Patients with Wegeners granulomatosis, other vasculitides, and rheumatic diseases who are treated with non-steroidal anti-inflammatory drugs (NSAIDs) and steroidal anti-inflammatory ones are at increased risk of developing upper gastrointestinal mucosal damage (6-8). This gastrointestinal damage varies from gastritis and erosions to gastric and duodenal ulcers and their life-threatening complication (bleeding and perforation). The role of H. pylori in the pathogenesis of NSAIDs and steroid-inducted gastrointestinal lesions remains controversial. Much evidence has accumulated for a pathogenetic role of H. pylori in the development of chronic gastritis.
The present study was undertaken to verify the potential impact of H. pylori infection on gastroduodenal lesions in patients with Wegeners granulomatosis. We addressed this issue by attempting to correlate H. pylori infection with clinical symptoms, laboratory data, and treatment used in Wegeners granulomatosis.
The study was approved by a local Ethics Committee. Informed consent was obtained from all patients examined. The study was performed in 36 patients with pulmonary Wegeners granulomatosis (24 female, 12 male, mean age 44.3 years, from the Primary Systemic Outpatients Clinic Czerniakowski Hospital, Warsaw Medical University in Warsaw, Poland. Wegeners granulomatosis has been diagnosed between 1998 and 2007 on the grounds of usually standing criteria, i.e., the assessment of disease process progression, serological assessment (ANCA antibodies titers), and histopathological examination.
The serological test for detection of ANCA antibodies has been performed using the indirect immunofluorescence (IIF) and immunoensimatic methods (ELISA). Treatment was introduced after obtaining consent. The patients were treated with immunosuppressive drugs (prednisone and cyclophosphamide orally). All patients fulfilled the American College of Rheumatology classification criteria, the Chapel Hill Consensus Conference definition, and also the EUVAS ANCA-associated vasculitis definition for Wegeners granulomatosis. At the time of the study 34 patients were ANCA positive. The patients were under Outpatient Clinic care and received follow-up clinical, laboratory, pulmonary function, and chest radiographic examinations. The clinical scoring (BVAS-Wegeners granulomatosis index and DEI index) and endoscopic examination were the basis of this study. All patients were treated with a typical induction regiment consisting of prednisolone and cyclophosphamide orally for at least 2 month; 25 patients took a single oral dose of NSAIDs (ibuprofen or naproxen). Upper gastrointestinal endoscopy was performed, regardless of gastrointestinal symptoms, after patients permission. The endoscopic examination was performed with a gastroscope (GIF E or GIF V2 Olympus, Japan) by two endoscopists. During each endoscopic examination a total of seven biopsy specimens were obtained from the gastroduodenal mucosa: three specimens from the greater curvature, three from the antrum and one from the duodenum. Six specimens were used for histological investigations and one for a rapid urease test (CLO-test). Biopsy specimens for histology were fixed in 10% buffered formalin, embedded in paraffin, and sectioned at 5 µm. The sections were stained with hematoxylin-eosin and Giemsa stain for the histological detection of H. pylori. H. pylori infection was determined to be positive when CLO-test and histology examination revealed positive results. Otherwise, the patients were regarded as negative for H. pylori infection. Histological analysis of the background gastric mucosa was based on the updated Sydney system, according to the degree of atrophic changes: none, mild, moderate, or marked. In each case, the most marked degree of atrophic changes from, at least, the two specimens was regarded as a representative grade of destruction.
In addition, we analyzed: age, gender, gastrointestinal symptoms, disease activity (DEI, BVAS-Wegeners granulomatosis indexes), biochemical and serological data, duration of corticosteroids treatment, duration of NSAIDs treatment, and the type of gastroprotective drugs - histamine H2 (H2) receptor antagonists or proton pomp inhibitors (PPI).
Statistical analysis
The Mann-Whitney U and Fisher tests were used for comparison between groups. P<0.05 was considered to be statistically significant. Statistical elaboration was performed with an SAS commercial software.
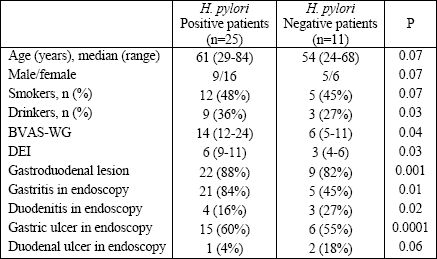
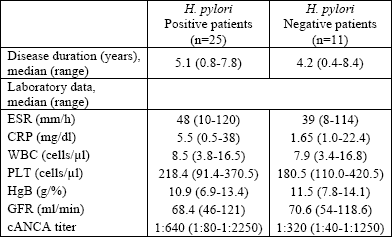
The overall prevalence of H. pylori infections in the Wegeners granulomatosis our patients was 71.4%; 25 patients were H. pylori positive and 11 patients were negative. Table 1 presents a comparison of clinical and endoscopy findings between H. pylori positive and negative patients. No gastric cancer was revealed. Table 2 summarizes the clinical features of Wegeners granulomatosis in both groups. Clinical activity (BVAS-Wegeners granulomatosis, DEI indexes) was higher in the H. pylori positive group. Laboratory parameters such as: WBC, ESR, CRP, PLT, and ANCA titers were all higher in this group. On the other side, hemoglobin concentration and the value of glomerular filtration rate were higher in the H. pylori negative group. Table 3 presents the medications used for Wegeners granulomatosis and the gastroprotective treatment in both group.
| Table 1. Comparison of baseline and endoscopy findings between H. pylori positive and negative patients with Wegeners granulomatosis. |
 |
| Table 2. Clinical and biochemical features in Wegeners granulomatosis patients. |
 |
| Table 3. Comparison of medications used between H. pylori positive and negative groups. |
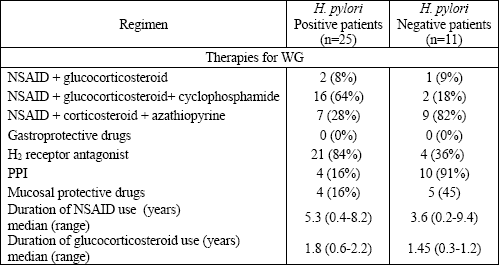 |
| PPI proton pump inhibitors. |
All patients from the H. pylori positive group received NSAIDs over a period of, at least, 1 month and H2 antagonist as a gastroprotective drug over a period of, at least, 1 month. Mild gastric mucosal atrophy in Wegeners granulomatosis patients with H. pylori positive infection was observed in 48.1% of patients, moderate in 35.3%, and marked in 7% of patients. In contrast, 92% of the patients negative for H. pylori infection had no gastric mucosal atrophy, and in the remaining negative for H. pylori patients, the signs of mucosal atrophy were never marked.
In the present study we observed that H. pylori infection caused clinical symptoms and gastroduodenal mucosal lesions in patients with Wegeners granulomatosis. The prevalence of H. pylori infection was 71.4% in Wegeners granulomatosis and 61.4% in rheumatoid arthritis patients (10). According to some epidemiological and clinical investigations, the difference in the prevalence of H. pylori infection among patients with Wegeners granulomatosis, with other types of vasculitis, and the general population is insignificant (7, 8). The possible interaction of NSAIDs and H. pylori in the pathogenesis of gastroduodenal lesions is still uncertain. NSAIDs can be a strong factor which causes the mucosal destruction, especially during concomitant treatment with steroids and cyclophosphamide, which was seen in the patients of this study. On the other side, there are reports that show that H. pylori infection does not influence the endoscopic grade of gastroduodenal mucosal lesions in patients using NSAIDs for a long time (9). A rapid development of selective COX-2 inhibitors (COXIB), as a safe alternative to NSAIDs might implications for the incidence of gastrointestinal damage. In fact, H. pylori infection might become the major cause of peptic ulceration in COXIB users.
In the present study, the incidence of dyspeptic syndrome and the intensity of gastroduodenal lesions were different between the H. pylori positive and negative groups, which is at variance with the rheumatoid arthritis patients described in a study by Ishikawa et al study (10). H. pylori infection contributes to the pathogenesis of gastroduodenal lesions and reflux oesophagitis in patients who receive NSAIDs. Zentilin et al (11) have reported the effect of H. pylori eradication on the severity of arthropathy in patient with rheumatoid arthritis. Ishikawa et al (10) have found no difference in the clinical severity of rheumatoid arthritis between H. pylori positive and negative patients, which is opposite to our present study.
A suppressive effect on H. pylori infection of glucocorticosteroids used in rheumatoid arthritis patients has been reported (12). Glucocorticosteroids might potentially act bacteriostatically against H. pylori, but our present results suggest that they do not decrease the prevalence of H. pylori infection in Wegeners granulomatosis patients. The reverse, the effect of drugs intended to eradicate H. pylori on the main disease activity has not been substantiated either (12).
Concluding, our results show that H. pylori infection seems associated with Wegeners granulomatosis activity and gastroduodenal mucosal lesions in patients who undergo a complex therapy with glucocorticosteroids, cyclophosphamide, and NSAIDs. There seems to be evidence to suggest the necessity for H. pylori eradication in patients with Wegeners granulomatosis, since the bacterium is a chronic source of infection during the disease course and can be a potential relapse factor (13-16).
Today, the standard triple therapy to eradicate H. pylori is amoxicillin, clarithromycin, and a proton pomp inhibitor (17, 18). A meta-analysis of randomized controlled trials suggests that supplementation with probiotics can improve eradication rates and reduce adverse events. Unfortunately, an increasing number of infected individuals are found with antibiotic-resistant bacteria. The resistance results in initial treatment failure and requires additional rounds of antibiotic therapy or alternative strategies (19, 20).
Conflicts of interest: No conflicts of interest were declared by the authors in regard to this work.
- Zycinska K, Wardyn KA, Zielonka TM, Otto M. The role of ANCA and anti-GBM antibodies in pulmonary-renal syndrom due to Wegener's granulomatosis. J Physiol Pharmacol 2007; 58 Suppl 5: 839-846.
- Pagnoux C, Teixeira L. Wegeners Granulomatosis. Presse Med 2007; 36: 860-874.
- Langford CA. Vasculitis in the geriatric population. Rheum Dis Clin North Am 2007; 33: 177-195.
- Taha AS, Sturrock RD, Russell RI. Helicobacter pylori and peptic ulcers in rheumatoid arthritis patients receiving gold, sulfasalazine and nonsteroidal anti-inflammatory drugs. Am J Gastroenterology 1992; 87: 1732-1735.
- Richter JE, Falk GW, Vaezi MF. Helicobacter pylori and gastroesophageal reflux disease: the bug may not be all bad. Am J Gastroenterology 1998; 93: 1800-1802.
- Apan TZ, Görsel R, Dolgun A. Increased seropositivity of Helicobacter pylori cytotoxin-associated gene-A in Behçets disease. Clin Rheumatol 2007; 26: 885-889.
- Gasbarrini A, Carloni E, Gasbarrini G, Chisholm SA. Helicobacter pylori and extragastric diseases-other Helicobacters. Helicobacter 2004; 9 Suppl 1: 57-66.
- Novak J, Szekenecz Z, Sebesi J et al. Elevated levels of anti-Helicobacter pylori antibodies in Henoch-Schönlein purpura. Autoimmunity 2003; 36: 307-311.
- de Leest HTJI, Steen KSS, van Wijngaarden S, Lems WF, van der Laar MAFJ, Dijkmans BAC. The prevalence of Helicobacter pylori is still substantial in rheumatic patients. Scan J Rheumatology 2002; 31: 94-96.
- Ishikawa N, Fuchigami T, Matsumoto T et al. Helicobacter pylori infection in rheumatoid arthritis: effect of drugs on prevalence and correlation with gastroduodenal lesion. Rheumatology 2002; 41: 72-77.
- Zentilin P, Savarino V, Garnero A, Accardo S, Seriolo B. Is Helicobacter pylori infection a risk factor for disease severity in rheumatoid arthritis? Gastroenterology 1999; 116: 503-504.
- Mizokami Y, Tamura K, Fukada Y, Yamamoto I, Shimoyama T. Non-steroidal anti-inflammatory drugs associated with gastroduodenal injury and Helicobacter pylori. Eur J Gastroenterol Hepatol 1994; 6 (Supp 1): 109-112.
- Bacon PA. The spectrum of Wegeners granulomatosis and disease relapse. N Engl J Med 2005; 352: 330-333.
- Nikkari S, Relman DA. Molecular approaches for identification of infectious agents in Wegeners granulomatosis and other vasculitides. Curr Opin Rheumatol 1999; 11: 11-16.
- Guilpain P, Guillevin L, Mouthon L. New insights into pathogenesis of ANCA-associated vasculitides. Presse Med 2007; 36: 854-859.
- Egermayer P. The role of infectious agents in pulmonary and systemic vascular disease. Expert Opin Pharmacother 2001; 2: 1093-1097.
- Tong JL, Ran ZH, Shen J, Zhang CX, Xiao SD. Meta-analysis: the effect of supplementation with probiotics on eradication rates and adverse events during Helicobacter pylori eradication therapy. Aliment Pharmacol Ther 2007; 25: 155-168.
- van Zanten SV, van der Knoop B. Gastric ulcer treatment: cure of Helicobacter pylori infection without subsequent acid-suppressive therapy: is it effective? Eur J Gastroenterol Hepatol 2008; 20: 489-491.
- Robinson K, Kenefeck R, Pidgeon E et al. Helicobacter pylori-induced peptic ulcer disease in associated with inadequate regulatory T-cell responses. Gut 2008; 57: 1375-1385.
- Umit H, Tezel A, Bukavaz S. The relationship between virulence factors of Helicobacter pylori and severity of gastritis in infected patients. Dig Dis Sci 2008 May 9 (Epub ahead of print).