Defective function of granulocytes has been implicated as an etiological factor of increased susceptibility to infections in diabetes mellitus (8, 9). Infection is a common and serious complication of diabetes mellitus and a well-recognized cause of morbidity and mortality (8, 9). It has been estimated that infections account for up to 22% of deaths of patients with diabetes (10). One explanation for this increased sensitivity to infections may be an impaired innate immune response in diabetic patients (11, 12). Neutrophils play an important role during the early host response to infection by a coordinated series of effector functions that include chemotaxis, phagocytosis, and the generation of reactive oxygen species (respiratory burst). Neutrophil chemotaxis is hampered in diabetic patients (8, 13). In addition, neutrophil phagocytic capacity of diabetic patients was reduced in some, but not all, investigations (9, 12).
A correct response of granulocytes to extrinsic stimuli depends on the function of cell signaling pathways (14, 15). Cytosolic free Ca2+ is believed to play a key role in the regulation of cellular function. Intracellular Ca2+ changes are essential for some enzymes in signal pathway activation. Therefore, monitoring its concentration could help in the assessment of stimuli-dependent granulocytes reactivity (16, 17). Neutrophil recruitment and activation in response to inflammatory stimuli is regulated through complex parallel and sequential events (18). The process begins after coupling of a specific ligand to the surface receptor, or by tight cell-cell contacts, and triggering a signaling cascade inside the cell. Molecular signals initiate directional cell movement, endocytosis, degranulation, superoxide generation, and chemiluminescence. All those functions initiated by chemotactic agonists, such as fMLP, are Ca2+-dependent (18, 19).
The aim of this study was to evaluated effects of insulin on non-activated and fMLP- induced intracellular Ca2+ changes in human neutrophils.
All enrolled subjects gave informed consent for participation in the study. The Ethics Committee of Warsaw Medical University in Warsaw, Poland approved the study protocol. Blood samples were collected from 30 healthy donors free from any metabolic and immunologically mediated disorders, aged 19-45 years (mean 29.9 ±7.2 years).
Neutrophils
Three milliliters of venous blood were taken from the ulnar vein to a tube containing heparin (10 U/ml). The blood count was determined using a Coulter HMX analyzer. The number of neutrophiles was identified microscopically in the blood smear after hematological staining. Neutrophils were isolated by 25 min centrifugation at 1200 x g on Gradisol G with d=1,115 ±0.002 g/cm3 (POLFA, Lodz, Poland) mixed with Histopaque in proportion (3:2). After isolation and final washing, cells were pelleted and resuspended in RPMI-1610 medium (Sigma Chemicals, Saint Louis, MO) and concentration was adjusted to 1-2 m/ml; 95% of the cells had the morphology of neutrophils. To the suspension of neutrophils, 5 µM of Fluo3 and Fura Red (Molecular Probes, Eugene, OR) were added and the aliquots were incubated in dark at 37°C. Then, the cells were washed and resuspended in RPMI-1610 at a concentration of 2 mln/ml and divided into three aliquots.
Flow cytometry
An analysis was performed on an FC500 flow cytometer (Beckman Coulter Hialeah, FL) according the method described earlier (16). Neutrophils were discriminated by flow cytometric measurements of cellular forward angle and right angle scatter. Fluo 3 and Fura Red were exited at 488 nm, Fluo 3 emission was detected at 515-535 nm and Fura Red emission was detected at 665-685 nm. The first 40 s of the analysis was considered as the initial, resting state. Then, the measurement was interrupted to add stimuli, after which it was continued for the next 60 s. As stimulants, 10-5 M fMLP (Sigma Chemicals) and 20 pM insulin (Bioton, Warsaw, Poland) were used. Ratio intensity of Fluo3/Fura Red vs. time was also calculated. To measure the effect of tyrosine kinase blockers on Ca2+ influx into the cells, tyrphostin 25 (100 nM) or genistein (100 nM) were added to examined samples.
Statistical analysis
Statistical analysis was performed with the use of Statistica 6.0 commercial package. Each result was calculated as a mean ±SD. Results were compared with the use of a non-parametric U Mann-Whitney test. The Spearman test was used for the analysis of correlations between different parameters. P<0.05 was considered statistically significant.
Isolated granulocytes were stimulated by fMLP or insulin alone, or by both substances added to the medium in combinations: fMLP + insulin (after 20 min) or insulin + fMLP (after 20 min). As a control, non-stimulated cells were used. fMLP evoked fast intracellular increase of free Ca2+ in neutrophils compared with the resting state (P<0.001). Similarly, the peak of fluorescence was significantly higher in neutrophils stimulated by insulin compared with control (P<0.001) (Table 1).
Insulin did not cause any changes in intracellular Ca2+, when added to the previously fMLP-stimulated cells (fMLP + insulin vs. fMLP) (Table 1). Prestimulation with insulin significantly decreased fMLP-induced intracellular free Ca2+ expressed as the Fluo3/Fura Red ratio compared with fMLP alone (P<0.01) (Table 1).
| Table 1. Fluorescence of granulocytes after fMLP and/or insulin stimulation. The results are expressed as means ±SE of fluorescence from Fluo 3, Fura Red, and index Fluo 3/ Fura Red (n = 20). |
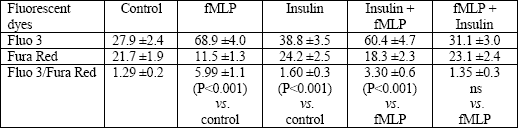 |
fMLP-induced emission of light in Ca2+-free medium was decreased (P<0.01) (data not shown). No relation between the initial intracellular Ca2+ in the resting state and the response to insulin was found (r=-0.19, P>0.05). Nor was the response to fMLP alone related to Ca2+ before stimulation (r=-0.256, P>0.05). A strong correlation was observed between the initial intracellular Ca2+ after incubation with insulin and the response to fMLP (r=0.90, P<0.0001, Fig. 1).
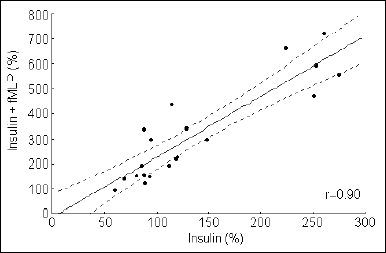 |
Fig. 1. Correlation between the initial intracellular Ca2+ level in granulocytes and the response to fMLP of insulin preincubated cells (n = 20, P<0.0001). The X and Y axis show a percent change in the Fluo 3/Fura Red index in samples with insulin and insulin plus fMLP. |
To explore the mechanism of intracellular Ca2+ concentration changes, the receptor signal transduction pathway was blocked by tyrosine kinase inhibitors: tyrphostin 25 and genistein. The tyrphostin 25 did not influence Ca2+ in control granulocytes but inhibited fMLP-induced Ca2+ increase, when added before fMLP (P<0.05) (Fig. 2). The tyrphostin added to the cells suspension after fMLP stimulation did not influence the Ca2+ level (Fig. 2).
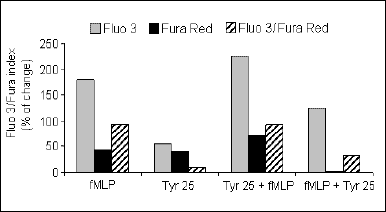 |
Fig. 2. Fluorescence of granulocytes after fMLP stimulation with and without the addition of tyrphostin 25. These results are means ± SE of the fluorescence values from Fluo 3, Fura Red and the Fluo 3/Fura Red index (n = 20). The results are expressed as a percent change in the Fluo 3/Fura index. |
In calcium-rich medium no correlation between the Ca2+ level and the response to fMLP after tyrphostin incubation was found (r=-0.3, P>0.05) (data not shown). In Ca2+-free medium, a strong relationship between the Ca2+ level and the response to fMLP after incubation with tyrphostin was found (r=0.92, P<0.001) (Fig. 3). The genistein did not influence the Ca2+ concentration in non-stimulated cells. However, it inhibited fMLP-induced Ca2+ increase, when added before fMLP (P<0.05) (Fig. 4). Genistein added to the suspension of cells after fMLP stimulation did not influence the Ca2+ level (Fig. 4).
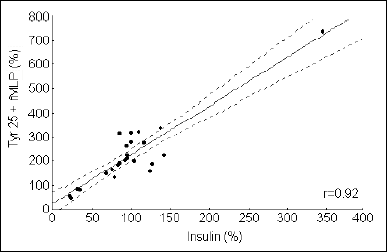 |
Fig. 3. Correlation between the initial intracellular Ca2+ level in granulocytes after incubation with tyrphostin 25 and fMLP stimulation, in Ca2+-free medium (n = 25, P<0.0001). The X and Y axis show a percent change in the Fluo 3/Fura Red index in samples with tyrphostin 25 and tyrphostin 25 plus fMLP. |
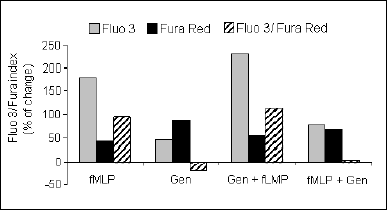 |
Fig. 4. Fluorescence of granulocytes after fMLP stimulation with and without the addition of genistein. These results are means ±SE of the fluorescence value from Fluo 3, Fura Red, and the Fluo 3/Fura Red index (n=20). The results are expressed as a percent change in the Fluo 3/Fura index. |
A positive correlation was found between the initial intracellular Ca2+ level and the response to fMLP of genistein-preincubated cells. This effect was seen in both Ca2+-rich (r=0.79, P<0.001 - data not shown), and Ca2+-free medium (r=0.75, P<0.001 - Fig. 5).
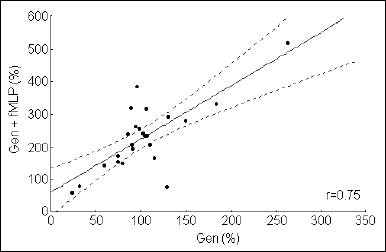 |
Fig. 5. Correlation between the initial intracellular Ca2+ level in granulocytes preincubated with genistein and fMLP stimulated, in Ca2+-free medium (n=25, P<0.0001). The X and Y axis show a percent change in the Fluo 3/Fura Red index in samples with genistein and genistein plus fMLP. |
The principal role of insulin under physiological conditions is to maintain metabolic homeostasis. Insulin exerts its effects through the insulin receptor (7, 20, 21). Signaling through this receptor is a complex process that involves activation of protein kinases and phosphatases (20, 21). The insulin receptor, which contains an intrinsic tyrosine kinase activity, undergoes tyrosyl autophosphorylation and is activated after insulin binding (22, 23). Insulin induces pleiotropic responses in many cell types, including granulocytes. Malfunctions in insulin signaling have been linked to increased susceptibility to infections. The development of these complications is dependent on the duration of diabetes and the quality of metabolic control. Type 2 diabetes has been associated with a number of neutrophil dysfunctions. Most investigations in type 2 diabetic patients have reported reduced neutrophil migration, phagocytic capacity, and respiratory burst (9, 11, 12). The present report was aimed to test the effects of insulin on resting and fMLP-induced intracellular Ca2+ concentration in human neutrophils, and to explore the mechanisms of those changes by blocking a potential pathway of signal transduction into these cells. The experiments were performed with the use of two fluorescent indicators Fluo3 and Fura Red. We showed that insulin increases the intracellular Ca2+ level in non-activated cells.
There are many studies regarding the involvement of Ca2+ in insulin signaling. However, large changes in intracellular levels of Ca2+ have not been reported in response to insulin (6). We did not observed dramatic changes of intracellular Ca2+ concentration after preincubation with insulin either. On the other hand, it is clear that an optimum Ca2+ concentration within cells is essential for insulin mediated events (5). We did not find any relationship between the initial intracellular Ca2+ concentration in the resting state and the response to insulin, although a strong correlation was observed between the initial intracellular Ca2+ after preincubation with insulin and the response to fMLP. It may partly explain how hyperinsulinemia impairs the innate response to infections. It has been suggested that small changes in Ca2+ concentration or Ca2+ fluxes may be important for insulin signaling (4, 5). We also observed dependency of fMLP response on the intracellular Ca2+ level in insulin-treated cells, but only when the response occurred in a Ca2+ free medium. In this situation, cytosolic Ca2+ increase derives only from the intracellular Ca2+ stores. Calcium can be mobilized through activation of inositol 1,4,5-trisphosphate receptors and ryanodine receptors. Both types of receptors can be gated by Ca2+ itself (25), resulting in release of Ca2+, a process called Ca2+-induced Ca2+ release (CICR). This process has been described in different cell types (26, 27). For example, an increase in Ca2+, as the underlying mechanism for the inotropic effect of insulin, has been demonstrated for rat heart preparations (28). To our knowledge, there are no reports assessing the effects of insulin on intracellular Ca2+ handling in human neutrophils. We confirmed the substantial influence of fMLP-stimulation on internal mobilization of Ca2+. This may indicate that calcium regulates respiratory burst in neutrophils after receptor-dependent stimulation. At first, an initial release of Ca2+ from an intracellular compartments and Ca2+ influx across the plasma membrane is observed (29). The final cellular response depends on a pathway of signal transduction, type the stimulator, and on the cell condition. We showed that bacterial peptide-induced granulocyte response is modified by insulin. This may explain neutrophil function impairment in type 2 diabetic patients.
The tyrphostins are a family of synthetic protein tyrosine kinase inhibitors that selectively inhibit receptor autophosphorylation (30) and represent an excellent tool to examine receptor function. It has been proposed that tyrphostins bind to the active center of the receptors, distorting it in such a way that in most cases neither the substrate nor ATP can bind to the receptor (31, 32). In this report, we evaluated tyrphostin for the inhibition of tyrosine kinase activity, to explore the mechanism of Ca2+ changes after cell stimulation. The tyrphostin 25 failed to alter intracellular Ca2+ in resting granulocytes, but significantly inhibited fMLP-induced Ca2+ increase, when added before fMLP.
Mobilization of Ca2+ is one of the early events triggered by binding of a chemoattractant to its receptor. The fMLP-stimulated neutrophil phosphorylation is dependent on phosphoinositide 3-kinase (PI3K), phospholipase D (PLD), and protein kinase C (PKC) activity (30-32). Two fMLP receptor subtypes have been identified in neutrophils, characterized by a distinct sensitivity to fMLP and antagonistic peptides. fMLP receptors involve an action of PI3K, PLD, and PKC isotypes. The possible mechanism may involve tyrosine kinase-mediated activation, which results in enhanced activation of Ca2+-dependent PKC by enhanced PLC activity, followed by intracellular Ca2+ release (29). In the light of this knowledge, it is not surprising that the tyrphostin 25, added to the cell suspension after fMLP stimulation, did not influence the Ca2+ level. In Ca2+-rich medium, no correlation between the intracellular Ca2+ level and the response to fMLP after incubation with tyrphostin was found. In many reports, tyrosine kinase signaling pathway was proved to play an important role in intracellular Ca2+ regulation. (33, 34). Tyrosine kinase inhibitors modulate influx of Ca2+ from extracellular space in fibroblasts and cardiac myocytes (33, 34). Tyrosine kinase inhibitors are believed to limit Ca2+ influx from extracellular space, but they mobilize Ca2+ from intracellular stores (33). In this report, we were not able to confirm this hypothesis. Our results indicate that blockade of tyrosine kinase signaling pathway depressed Ca2+ mobilization only from the intracellular pool and did not influence Ca2+ influx from outside the cell. The mechanism of this phenomenon has to be identified.
Genistein, a major soy isoflavone, is also a potent tyrosine kinase inhibitor (30). Genistein failed to alter the intracellular Ca2+ concentration in non-stimulated granulocytes, but decreased fMLP-induced calcium peak. Genistein added to the cell suspension after fMLP stimulation did not influence the Ca2+ level. Thus, genistein reproduced the tyrphostin inhibitory effects. Opposite to tyrphostin, irrespective of the presence or absence of Ca2+ in extracellular fluid, a positive correlation was found between the initial intracellular Ca2+ level and the response to fMLP in genistein-preincubated cells. That could be explained by suppression of a wider range of phosphorylation processes by genistein compared with tyrphostin. These differential responses may be attributable to the fact that genistein has some effect on other protein kinases and signal transduction systems besides tyrosine kinases, whereas tyrphostin selectively inhibits tyrosine kinases (35). Yang et al (36) have demonstrated variable inhibitory effects of genistein and tyrphostin. In guinea pig gastric muscle, genistein partially inhibits prostaglandin F2a-induced contraction, whereas tyrphostin has no effect; and in Ang II-stimulated muscle, both genistein and tyrphostin inhibit contraction by only 43% (36). Unlike the present findings and those of Yang et al (36), Marrero et al (37) recently reported that Ang II-mediated inositol triphosphate production, and probably Ca2+ level modulation are inhibited by genistein.
Our findings strongly argue for an acute effect of hyperinsulinemia on neutrophil functions that are considered important for antibacterial defense. Furthermore, the present study opens up the possibility that Ca2+ influx might be one of the mechanisms of insulin action on immune cells and that some of those processes are tyrosine kinase-related. To fully elucidate this phenomenon, further investigations are needed. We draw the following conclusions:
- Chronic hyperinsulinemia has an impact on neutrophil functions;
- The insulin effects at the cellular level are related to intracellular Ca2+ changes;
- The process of intracellular Ca2+ changes following insulin signaling is, at least partly, tyrosine kinase-related;
- Derangements in the concentration of intracellular Ca2+ may represent a link between the mechanisms of insulin resistance in diabetes.
Conflicts of interest: The authors declared no conflicts of interest regarding this work.
- Krogh-Madsen R, Plomgaard P, Keller P, Keller C, Pedersen BK. Insulin stimulates interleukin-6 and tumor necrosis factor-alpha gene expression in human subcutaneous adipose tissue. Am J Physiol Endocrinol Metab 2004; 286: E234-E238.
- Iida KT, Shimano H, Kawakami Y et al. Insulin up-regulates tumor necrosis factor-alpha production in macrophages through an extracellular-regulated kinase-dependent pathway. J Biol Chem 2001; 276: 32531-32537.
- Kirwan JP, Krishnan RK, Weaver JA et al. Human aging is associated with altered TNF-alpha production during hyperglycemia and hyperinsulinemia. Am J Physiol Endocrinol Metab 2001; 281: E1137-E1143.
- Clausen T, Elbrink J, Martin BR. Insulin controlling calcium distribution in muscle and fat cells. Acta Endocrinol 1974; 77: 137-143.
- Draznin B, Sussman K, Kao M et al. The existence of an optimal range of cytosolic free calcium for insulin-stimulated glucose transport in rat adipocytes. J Biol Chem 1987; 262: 14385-1488.
- Kelly KL, Deeney JT, Corkey BE. Cytosolic free calcium in adipocytes. Distinct mechanisms of regulation and effects on insulin action. J Biol Chem 1989; 264: 12754-12757.
- Worrall DS, Olefsky JM. The effects of intracellular calcium depletion on insulin signaling in 3T3-L1 adipocytes. Mol Endocrinol 2002; 16: 378-389.
- Geerlings SE, Hoepelman AI. Immune dysfunction in patients with diabetes mellitus (DM). FEMS Immunol Med Microbiol 1999; 26: 259-265.
- Delamaire M, Maugendre D, Moreno M, Le Goff MC, Allannic H, Genetet B. Impaired leucocyte functions in diabetic patients. Diabet Med 1997; 14: 29-34.
- Tater D, Tepaut B, Bercovici JP, Youinou P. Polymorphonuclear cell derangements in type I diabetes. Horm Metab Res 1987; 19: 642-647.
- Joshi N, Caputo GM, Weitekamp MR, Karchmer AW. Infections in patients with diabetes mellitus. N Engl J Med 1999; 341: 1906-1912.
- Marhoffer W, Stein M, Maeser E, Federlin K. Impairment of polymorphonuclear leukocyte function and metabolic control of diabetes. Diabetes Care 1992; 15: 256-260.
- Balasoiu D, van Kessel KC, van Kats-Renaud HJ et al. Granulocyte function in women with diabetes and asymptomatic bacteriuria. Diabetes Care 1997; 20: 392-395.
- Stegenga ME, van der Crabben SN, Dessing MC et al. Effect of acute hyperglycaemia and/or hyperinsulinaemia on proinflammatory gene expression, cytokine production and neutrophil function in humans. Diabet Med 2008; 25: 157-164.
- Popko K, Winklewski P, Jakubczak B, Wasilewski R, W¹sik M. Changes in intracellular Ca2+-free and calcium stored balance in children granulocytes after stimulation. Preliminary results. Central European Journal of Immunology 2003; 28: 62-66
- Tianhui Hu, Ling Bei, Zhong- Ming Qian, Xun Shen. Intracellular free calcium regulates the onset respiratory burst in human neutrophils activated by phorbol myristate acetate. Cell Signal 1999; 11: 355-360.
- Jakubczak B, Wasik M, Popko K, Demkow U. Kinetics of calcium ion concentration accompanying signal transduction in neutrophils from children with increased susceptibility to infections. J Physiol Pharmacol 2006; 57 Suppl 4: 131-137.
- Hu TH, Bei L, Qian ZM, Shen X. Intracellular free calcium regulates the onset of respiratory burst in human neutrophils activated by phorbol myristate acetate. Cell Signal 1999; 11: 355-360.
- Foyouzi-Youssefi R, Petersson F, Lew D, Krause KH, Nusse O. Chemoattractant-induced respiratory burst: increases in cytosolic Ca2+ concentrations are essential and synergize with a kinetically distinct second signal. J Biochem 1997; 322: 709-718.
- Vlahos CJ, Matter WF, Brown RF et al. Investigation of neutrophil signal transduction using a specific inhibitor of phosphatidylinositol 3-kinase. J Immunol 1995; 154: 2413-2422.
- Draznin B, Leitner JW, Sussman KE, Sherman NA. Insulin and glucose modulate protein kinase C activity in rat adipocytes. Biochem Biophys Res Commun 1988; 156: 570-575.
- Kim SK, Novak RF. The role of intracellular signaling in insulin-mediated regulation of drug metabolizing enzyme gene and protein expression. Pharmacol Ther 2007; 113: 88-120.
- Haber EP, Hirabara SM, Gomes AD et al. Palmitate modulates the early steps of insulin signalling pathway in pancreatic islets. FEBS Lett 2003; 1-3: 185-188.
- Shechter Y, Yaish P, Chorev M et al. Inhibition of insulin-dependent lipogenesis and anti-lipolysis by protein tyrosine kinase inhibitors. EMBO J 1989; 8: 1671-1676.
- Krogh-Madsen R, Moller K, Dela F et al. Effect of hyperglycemia and hyperinsulinemia on the response of IL-6, TNF-alpha, and FFAs to low-dose endotoxemia in humans. Am J Physiol Endocrinol Metab 2004; 286: E766-E772.
- Zucchi R, Ronca-Testoni S. The sarcoplasmic reticulum Ca2+ channel/ryanodine receptor: modulation by endogenous effectors, drugs and disease states. Pharmacol Rev 1997; 49: 1-51.
- Bruton JD, Lemmens R, Shi CL, Persson-Sjogren S et al. Ryanodine receptors of pancreatic ß-cells mediate a distinct context-dependent signal for insulin secretion. FASEB J 2003; 17: 301-303.
- Dyachok O, Tufveson G, Gylfe E. Ca2+-induced Ca2+ release by activation of inositol 1,4,5-trisphosphate receptors in primary pancreatic beta-cells. Cell Calcium 2004; 36: 1-9. 28.
- Ren J, Sowers JR, Walsh MF, Brown RA. Reduced contractile response to insulin and IGF-I in ventricular myocytes from genetically obese Zucker rats. Am J Physiol Heart Circ Physiol 2000; 279: H1708-H1714.
- Dewitt S, Laffafian I, Hallett MB. Phagosomal oxidative activity during ß2 integrin (CR3)-mediated phagocytosis by neutrophils is triggered by a non-restricted Ca2+ signal: Ca2+ controls time not space. J Cell Sci 2003; 116: 2857-2865.
- Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science 1995; 267: 1782-1788.
- Posner I, Engel M, Gazit A, Levitzki A. Kinetics of inhibition by tyrphostins of the tyrosine kinase activity of the epidermal growth factor receptor and analysis by a new computer program. Mol Pharmacol 1994; 45: 673-683.
- Zhu HL, Ito Y, Teramoto N. Effects of tyrosine kinase inhibitors on voltage-dependent Ba2+ currents in the guinea-pig gastric antrum. J Physiol Pharmacol. 2006; 57: 415-424.
- Salomonsson M, Arendshorst WJ. Effect of tyrosine kinase blockade on norepinephrine-induced cytosolic calcium response in rat afferent arterioles. Am J Physiol Renal Physiol 2004; 286: F866-F874.
- Lee KM, Toscas K, Villereal ML. Inhibition of bradykinin- and thapsigardin-induced Ca2+ entry by tyrosine kinase inhibitors. J Biol Chem 1993; 268: 9945-9948.
- Levitzki A, Gilon C. Tyrphostins as molecular tools and potential antiproliferative drugs. Trends Pharmacol Sci 1991; 12: 171-174.
- Yang S-G, Saifeddine M, Hollenberg MD. Tyrosine kinase inhibitors and the contractile action of epidermal growth factor-urogastrone and other agonists in gastric smooth muscle. Can J Physiol Pharmacol 1992; 70: 85-93.
- Marrero MB, Paxton WG, Duff JL, Berk BC, Bernstein KE. Angiotensin II stimulates tyrosine phosphorylation of phospholipase C- 1 in vascular smooth muscle cells. J Biol Chem 1994; 269: 10935-10939.