via Spiros dry powder inhaler is consistent among user inhalation techniques. Diabetologia 2001; 44 (Suppl 2): A211 (Abstract).
- Rave K, Muchmore D, Gonzales C. Inhaled insulin with an improved Spiros® dry powder inhaler: Dose response and time-action profiles. Diabetologia 2001; 44 (Suppl 2): A211 (Abstract).
- Steiner S, Rave KM, Heise T. Bioavailability and pharmacokinetic properties of inhaled dry powder Technosphere/Insulin. Diabetes 2000; 49 (Suppl 1): A126 (Abstract).
- Heinemann L, Traut T, Heise T. Time-action profile of inhaled insulin. Diabet Med 1997; 14: 63-72.
- Brunner GA, Balent B, Ellmerer M et al. Dose-response relation of liquid aerosol inhaled insulin in type I diabetic patients. Diabetologia 2001; 44: 305-308.
- Kim D, Mudaliar S, Plodkowski R. Dose-response relationships of inhaled and subcutaneous insulin in type 2 diabetic patients. Diabetes 2002; 51 (Suppl 2): A47 (Abstract).
- Rave KM, Heise T, Pfützner A. Results of a dose-response study with a new pulmonary insulin formulation and inhaler. Diabetes 2000; 49 (Suppl 1): A75 (Abstract).
- Barnett AH. Exubera inhaled insulin: A review. Int J Clin Pract 2004; 58: 394-401.
- Heinemann L, Weyer C, Rauhaus M, Heinrichs S, Heise T. Variability of the metabolic effect of soluble insulin and the rapid-acting insulin analog insulin aspart. Diabetes Care 1998; 21: 1910-1914.
- Himmelmann A, Jendle J, Mellen A, Petersen AH, Dahl UL, Wollmer P. The impact of smoking on inhaled insulin. Diabetes Care 2003; 26: 677-682.
- Valente AXCN, Langer R, Stone HA, Edwards DA. Recent advances in the development of an inhaled insulin product. Biodrugs 2003; 17: 9-17.
- Heinemann L. Variability of insulin absorption and insulin action. Diabetes Technol Ther 2002; 4: 673-682.
- Gelfand RA, Schwartz S, Horton M. Pharmacological reproducibility of inhaled human insulin pre-meal dosing in patients with type 2 diabetes mellitus (NIDDM). Diabetes 1998; 47 (Suppl 1): A99 (Abstract).
- Hompesch M, Kapitza C, Scharling B. Intra-subject variability of pulmonary insulin via the AERx® insulin diabetes management system versus subcutaneous insulin. Diabetologia 2001; 44 (Suppl 2): A212 (Abstract).
- Pfutzner A, Heise T, Steiner S. Inhaled Technosphere/Insulin shows a low variability in metabolic action in type 2 diabetic patients. Diabetes 2000; 49 (Suppl 1): A121 (Abstract).
- Sha S, Becker RHA, Willavise SA. The effect of smoking cessation on the absorption of inhaled insulin (Exubera®). Diabetes 2002; 51 (Suppl 2): A133 (Abstract).
- Becker RHA, Sha S, Frick AD, Fountaine RJ. The effect of smoking cessation and subsequent resumption on absorption of inhaled insulin. Diabetes Care 2006; 29: 277-282.
- McElduff A, Mather LE, Kam PC, Clauson P. Influence of acute upper respiratory tract infection on the absorption of inhaled insulin using the AERx® insulin Diabetes Management system. Br J Clin Pharmacol 2005; 59: 546-551.
- Henry RR, Mudaliar SRD, Howland WC et al. Inhaled insulin using the AERx insulin diabetes management system in healthy and asthmatic subjects. Diabetes Care 2003; 26: 764-769.
- Henry RR, Mudaliar S, Chu N et al. Young and elderly type 2 diabetic patients inhaling insulin with the AERx® insulin diabetes management system: A pharmacokinetic and pharmacodynamic comparison. J Clin Pharmacol 2003; 43: 1228-1234.
- Jones JG, Minty BD, Lawler P, Hulands GL, Crawley JCW, Veall N. Increased alveolar epithelial permeability in cigarette smokers. Lancet 1980; 1: 66-68.
- Minty BD, Jordan C, Jones JG. Rapid improvement in abnormal pulmonary epithelial permeability after stopping cigarettes. Br Med J 1981; 282: 1183-1186.
- Lehr HA. Microcirculatory dysfunction induced by cigarette smoking. Microcirculation 2000; 7: 367-384.
- Nieman GF, Clark WR, Paskanik AM, Bredenberg CE, Hakim TS. Unilateral smoke inhalation increases pulmonary blood flow to the injured lung. J Trauma 1994; 36: 617-623.
- Li XY, Rahman I, Donaldson K, MacNee W. Mechanisms of cigarette smoke induced increased airspace permeability. Thorax 1996; 51: 465-471.
- Schmekel B, Bos JA, Khan AR, Wohlfart B, Lachmann B, Wollmer W. Integrity of the alveolar-capillary barrier and alveolar surfactant system in smokers. Thorax 1992; 47: 603-608.
- Miller LG. Cigarettes and drug therapy: Pharmacokinetic and pharmacodynamic considerations. Clin Pharm 1990; 9: 125-135.
- Attvall S, Fowelin J, Lager I, von Schenck H, Smith U. Smoking induces insulin resistance: A potential link with the insulin resistance syndrome. J Intern Med 1992; 233: 327-332.
- Facchini FS, Hollenbeck CB, Jeppesen J, Chen YDI, Reaven GM. Insulin resistance and cigarette smoking. Lancet 1992; 339: 1128-1130.
- Nakanishi N, Nakamura K, Matsuo Y, Suzuki K, Tatara K. Cigarette smoking and risk for impaired fasting glucose and type 2 diabetes in middle-aged Japanese men. Ann Intern Med 2000; 133: 181-191.
- Targher G, Alberiche M, Zenere MB, Bonadonna RC, Muggeo M, Bonora E: Cigarette smoking and insulin resistance in patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1997; 82: 3619-3624.
- Klemp P, Staberg B, Madsbad S, Kolendorf K. Smoking reduces insulin absorption from subcutaneous tissue. Br Med J 1982; 284: 237.
- Fountaine R, Milton A, Checchio T et al. Acute passive cigarette smoke exposure and inhaled human insulin (Exubera) pharmacokinetics. Br J Clin Pharmacol 2008; 65: 864-870.
- Davis TME, Davis WA. An assessment of eligibility for inhaled insulin (Exubera). Diabetes Care 2007; 30: 360-361.
- Mudaliar S, Henry RR. Inhaled insulin in patients with asthma and chronic obstructive pulmonary disease. Diabetes Technol Ther 2007; 9 (Suppl 1): S83-S92.
- Rave K, de la Pena A, Tibaldi FS et al. AIR inhaled insulin in subjects with chronic obstructive pulmonary disease: Pharmacokinetics, glucodynamics, safety, and tolerability. Diabetes Care 2007; 30: 1777-1782.
- Barnett AH, Dreyer M, Lange P, Serdarevic-Pehar M on behalf of the Exubera Phase III Study Group. An open, randomized, parallel-group study to compare the efficacy and safety profile of inhaled human insulin (Exubera) with metformin as adjunctive therapy in patients with type 2 diabetes poorly controlled on a sulfonylurea. Diabetes Care 2006; 29: 1282-1287.
- Barnett AH, Dreyer M, Lange P, Serdarevic-Pehar M on behalf of the Exubera Phase III Study Group. An open, randomized, parallel-group study to compare the efficacy and safety profile of inhaled human insulin (Exubera) with glibenclamide as adjunctive therapy in patients with type 2 diabetes poorly controlled on metformin. Diabetes Care 2006; 29: 1818-1825.
- Barnett AH, Lange P, Dreyer M, Serdarevic-Pehar M on behalf of the Exubera® Phase 3 Study Group. Long-term tolerability of inhaled human insulin (Exubera®) in patients with poorly controlled type 2 diabetes. Int J Clin Pract 2007; 61: 1614-1625.
- Ceglia L, Lau J, Pittas AG. Meta-analysis: Efficacy and safety of inhaled insulin therapy in adults with diabetes mellitus. Ann Intern Med 2006; 145: 665-675.
- DeFronzo RA, Bergenstal RM, Cefalu WT et al. for the Exubera Phase III Study group. Efficacy of inhaled insulin in patients with type 2 diabetes not controlled with diet and exercise: A 12-week, randomized, comparative trial. Diabetes Care 2005; 28: 1922-1928.
- Rosenstock J, Zinman B, Murphy LJ et al. Inhaled insulin improves glycemic control when substituted for or added to oral combination therapy in type 2 diabetes: A randomized, controlled trial. Ann Intern Med 2005; 143: 549-558.
- Skyler JS, Jovanovic L, Klioze S, Reis J, Duggan W for the Inhaled Human Insulin Type 1 Diabetes Study Group. Two-year safety and efficacy of inhaled human insulin (Exubera) in adult patients with type 1 diabetes. Diabetes Care 2007; 30: 579-585.
- Black C, Cummins E, Royle P, Philip S, Waugh N. The clinical effectiveness and cost-effectiveness of inhaled insulin in diabetes mellitus: A systematic review and economic evaluation. Health Technol Assess 2007; 11 (33): 1-126.
- Cefalu WT, Skyler JS, Kourides IA et al. for the Inhaled Insulin Study Group. Inhaled human insulin treatment in patients with type 2 diabetes mellitus. Ann Intern Med 2001; 134: 203-207.
- Hermansen K, Ronnemaa T, Petersen AH, Bellaire S, Adamson U. Intensive therapy with inhaled insulin via the AERx insulin diabetes management system: A 12-week proof-of-concept trial in patients with type 2 diabetes. Diabetes Care 2004; 27: 162-167.
- Hollander PA, Blonde L, Rowe R et al. for the Exubera Phase III Study Group. Efficacy and safety of inhaled insulin (Exubera) compared with subcutaneous insulin therapy in patients with type 2 diabetes: Results of a 6-month, randomized, comparative trial. Diabetes Care 2004; 27: 2356-2362.
- Quattrin T, Belanger A, Bohannon NJV, Schwartz SL for the Exubera Phase III Study Group. Efficacy and safety of inhaled insulin (Exubera) compared with subcutaneous insulin therapy in patients with type 1 diabetes. Diabetes Care 2004; 27: 2622-2627.
- Skyler JS, Cefalu WT, Kourides IA et al. Efficacy of inhaled human insulin in type 1 diabetes mellitus: A randomised proof-of-concept study. Lancet 2001; 357: 331-335.
- Skyler JS, Weinstock RS, Raskin P et al. the Inhaled Insulin Phase III Type 1 Diabetes Study Group. Use of inhaled insulin in a basal/bolus insulin regimen in type 1 diabetic subjects: A 6-month, randomized, comparative trial. Diabetes Care 2005; 28: 1630-1635.
- NDA 21-868/EXUBERA: U.S. package insert, 2006. New York, NY, Pfizer Labs. Available from http://www.pfizer.com/pfizer/download/uspi_exubera.pdf.
- FDA MedWatch Alert. Exubera (insulin human rDNA origin) Inhalation Powder. Available from http://www.drugs.com/fda/exubera-insulin-human-rdna-origin-inhalation-powder-12372.html.
- Fineberg SE, Kawabata T, Finco-Kent D, Liu C, Krasner A. Antibody response to inhaled insulin in patients with type 1 or type 2 diabetes. An analysis of initial phase II and III inhaled insulin (Exubera) trials and a two-year extension trial. J Clin Endocrinol Metab 2005; 90: 3287-3394.
- Stoever JA, Palmer JP. Inhaled insulin and insulin antibodies: A new twist to an old debate. Diabetes Technol Ther 2002; 4: 157-161.
- Hauner H, Stockamp B, Haastert B. Prevalence of lipohypertrophy in insulin-treated diabetic patients and predisposing factors. Exp Clin Endocrinol Diabetes 1996; 104: 106-110.
- Lawrence RD. Local insulin reactions. Lancet 1925; 1: 1125-1126.
- Radermecker RP, Pierard GE, Scheen AJ. Lipodystrophy reactions to insulin. Effects of continuous insulin infusion and new insulin analogs. Am J Clin Dermatol 2007; 8: 21-28.
- Ghosh S, Collier A. Inhaled insulins. Postgrad Med J 2007; 83: 178-181.
- Royle P, Waugh N, McAuley L, McIntyre L, Thomas S. Inhaled insulin in diabetes mellitus. Cochrane Database Syst Rev 2004; CD003890.
- Hamilton JG. Needle phobia: A neglected diagnosis. J Fam Pract 1995; 41: 169-175.